Antioxidant Effect of Lycium barbarum Leaf through Inflammatory and Endoplasmic Reticulum Stress Mechanism



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Liquid Chromatography (LC)–Mass Spectrometry
2.3. Cell Culture
2.4. Experimental Animals
2.4.1. Mice
2.4.2. Zebrafish
2.5. Cell Viability Assay
2.6. Nitric Oxide (NO) Assay
2.7. Paracellular Permeability
2.8. RNA Extraction and cDNA Synthesis
2.9. Real-Time Polymerase Chain Reaction
2.10. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
2.11. Enzyme-Linked Immunosorbent Assay (ELISA)
2.12. Tissue Histology
2.13. Flow Cytometry for Cell Cycle
2.14. Statistical Analysis
3. Results
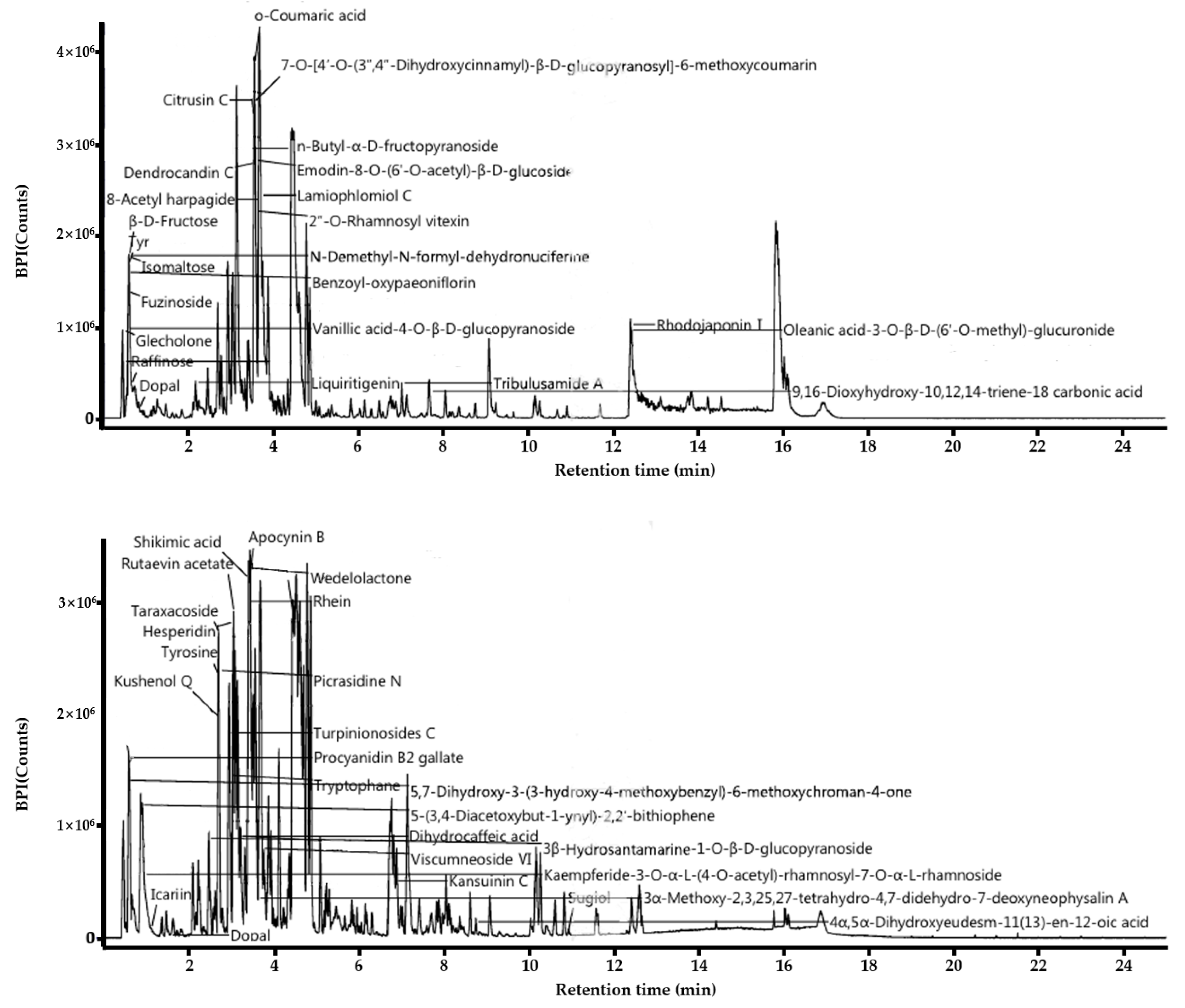
3.1. LC-MS Analysis of L. barbarum Leaf
3.2. In Vitro and In Vivo Toxicity Assessment
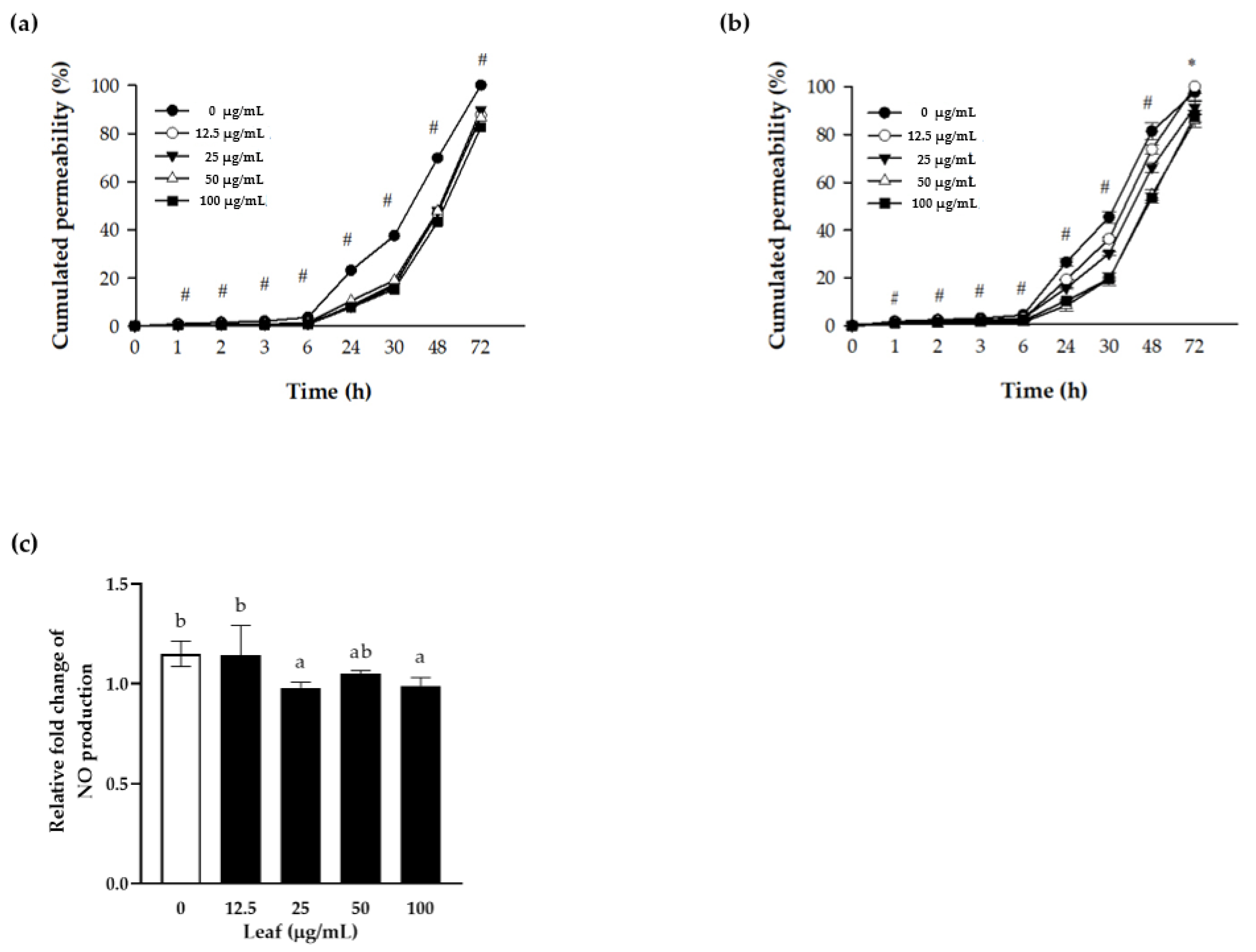
3.3. Nitric Oxide (NO) Production and Paracellular Permeability of Polarized Caco-2 Cells
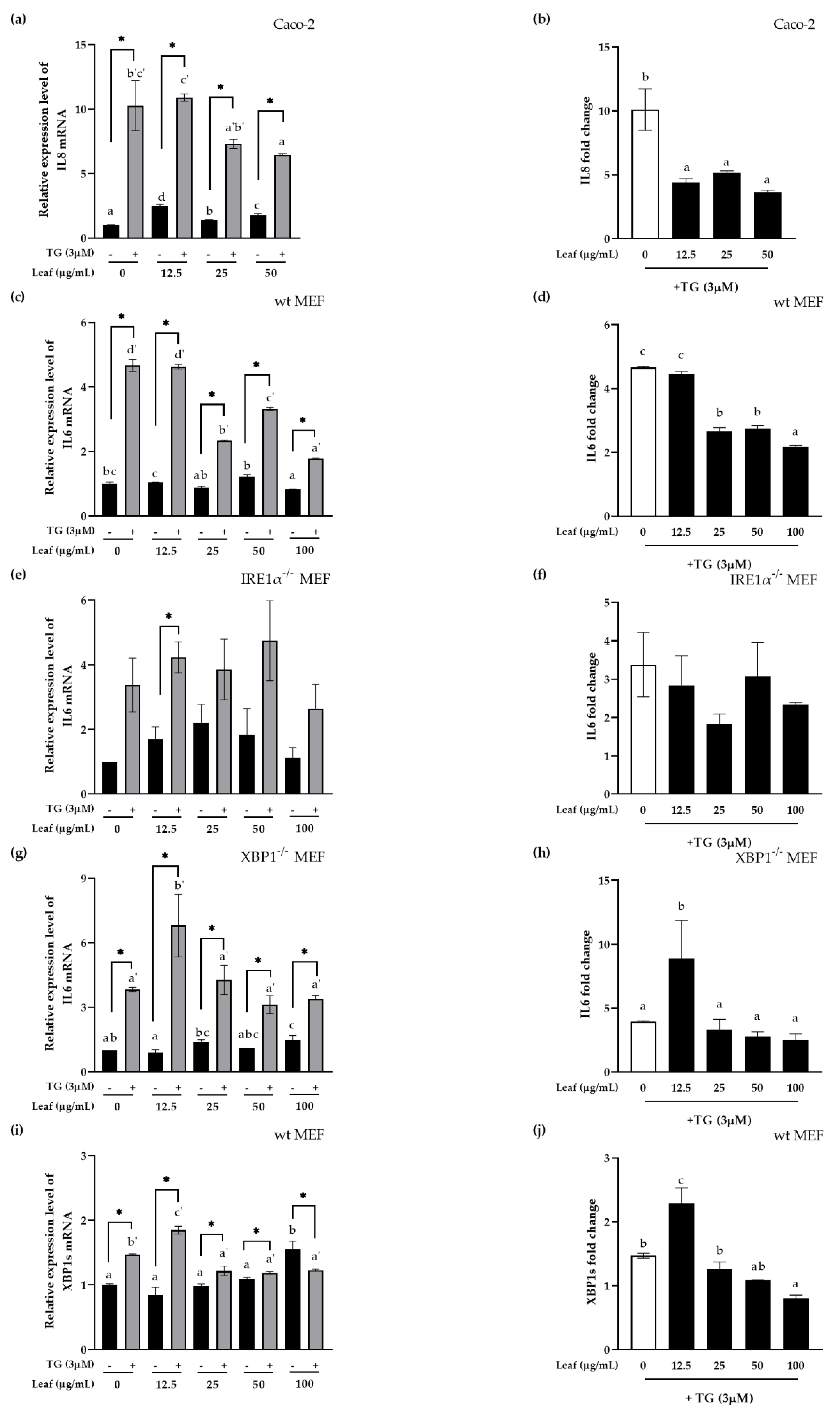
3.4. Inhibitory Effect of LL on Inflammatory Response and XBP1 Splicing In Vitro
3.5. Inhibitory Effect of LL on Inflammatory Response and ER Stress In Vivo
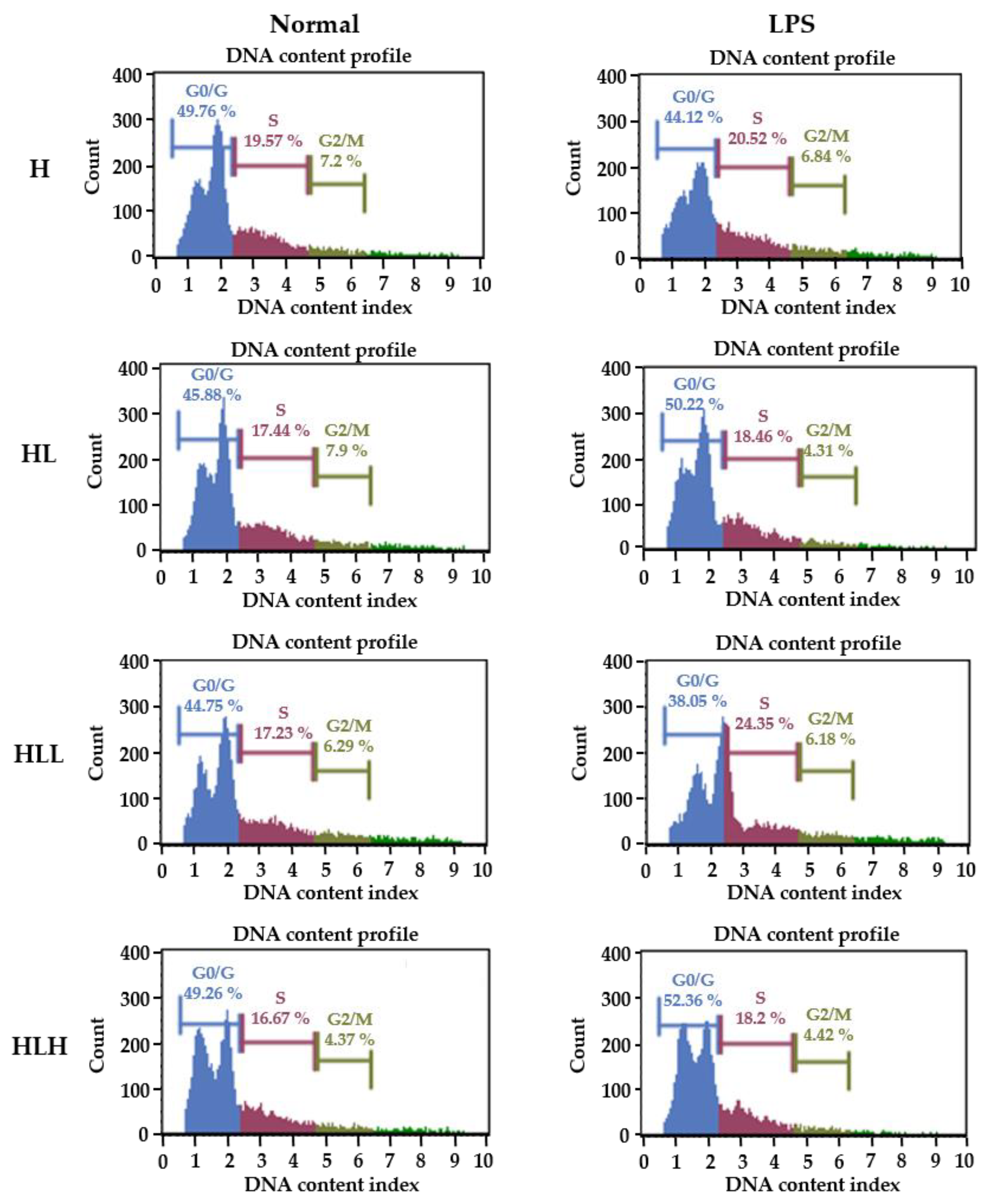
3.6. LPS-Induced Splenocytes Proliferation In Vivo
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, K. Integration of ER stress, oxidative stress and the inflammatory response in health and disease. Clin. Exp. Med. 2010, 3, 33. [Google Scholar]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA 2008, 105, 18525–18530. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J.J.N. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef]
- Popat, A.; Patel, A.; Warnes, G. A flow cytometric study of ER stress and autophagy. Cytom. A 2019, 95, 672–682. [Google Scholar] [CrossRef]
- Okamoto, R.; Watanabe, M. Role of epithelial cells in the pathogenesis and treatment of inflammatory bowel disease. J. Gastroenterol. 2016, 51, 11–21. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.J.C. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Bettigole, S.E.; Lis, R.; Adoro, S.; Lee, A.-H.; Spencer, L.A.; Weller, P.F.; Glimcher, L. The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat. Immunol. 2015, 16, 829–837. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef]
- Surawicz, C.M.; Haggitt, R.C.; Husseman, M.; McFarland, L.V. Mucosal biopsy diagnosis of colitis: Acute self-limited colitis and idiopathic inflammatory bowel disease. Gastroenterology 1994, 107, 755–763. [Google Scholar] [CrossRef]
- Reif, S.; Klein, I.; Lubin, F.; Farbstein, M.; Hallak, A.; Gilat, T. Pre-illness dietary factors in inflammatory bowel disease. Gut 1997, 40, 754–760. [Google Scholar] [CrossRef]
- Larabi, A.; Barnich, N.; Nguyen, H. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef]
- Birchenough, G.M.; Johansson, M.E.; Gustafsson, J.K.; Bergström, J.H.; Hansson, G. New developments in goblet cell mucus secretion and function. Mucosal Immunol. 2015, 8, 712–719. [Google Scholar] [CrossRef]
- Povolo, C.; Foschini, A.; Ribaudo, G. Optimization of the extraction of bioactive molecules from Lycium barbarum fruits and evaluation of the antioxidant activity: A combined study. Nat. Prod. Res. 2019, 33, 2694–2698. [Google Scholar] [CrossRef]
- Mocan, A.; Vlase, L.; Vodnar, D.C.; Bischin, C.; Hanganu, D.; Gheldiu, A.-M.; Oprean, R.; Silaghi-Dumitrescu, R.; Crișan, G.J.M. Polyphenolic content, antioxidant and antimicrobial activities of Lycium barbarum L. and Lycium chinense Mill. leaves. Molecules 2014, 19, 10056–10073. [Google Scholar] [CrossRef]
- Hempel, J.; Schädle, C.N.; Sprenger, J.; Heller, A.; Carle, R.; Schweiggert, R. Ultrastructural deposition forms and bioaccessibility of carotenoids and carotenoid esters from goji berries (Lycium barbarum L.). Food Chem. 2017, 218, 525–533. [Google Scholar] [CrossRef]
- Lin, C.; Wang, C.; Chang, S.; Inbaraj, B.S.; Chen, B.J. Antioxidative activity of polysaccharide fractions isolated from Lycium barbarum Linnaeus. Int. J. Biol. Macromol. 2009, 45, 146–151. [Google Scholar] [CrossRef]
- Li, X.; Ma, Y.; Liu, X. Effect of the Lycium barbarum polysaccharides on age-related oxidative stress in aged mice. J. Ethnopharmacol. 2007, 111, 504–511. [Google Scholar] [CrossRef]
- Zou, S.; Zhang, X.; Yao, W.; Niu, Y.; Gao, X. Structure characterization and hypoglycemic activity of a polysaccharide isolated from the fruit of Lycium barbarum L. Carbohydr. Polym. 2010, 80, 1161–1167. [Google Scholar] [CrossRef]
- Wawruszak, A.; Czerwonka, A.; Okła, K.; Rzeski, W. Anticancer effect of ethanol Lycium barbarum (Goji berry) extract on human breast cancer T47D cell line. Nat. Prod. Res. 2016, 30, 1993–1996. [Google Scholar] [CrossRef]
- Pricolo, V.E.; Madhere, S.M.; Finkelstein, S.D.; Reichner, J. Effects of lambda-carrageenan induced experimental enterocolitis on splenocyte function and nitric oxide production. J. Surg. Res. 1996, 66, 6–11. [Google Scholar] [CrossRef]
- Hsu, C.-L.; Huang, S.-L.; Yen, G.-C. Inhibitory effect of phenolic acids on the proliferation of 3T3-L1 preadipocytes in relation to their antioxidant activity. J. Agric. Food Chem. 2006, 54, 4191–4197. [Google Scholar] [CrossRef]
- Hwang, Y.-J.; Nam, S.-J.; Chun, W.; Kim, S.I.; Park, S.C.; Kang, C.D.; Lee, S. Anti-inflammatory effects of apocynin on dextran sulfate sodium-induced mouse colitis model. PLoS ONE 2019, 14, e0217642. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, C.-L.; Wang, Y.-J.; Guo, S.-X.; Yang, J.-S.; Chen, X.-M.; Xiao, P.-G. Three new bibenzyl derivatives from Dendrobium candidum. Chem. Pharm. Bull. 2009, 57, 218–219. [Google Scholar] [CrossRef] [PubMed]
- Ninfali, P.; Antonini, E.; Frati, A.; Scarpa, E.S. C-glycosyl flavonoids from beta vulgaris cicla and betalains from beta vulgaris rubra: Antioxidant, anticancer and antiinflammatory activities—A review. Phytother. Res. 2017, 31, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Matsui, R.; Sugiyama, M.; Asami, T.; Inaba, C.; Kobayashi, S.; Makabe, H.; Tanaka, S. Procyanidin B2 gallate regulates TNF-α production from T cells through inhibiting glycolytic activity via mTOR-HIF-1 pathway. Biochem. Pharmacol. 2020, 177, 113952. [Google Scholar] [CrossRef]
- Yang, J.; Shimogonya, Y.; Ishikawa, T. Mixing and pumping functions of the intestine of zebrafish larvae. J. Theor. Biol. 2017, 419, 152–158. [Google Scholar] [CrossRef]
- Vliegenthart, A.B.; Tucker, C.S.; Del Pozo, J.; Dear, J. Zebrafish as model organisms for studying drug-induced liver injury. Br. J. Clin. Pharmacol. 2014, 78, 1217–1227. [Google Scholar] [CrossRef]
- Kaser, A.; Lee, A.-H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession Number | Primer | Sequence (5′ → 3′) |
|---|---|---|---|
| Human | |||
| GAPDH | NM_001357943.2 | Forward | ATG GGG AAG GTG AAG GTC G |
| Reverse | GGG GTC ATT GAT GGC AAC AAT A | ||
| IL8 | NM_001354840.3 | Forward | GGG GTC ATT GAT GGC AAC AAT A |
| Reverse | CAT GAA GTG TTG AAG TAG ATT TGC TTG | ||
| Mouse | |||
| β-actin | NM_007393.5 | Forward | TAC CAC CAT GTA CCC AGG CA |
| Reverse | CTC AGG AGG AGC AAT GAT CTT GAT | ||
| XBP1s | NM_001271730.1 | Forward | ACA CGC TTG GGA ATG GAC AC |
| Reverse | CCA TGG GAA GAT GTT CTG GG | ||
| IL6 | NM_001314054.1 | Forward | CTG CAA GAG ACT TCC ATC CAG |
| Reverse | AGT GGT ATA GAC AGG TCT GTT GG | ||
| IL1β | NM_008361.4 | Forward | GAAATGCCACCTTTTGACAGTG |
| Reverse | TGGATGCTCTCATCAGGACAG | ||
| CXCL1 | NM_008176.3 | Forward | ACT GCA CCC AAA CCG AAG TC |
| Reverse | TGG GGA CAC CTT TTA GCA TCT T | ||
| IL4 | NM_021283.2 | Forward | ACA GGA GAA GGG ACG CCA T |
| Reverse | GAA GCC CTA CAG ACG AGC TCA | ||
| IL12p40 | NM_001303244.1 | Forward | AGC AGT AGC AGT TCC CCT GA |
| Reverse | AGT CCC TTT GGT CCA GTG TG | ||
| IFNγ | NM_008337.4 | Forward | TCA AGT GGC ATA GAT GTG GAA GAA |
| Reverse | TGG CTC TGC AGGATTTTCATG |
| Gene | Accession Number | Primer | Sequence (5′ → 3′) | Base Pair (bp) |
|---|---|---|---|---|
| β-actin | NM_007393.5 | Forward | TCTCCAGCAACGAGGAGAAT | 348 |
| Reverse | TGTGATCTGAAACCTGCTGC | |||
| IL6 | NM_001314054.1 | Forward | CCGGAGAGGAGACTTCACAG | 421 |
| Reverse | GGAAATTGGGGTAGGAAGGA | |||
| BiP | NM_001163434.1 | Forward | CTG GGT ACA TTT GAT CTG ACT GG | 398 |
| Reverse | GCA TCC TGG TGG CTT TCC AGC CAT TC | |||
| CHOP | NM_007837.4 | Forward | CAC ATC CCA AAG CCC TCG CTC TC | 286 |
| Reverse | TCA TGC TTG GTG CAG GCT GAC CAT |
| Group. | Percentage of Cell Counts in Different Phases (%) | |||||
|---|---|---|---|---|---|---|
| Non-Stimulation | LPS Stimulation | |||||
| G0/G1 | S | G2/M | G0/G1 | S | G2/M | |
| H | 48.47 ± 3.28 | 19.08 ± 0.95 b | 5.73 ± 0.66 b | 46.14 ± 1.89 a | 20.02 ± 1.46 a | 6.5 ± 0.7 c |
| HL | 48.47 ± 2 | 18.47 ± 1.5 b | 5.25 ± 0.62 b | 47.27 ± 2.43 ab | 18.63 ± 1.65 ab | 5.25 ± 0.68 ab |
| HLL | 48.47 ± 3.7 | 16.23 ± 1.9 a | 5.01 ± 1.09 b | 44.99 ± 4.59 a | 19.92 ± 2.44 b | 5.8 ± 0.78 bc |
| HLH | 48.47 ± 2.3 | 16.13 ± 1.5 a | 4.26 ± 0.54 a | 49.99 ± 3.19 b | 16.73 ± 2.1 b | 4.64 ± 1.12 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.R.; An, M.-Y.; Hwang, H.-J.; Yoon, J.-G.; Cho, J.A. Antioxidant Effect of Lycium barbarum Leaf through Inflammatory and Endoplasmic Reticulum Stress Mechanism. Antioxidants 2021, 10, 20. https://doi.org/10.3390/antiox10010020
Lee SR, An M-Y, Hwang H-J, Yoon J-G, Cho JA. Antioxidant Effect of Lycium barbarum Leaf through Inflammatory and Endoplasmic Reticulum Stress Mechanism. Antioxidants. 2021; 10(1):20. https://doi.org/10.3390/antiox10010020
Chicago/Turabian StyleLee, So Rok, Mi-Yeong An, Hye-Jeong Hwang, Ju-Gyeong Yoon, and Jin Ah Cho. 2021. "Antioxidant Effect of Lycium barbarum Leaf through Inflammatory and Endoplasmic Reticulum Stress Mechanism" Antioxidants 10, no. 1: 20. https://doi.org/10.3390/antiox10010020
APA StyleLee, S. R., An, M.-Y., Hwang, H.-J., Yoon, J.-G., & Cho, J. A. (2021). Antioxidant Effect of Lycium barbarum Leaf through Inflammatory and Endoplasmic Reticulum Stress Mechanism. Antioxidants, 10(1), 20. https://doi.org/10.3390/antiox10010020