Simultaneous Quantification of Antioxidants Paraxanthine and Caffeine in Human Saliva by Electrochemical Sensing for CYP1A2 Phenotyping


 , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Apparatus
2.3. Preparation of Working Standards
2.4. Electrochemical Measurements
2.5. Assay Validation for Real Application
2.6. Clinical Study
2.7. Data Analysis and Statistical Evaluation
3. Results
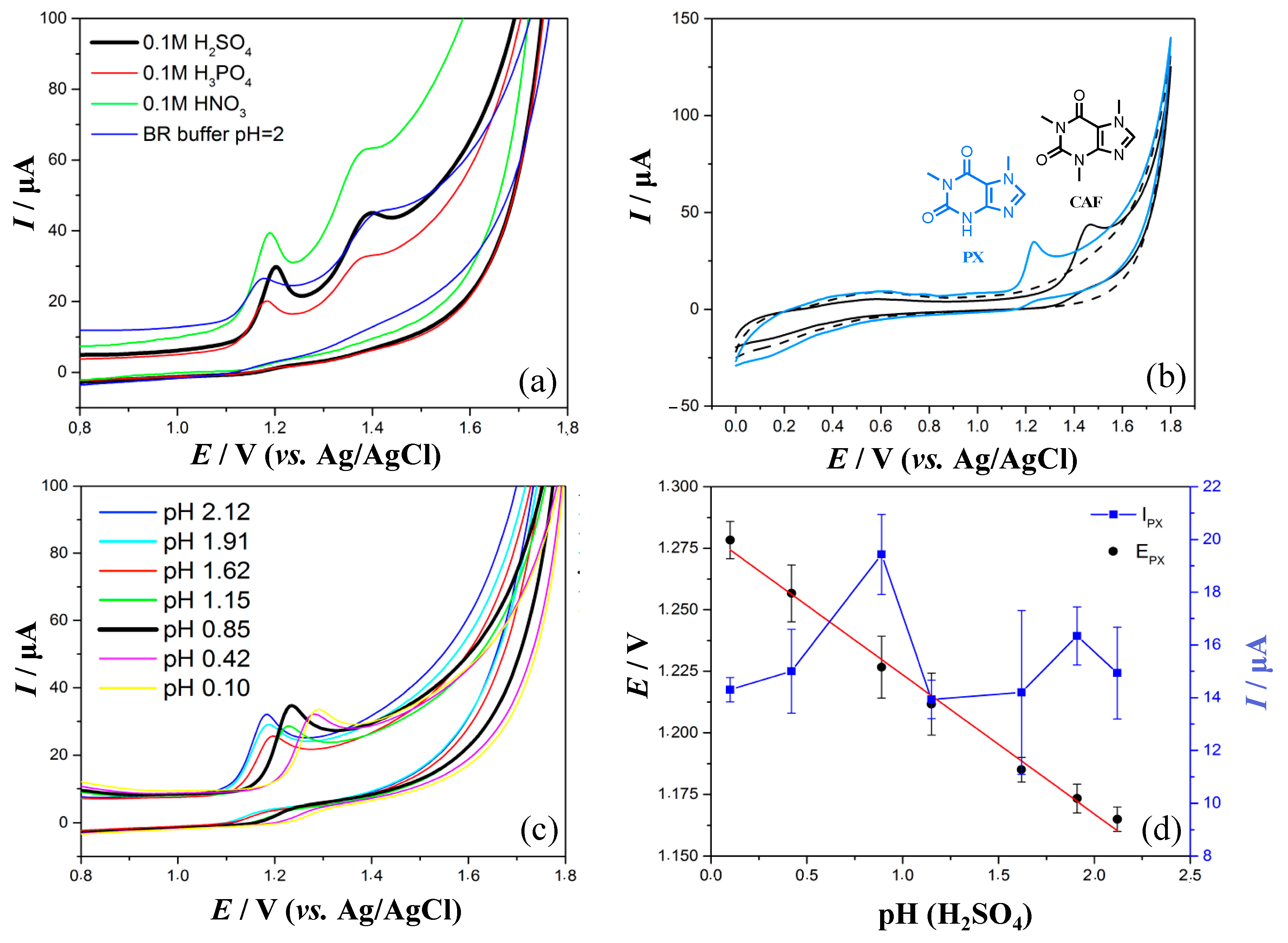
3.1. Influence of Supporting Electrolyte on Voltammetric Behavior of PX and CAF
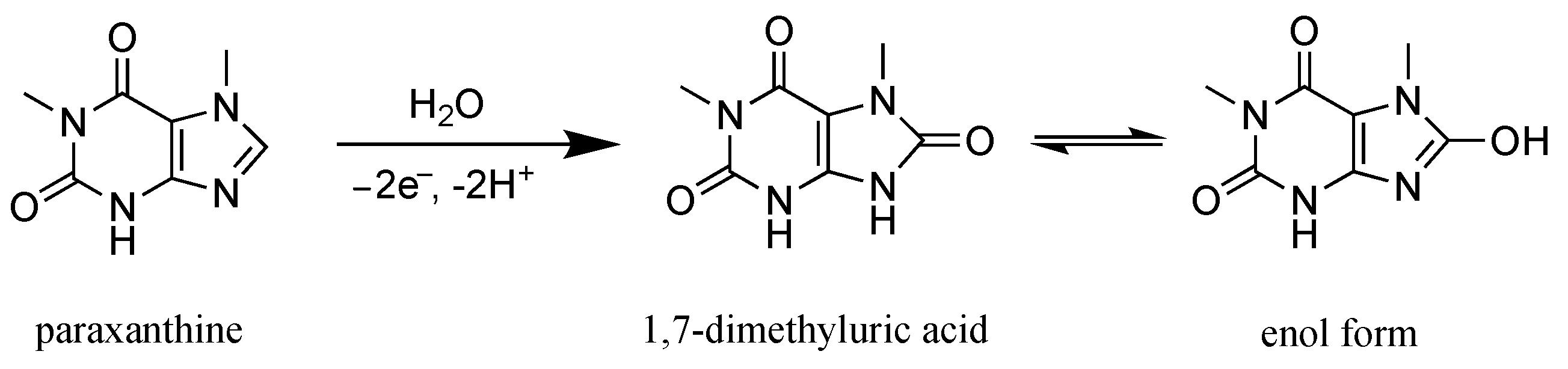
3.2. Oxidation Mechanism of Paraxanthine on the GC Electrode
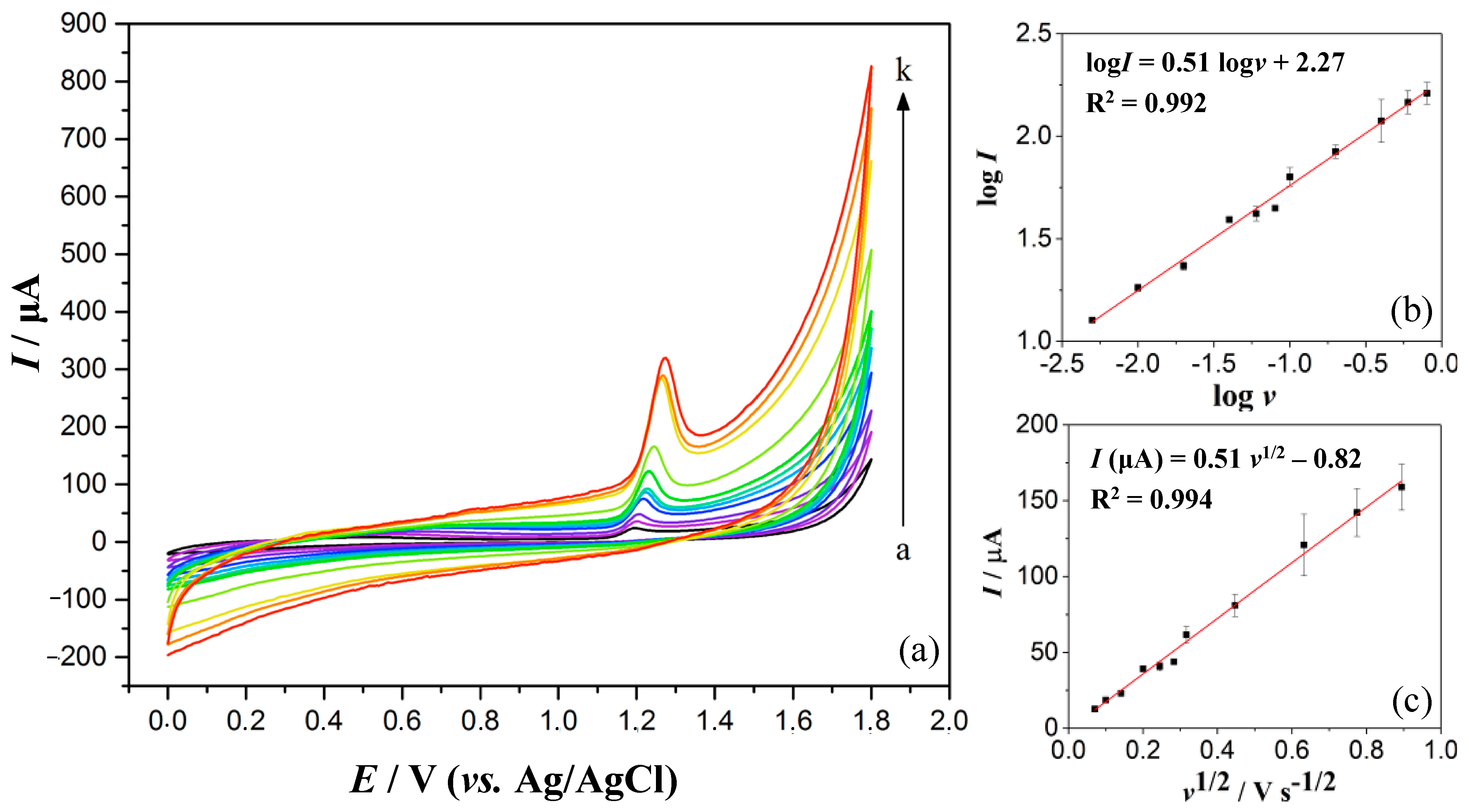
3.3. Optimisation of DPV Operating Parameters
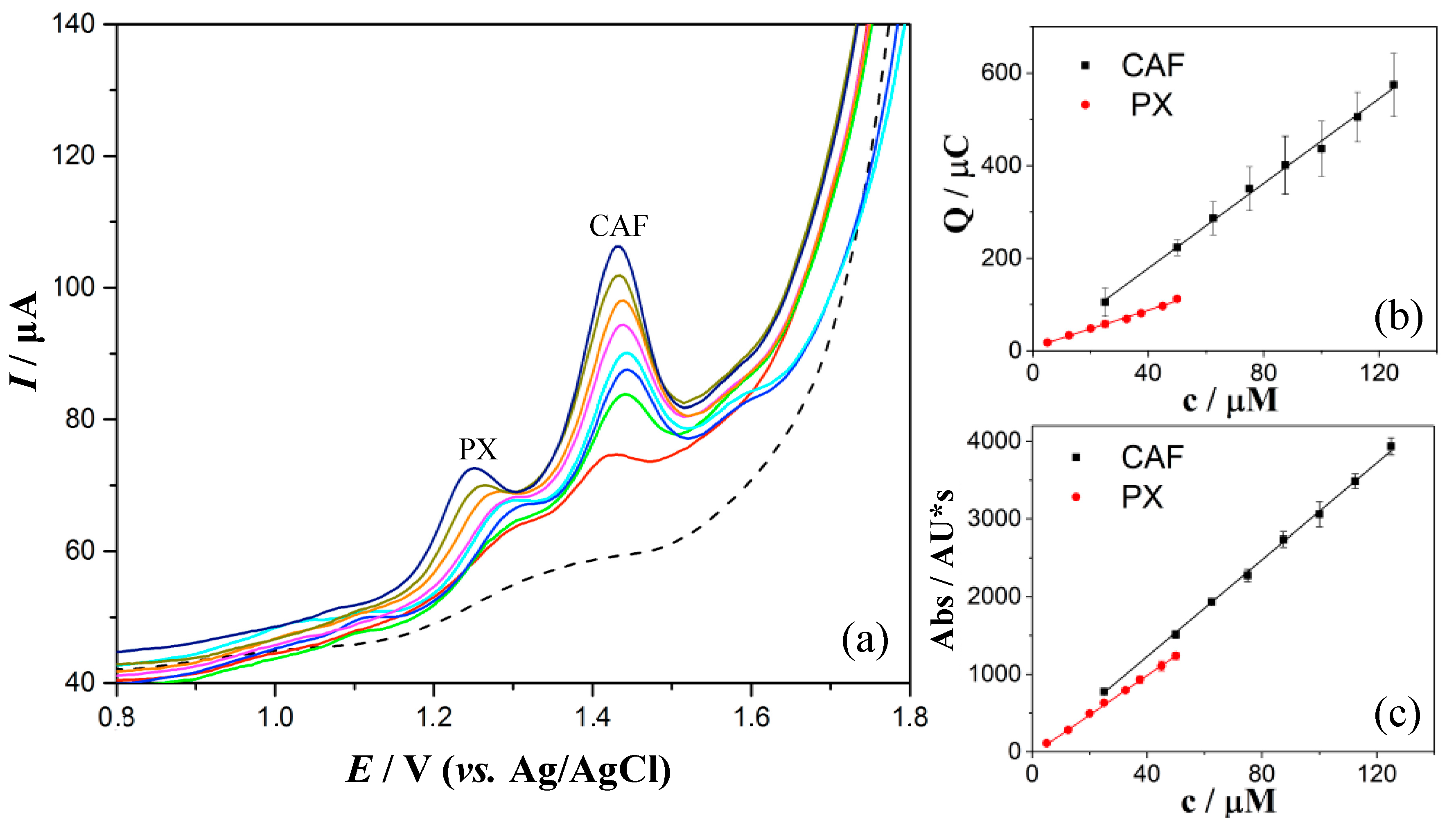
3.4. Quantification of PX and CAF in 0.1 M H2SO4 by DPV
3.5. Selectivity Study
3.6. Quantification of PX and CAF in Spiked Human Saliva
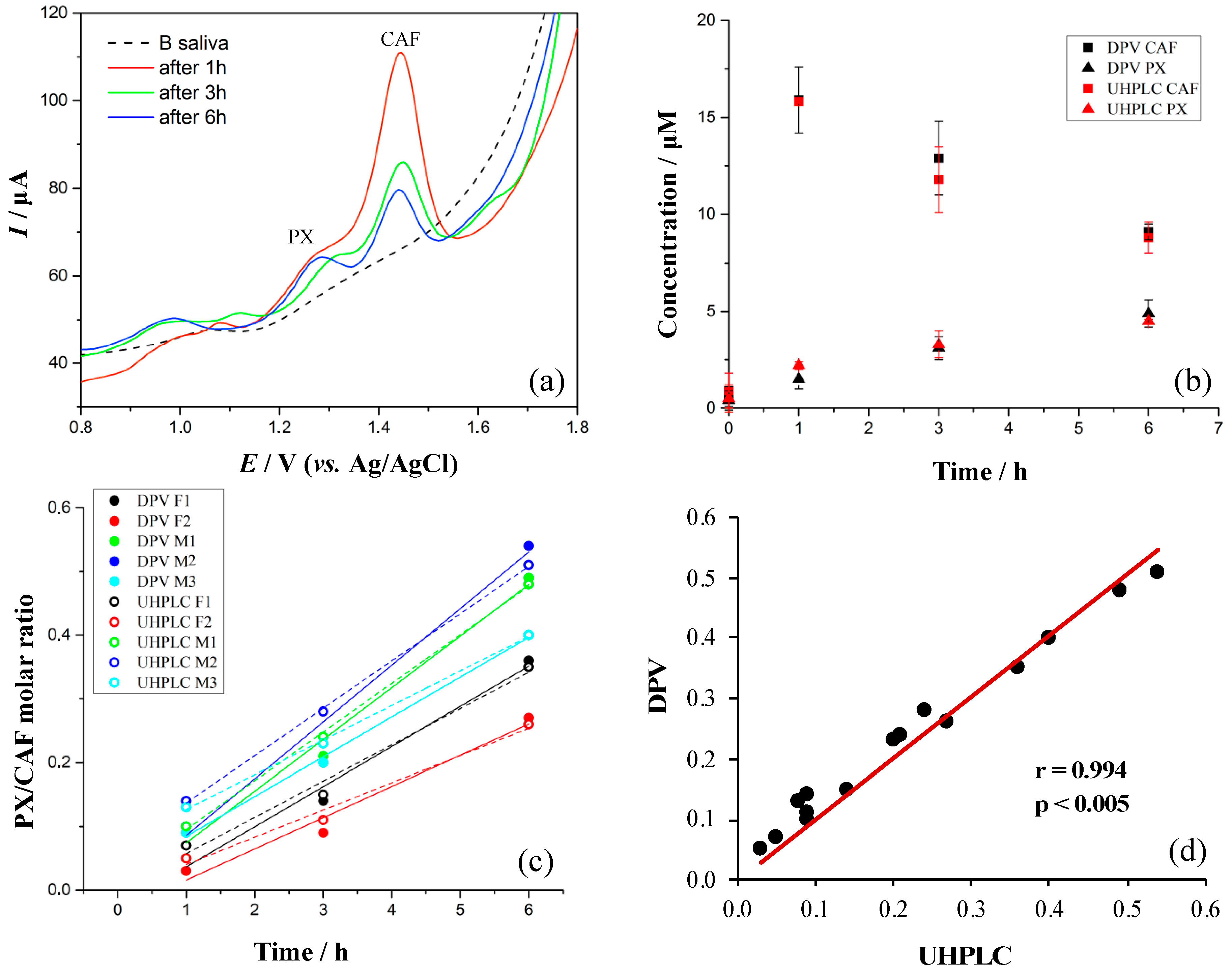
3.7. Evaluation of Method with Five Volunteers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Monteiro, J.P.; Alves, M.G.; Oliveira, P.F.; Silva, B.M. Structure-bioactivity relationships of methylxanthines: Trying to make sense of all the promises and the drawbacks. Molecules 2016, 21, 974. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Dalal, N.S.; Jain, A.C. Antioxidant behaviour of caffeine: Efficient scavenging of hydroxyl radicals. Food Chem. Toxicol. 1991, 29, 1–6. [Google Scholar] [CrossRef]
- Liang, N.; Kitts, D.D. Antioxidant property of coffee components: Assessment of methods that define mechanism of action. Molecules 2014, 19, 19180–19208. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Roxo, M.; Cheng, X.; Zhang, S.; Cheng, H.; Wink, M. Pro-oxidant and lifespan extension effects of caffeine and related methylxanthines in Caenorhabditis elegans. Food Chem. X 2019, 1, 1–9. [Google Scholar] [CrossRef]
- Nehlig, A. Is caffeine a cognitive enhancer? J. Alzheimer’s Dis. 2010, 20, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Ullah, F.; Ali, T.; Ullah, N.; Kim, M.O. Caffeine prevents d-galactose-induced cognitive deficits, oxidative stress, neuroinflammation and neurodegeneration in the adult rat brain. Neurochem. Int. 2015, 90, 114–124. [Google Scholar] [CrossRef]
- Ikram, M.; Park, T.J.; Ali, T.; Kim, M.O. Antioxidant and Neuroprotective Effects of Caffeine against Alzheimer’s and Parkinson’s Disease: Insight into the Role of Nrf-2 and A2AR Signaling. Antioxidants 2020, 9, 902. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Ashihara, H.; Misako, K.; Crozier, A.; Francis, S.H.; Arnaud, M.J.; Sekhar, K.R. Methylxanthines; Springer International Publishing: Heidelberg, Germany, 2011; Volume 200, ISBN 9783642134425. [Google Scholar]
- Doré, A.S.; Robertson, N.; Errey, J.C.; Ng, I.; Hollenstein, K.; Tehan, B.; Hurrell, E.; Bennett, K.; Congreve, M.; Magnani, F.; et al. Structure of the adenosine A 2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 2011, 19, 1283–1293. [Google Scholar] [CrossRef]
- Redivo, L.; Anastasiadi, R.M.; Pividori, M.; Berti, F.; Peressi, M.; Di Tommaso, D.; Resmini, M. Prediction of self-assembly of adenosine analogues in solution: A computational approach validated by isothermal titration calorimetry. Phys. Chem. Chem. Phys. 2019, 21, 4258–4267. [Google Scholar] [CrossRef]
- Chavez-Valdez, R.; Wills-Karp, M.; Ahlawat, R.; Cristofalo, E.A.; Nathan, A.; Gauda, E.B. Caffeine modulates TNF-α production by cord blood monocytes: The role of adenosine receptors. Pediatr. Res. 2009, 65, 203–208. [Google Scholar] [CrossRef]
- Postuma, R.B.; Lang, A.E.; Munhoz, R.P.; Charland, K.; Pelletier, A.; Moscovich, M.; Filla, L.; Zanatta, D.; Romenets, S.R.; Altman, R.; et al. Caffeine for treatment of Parkinson disease: A randomized controlled trial. Neurology 2012, 79, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Arendash, G.W.; Cao, C. Caffeine and coffee as therapeutics against Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.; De Mendonça, A. Does caffeine intake protect from Alzheimer’s disease? Eur. J. Neurol. 2002, 9, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Stavric, B. Methylxanthines: Toxicity to humans. 3. Theobromine, paraxanthine and the combined effects of methylxanthines. Food Chem. Toxicol. 1988, 26, 725–733. [Google Scholar] [CrossRef]
- Zhou, S.-F.; Wang, B.; Yang, L.-P.; Liu, J.-P. Structure, function, regulation and polymorphism and the clinical significance of human cytochrome P450 1A2. Drug Metab. Rev. 2010, 42, 268–354. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S. Summary of Information on Human Cyp Enzymes: Human P450 Metabolism Data. Drug Metab. Rev. 2002, 34, 83–448. [Google Scholar] [CrossRef]
- Dalla Libera, A.; Scutari, G.; Boscolo, R.; Rigobello, M.P.; Bindoli, A. Antioxidant properties of clozapine and related neuroleptics. Free Radic. Res. 1998, 29, 151–157. [Google Scholar] [CrossRef]
- Brinholi, F.F.; de Farias, C.C.; Bonifácio, K.L.; Higachi, L.; Casagrande, R.; Moreira, E.G.; Barbosa, D.S. Clozapine and olanzapine are better antioxidants than haloperidol, Quetiapine, Risperidone and ziprasidone in in vitro models. Biomed. Pharmacother. 2016, 81, 411–415. [Google Scholar] [CrossRef]
- Sala, G.; Arosio, A.; Conti, E.; Beretta, S.; Lunetta, C.; Riva, N.; Ferrarese, C.; Tremolizzo, L. Riluzole selective antioxidant effects in cell models expressing amyotrophic lateral sclerosis endophenotypes. Clin. Psychopharmacol. Neurosci. 2019, 17, 438–442. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Dobrinas, M.; Cornuz, J.; Oneda, B.; Kohler Serra, M.; Puhl, M.; Eap, C.B. Impact of smoking, smoking cessation, and genetic polymorphisms on CYP1A2 activity and inducibility. Clin. Pharmacol. Ther. 2011, 90, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M.; Oscarson, M.; McLellan, R.A. Polymorphic human cytochrome P450 enzymes: An opportunity for individualized drug treatment. Trends Pharmacol. Sci. 1999, 20, 342–349. [Google Scholar] [CrossRef]
- De Kesel, P.M.M.; Lambert, W.E.; Stove, C.P. Alternative Sampling Strategies for Cytochrome P450 Phenotyping. Clin. Pharmacokinet. 2016, 55, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Fuhr, U.; Jetter, A.; Kirchheiner, J. Appropriate phenotyping procedures for drug metabolizing enzymes and transporters in humans and their simultaneous use in the “cocktail” approach. Clin. Pharmacol. Ther. 2007, 81, 270–283. [Google Scholar] [CrossRef]
- Holmes, E.; Wilson, I.D.; Nicholson, J.K. Metabolic Phenotyping in Health and Disease. Cell 2008, 134, 714–717. [Google Scholar] [CrossRef]
- Faber, M.S.; Jetter, A.; Fuhr, U. Assessment of CYP1A2 activity in clinical practice: Why, how, and when? Basic Clin. Pharmacol. Toxicol. 2005, 97, 125–134. [Google Scholar] [CrossRef]
- Doude Van Troostwijk, L.J.A.E.; Koopmans, R.P.; Guchelaar, H.J. Two novel methods for the determination of CYP1A2 activity using the paraxanthine/caffeine ratio. Fundam. Clin. Pharmacol. 2003, 17, 355–362. [Google Scholar] [CrossRef]
- Nehlig, A. Interindividual differences in caffeine metabolism and factors driving caffeine consumption. Pharmacol. Rev. 2018, 70, 384–411. [Google Scholar] [CrossRef]
- Schievano, E.; Finotello, C.; Navarini, L.; Mammi, S. Quantification of caffeine in human saliva by nuclear magnetic resonance as an alternative method for cytochrome CYP1A2 phenotyping. Talanta 2015, 140, 36–41. [Google Scholar] [CrossRef]
- Caporossi, L.; Santoro, A.; Papaleo, B. Saliva as an analytical matrix: State of the art and application for biomonitoring. Biomarkers 2010, 15, 475–487. [Google Scholar] [CrossRef]
- Chiappin, S.; Antonelli, G.; Gatti, R.; De Palo, E.F. Saliva specimen: A new laboratory tool for diagnostic and basic investigation. Clin. Chim. Acta 2007, 383, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Jordan, N.Y.; Mimpen, J.Y.; van den Bogaard, W.J.M.; Flesch, F.M.; van de Meent, M.H.M.; Torano, J.S. Analysis of caffeine and paraxanthine in human saliva with ultra-high-performance liquid chromatography for CYP1A2 phenotyping. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 995–996, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Perera, V.; Gross, A.S.; McLachlan, A.J. Caffeine and paraxanthine HPLC assay for CYP1A2 phenotype assessment using saliva and plasma. Biomed. Chromatogr. 2010, 24, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Fuhr, U.; Rost, K.L. Simple and reliable CYP1A2 phenotyping by the paraxanthine/caffeine ratio in plasma and saliva. Pharmacogenetics 1994, 4, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Begas, E.; Kouvaras, E.; Tsakalof, A.K.; Bounitsi, M.; Asprodini, E.K. Development and validation of a reversed-phase HPLC method for CYP1A2 phenotyping by use of a caffeine metabolite ratio in saliva. Biomed. Chromatogr. 2015, 29, 1657–1663. [Google Scholar] [CrossRef]
- Lopez-Sanchez, R.D.C.; Lara-Diaz, V.J.; Aranda-Gutierrez, A.; Martinez-Cardona, J.A.; Hernandez, J.A. HPLC Method for Quantification of Caffeine and Its Three Major Metabolites in Human Plasma Using Fetal Bovine Serum Matrix to Evaluate Prenatal Drug Exposure. J. Anal. Methods Chem. 2018, 2018. [Google Scholar] [CrossRef]
- Martínez-López, S.; Sarriá, B.; Baeza, G.; Mateos, R.; Bravo-Clemente, L. Pharmacokinetics of caffeine and its metabolites in plasma and urine after consuming a soluble green/roasted coffee blend by healthy subjects. Food Res. Int. 2014, 64, 125–133. [Google Scholar] [CrossRef]
- Yin, O.Q.P.; Lam, S.S.L.; Lo, C.M.Y.; Chow, M.S.S. Rapid determination of five probe drugs and their metabolites in human plasma and urine by liquid chromatography/tandem mass spectrometry: Application to cytochrome P450 phenotyping studies. Rapid Commun. Mass Spectrom. 2004, 18, 2921–2933. [Google Scholar] [CrossRef]
- Idili, A.; Parolo, C.; Ortega, G.; Plaxco, K.W. Calibration-Free Measurement of Phenylalanine Levels in the Blood Using an Electrochemical Aptamer-Based Sensor Suitable for Point-of-Care Applications. ACS Sens. 2019, 4, 3227–3233. [Google Scholar] [CrossRef]
- Benbouguerra, N.; Richard, T.; Saucier, C.; Garcia, F. Voltammetric Behavior, Flavanol and Anthocyanin Contents, and Antioxidant Capacity of Grape Skins and Seeds during Ripening (Vitis vinifera var. Merlot, Tannat, and Syrah). Antioxidants 2020, 9, 800. [Google Scholar] [CrossRef]
- Jara-Palacios, M.J.; Gonçalves, S.; Heredia, F.J.; Hernanz, D.; Romano, A. Extraction of antioxidants from winemaking byproducts: Effect of the solvent on phenolic composition, antioxidant and anti-cholinesterase activities, and electrochemical behaviour. Antioxidants 2020, 9, 675. [Google Scholar] [CrossRef] [PubMed]
- McCreery, R.L. ChemInform Abstract: Advanced Carbon Electrode Materials for Molecular Electrochemistry. Chem. Rev. 2008, 108, 2646–2687. [Google Scholar] [CrossRef] [PubMed]
- Scholz, F. Voltammetric techniques of analysis: The essentials. ChemTexts 2015, 1, 1–24. [Google Scholar] [CrossRef]
- Karikalan, N.; Karthik, R.; Chen, S.M.; Chen, H.A. A voltammetric determination of caffeic acid in red wines based on the nitrogen doped carbon modified glassy carbon electrode. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Udomsap, D.; Branger, C.; Culioli, G.; Dollet, P.; Brisset, H. A versatile electrochemical sensing receptor based on a molecularly imprinted polymer. Chem. Commun. 2014, 50, 7488–7491. [Google Scholar] [CrossRef]
- Li, J.; Lin, X. Glucose biosensor based on immobilization of glucose oxidase in poly(o-aminophenol) film on polypyrrole-Pt nanocomposite modified glassy carbon electrode. Biosens. Bioelectron. 2007, 22, 2898–2905. [Google Scholar] [CrossRef] [PubMed]
- Zoski, C. (Ed.) Handbook of Electrochemistry; Elsevier: Amsterdam, The Netherlands, 2007; ISBN 9780444519580. [Google Scholar]
- Ngamchuea, K.; Chaisiwamongkhol, K.; Batchelor-Mcauley, C.; Compton, R.G. Chemical analysis in saliva and the search for salivary biomarkers-a tutorial review. Analyst 2018, 143, 81–99. [Google Scholar] [CrossRef]
- Campuzano, S.; Yánez-Sedeño, P.; Pingarrón, J.M. Electrochemical bioaffinity sensors for salivary biomarkers detection. TrAC Trends Anal. Chem. 2017, 86, 14–24. [Google Scholar] [CrossRef]
- Ngamchuea, K.; Batchelor-McAuley, C.; Compton, R.G. Understanding electroanalytical measurements in authentic human saliva leading to the detection of salivary uric acid. Sens. Actuators B Chem. 2018, 262, 404–410. [Google Scholar] [CrossRef]
- Jesny, S.; Girish Kumar, K. Electrocatalytic resolution of guanine, adenine and cytosine along with uric acid on poly (4-amino-3-hydroxy naphthalene-1-sulfonic acid) modified glassy carbon electrode. J. Electroanal. Chem. 2017, 801, 153–161. [Google Scholar] [CrossRef]
- Dhull, N.; Kaur, G.; Gupta, V.; Tomar, M. Highly sensitive and non-invasive electrochemical immunosensor for salivary cortisol detection. Sens. Actuators B Chem. 2019, 293, 281–288. [Google Scholar] [CrossRef]
- Dos Santos, W.T.P.; Amin, H.M.A.; Compton, R.G. A nano-carbon electrode optimized for adsorptive stripping voltammetry: Application to detection of the stimulant selegiline in authentic saliva. Sens. Actuators B Chem. 2019, 279, 433–439. [Google Scholar] [CrossRef]
- Kesavan, S.; Gowthaman, N.S.K.; Alwarappan, S.; John, S.A. Real time detection of adenosine and theophylline in urine and blood samples using graphene modified electrode. Sens. Actuators B Chem. 2019, 278, 46–54. [Google Scholar] [CrossRef]
- Güney, S.; Cebeci, F. Selective electrochemical sensor for theophylline based on an electrode modified with imprinted sol-gel film immobilized on carbon nanoparticle layer. Sens. Actuators B Chem. 2015, 208, 307–314. [Google Scholar] [CrossRef]
- Carolina Torres, A.; Barsan, M.M.; Brett, C.M.A. Simple electrochemical sensor for caffeine based on carbon and Nafion-modified carbon electrodes. Food Chem. 2014, 149, 215–220. [Google Scholar] [CrossRef]
- Santos, W.D.J.R.; Santhiago, M.; Yoshida, I.V.P.; Kubota, L.T. Electrochemical sensor based on imprinted sol-gel and nanomaterial for determination of caffeine. Sens. Actuators B Chem. 2012, 166–167, 739–745. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, Y.; Li, L.; Hu, P. Nitrogen-doped carbon nanotubes decorated poly (L-Cysteine) as a novel, ultrasensitive electrochemical sensor for simultaneous determination of theophylline and caffeine. Talanta 2018, 178, 449–457. [Google Scholar] [CrossRef]
- Chojnowska, S.; Baran, T.; Wilińska, I.; Sienicka, P.; Cabaj-Wiater, I.; Knaś, M. Human saliva as a diagnostic material. Adv. Med. Sci. 2018, 63, 185–191. [Google Scholar] [CrossRef]
- Spãtaru, N.; Sarada, B.V.; Tryk, D.A.; Fujishima, A. Anodic voltammetry of xanthine, theophylline, theobromine and caffeine at conductive diamond electrodes and its analytical application. Electroanalysis 2002, 14, 721–728. [Google Scholar] [CrossRef]
- Cinková, K.; Zbojeková, N.; Vojs, M.; Marton, M.; Samphao, A. Švorc Electroanalytical application of a boron-doped diamond electrode for sensitive voltammetric determination of theophylline in pharmaceutical dosages and human urine. Anal. Methods 2015, 7, 6755–6763. [Google Scholar] [CrossRef]
- Jesny, S.; Girish Kumar, K. Non-enzymatic Electrochemical Sensor for the Simultaneous Determination of Xanthine, its Methyl Derivatives Theophylline and Caffeine as well as its Metabolite Uric Acid. Electroanalysis 2017, 29, 1828–1837. [Google Scholar] [CrossRef]
- Compton, R.G.; Banks, C.E. Understanding Voltammetry, 2nd ed.; World Scientific: Singapore, 2010; ISBN 9781848165878. [Google Scholar]
- Petrucci, R.; Zollo, G.; Curulli, A.; Marrosu, G. A new insight into the oxidative mechanism of caffeine and related methylxanthines in aprotic medium: May caffeine be really considered as an antioxidant? Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.H.; Dryhurst, G. Electrochemical oxidation of theophylline at the pyrolytic graphite electrode. J. Electroanal. Chem. 1971, 32, 405–414. [Google Scholar] [CrossRef]
- Wang, T.; Randviir, E.P.; Banks, C.E. Detection of theophylline utilising portable electrochemical sensors. Analyst 2014, 139, 2000–2003. [Google Scholar] [CrossRef] [PubMed]
- Sunil Paul, M.M.; Aravind, U.K.; Pramod, G.; Saha, A.; Aravindakumar, C.T. Hydroxyl radical induced oxidation of theophylline in water: A kinetic and mechanistic study. Org. Biomol. Chem. 2014, 12, 5611–5620. [Google Scholar] [CrossRef] [PubMed]
- Vinchurkar, M.S.; Rao, B.S.M.; Mohan, H.; Mittal, J.P. Kinetics, spectral and redox behaviour of OH adducts of methylxanthines: A radiation chemical study 1. J. Chem. Soc. Perkin Trans. 1999, 2, 609–618. [Google Scholar] [CrossRef]
- Santos, P.M.P.; Silva, S.A.G.; Justino, G.C.; Vieira, A.J.S.C. Demethylation of theophylline (1,3-dimethylxanthine) to 1-methylxanthine: The first step of an antioxidising cascade. Redox Rep. 2010, 15, 138–144. [Google Scholar] [CrossRef]
- Sun, S.; Jiang, J.; Pang, S.; Ma, J.; Xue, M.; Li, J.; Liu, Y.; Yuan, Y. Oxidation of theophylline by Ferrate (VI) and formation of disinfection byproducts during subsequent chlorination. Sep. Purif. Technol. 2018, 201, 283–290. [Google Scholar] [CrossRef]
- Zhu, Y.H.; Zhang, Z.L.; Pang, D.W. Electrochemical oxidation of theophylline at multi-wall carbon nanotube modified glassy carbon electrodes. J. Electroanal. Chem. 2005, 581, 303–309. [Google Scholar] [CrossRef]
- Geremia, K.L.; Seybold, P.G. Computational estimation of the acidities of purines and indoles. J. Mol. Model. 2019, 25. [Google Scholar] [CrossRef]
- Santos, P.M.P.; Telo, J.P.; Vieira, A.J.S.C. Structure and redox properties of radicals derived from one-electron oxidised methylxanthines. Redox Rep. 2008, 13, 123–133. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications; Wiley: New York, NY, USA, 2000; ISBN 9788181892638. [Google Scholar]
- Compton, R.G.; Laborda, E.; Ward, K.R. Understanding Voltammetry: Simulation of Electrode Processes; World Scientific: Singapore, 2013; ISBN 9781783263257. [Google Scholar]
- Guidelli, R.; Compton, R.G.; Feliu, J.M.; Gileadi, E.; Lipkowski, J.; Schmickler, W.; Trasatti, S. Defining the transfer coefficient in electrochemistry: An assessment (IUPAC Technical Report). Pure Appl. Chem. 2014, 86, 245–258. [Google Scholar] [CrossRef]
- Batchelor-Mcauley, C.; Kätelhön, E.; Barnes, E.O.; Compton, R.G.; Laborda, E.; Molina, A. Recent Advances in Voltammetry. ChemistryOpen 2015, 4, 224–260. [Google Scholar] [CrossRef] [PubMed]
- Samiec, P.; Švorc, Ľ.; Stanković, D.M.; Vojs, M.; Marton, M.; Navrátilová, Z. Mercury-free and modification-free electroanalytical approach towards bromazepam and alprazolam sensing: A facile and efficient assay for their quantification in pharmaceuticals using boron-doped diamond electrodes. Sens. Actuators B Chem. 2017, 245, 963–971. [Google Scholar] [CrossRef]
- Pysarevska, S.; Dubenska, L.; Plotycya, S.; Švorc, Ľ. A state-of-the-art approach for facile and reliable determination of benzocaine in pharmaceuticals and biological samples based on the use of miniaturized boron-doped diamond electrochemical sensor. Sens. Actuators B Chem. 2018, 270, 9–17. [Google Scholar] [CrossRef]
- Ojani, R.; Alinezhad, A.; Abedi, Z. A highly sensitive electrochemical sensor for simultaneous detection of uric acid, xanthine and hypoxanthine based on poly(L-methionine) modified glassy carbon electrode. Sens. Actuators B Chem. 2013, 188, 621–630. [Google Scholar] [CrossRef]
- Švorc, Ľ.; Haššo, M.; Sarakhman, O.; Kianičková, K.; Stanković, D.M.; Otřísal, P. A progressive electrochemical sensor for food quality control: Reliable determination of theobromine in chocolate products using a miniaturized boron-doped diamond electrode. Microchem. J. 2018, 142, 297–304. [Google Scholar] [CrossRef]
- Raymundo-Pereira, P.A.; Shimizu, F.M.; Coelho, D.; Piazzeta, M.H.O.; Gobbi, A.L.; Machado, S.A.S.; Oliveira, O.N. A Nanostructured Bifunctional platform for Sensing of Glucose Biomarker in Artificial Saliva: Synergy in hybrid Pt/Au surfaces. Biosens. Bioelectron. 2016, 86, 369–376. [Google Scholar] [CrossRef]
- Gupta, V.K.; Jain, A.K.; Shoora, S.K. Multiwall carbon nanotube modified glassy carbon electrode as voltammetric sensor for the simultaneous determination of ascorbic acid and caffeine. Electrochim. Acta 2013, 93, 248–253. [Google Scholar] [CrossRef]
- Ren, W.; Luo, H.Q.; Li, N.B. Simultaneous voltammetric measurement of ascorbic acid, epinephrine and uric acid at a glassy carbon electrode modified with caffeic acid. Biosens. Bioelectron. 2006, 21, 1086–1092. [Google Scholar] [CrossRef]
- Perera, V.; Gross, A.S.; Xu, H.; McLachlan, A.J. Pharmacokinetics of caffeine in plasma and saliva, and the influence of caffeine abstinence on CYP1A2 metrics. J. Pharm. Pharmacol. 2011, 63, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.; Broughton, L.J.; Lind, M.J.; Morrison, P.J.; Rogers, H.J.; Bradbrook, I.D. Plasma and Salivary Pharmacokinetics of Caffeine in Man. Eur. J. Clin. Pharmacol. 1981, 21, 45–52. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytical Parameter | DPV | |
|---|---|---|
| PX | CAF | |
| Sensitivity (μC/μM) | 2.02 ± 0.06 | 4.55 ± 0.10 |
| Intercept (μC) | 7.37 ± 1.95 | −3.67 ± 8.80 |
| R2 | 0.994 | 0.996 |
| LOD (μM) | 2.89 | 5.80 |
| Linear range (μM) | 10–60 | 19–125 |
| Analytical Parameter | UHPLC-UV | |
|---|---|---|
| PX | CAF | |
| Sensitivity (AU/μM) | 25.01 ± 0.28 | 31.41 ± 0.41 |
| Intercept (AU) | −14.01 ± 9.01 | −39.01 ± 35.01 |
| R2 | 0.999 | 0.999 |
| LOD (μM) | 1.07 | 3.34 |
| Linear range (μM) | 4–60 | 11–125 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anastasiadi, R.-M.; Berti, F.; Colomban, S.; Tavagnacco, C.; Navarini, L.; Resmini, M. Simultaneous Quantification of Antioxidants Paraxanthine and Caffeine in Human Saliva by Electrochemical Sensing for CYP1A2 Phenotyping. Antioxidants 2021, 10, 10. https://doi.org/10.3390/antiox10010010
Anastasiadi R-M, Berti F, Colomban S, Tavagnacco C, Navarini L, Resmini M. Simultaneous Quantification of Antioxidants Paraxanthine and Caffeine in Human Saliva by Electrochemical Sensing for CYP1A2 Phenotyping. Antioxidants. 2021; 10(1):10. https://doi.org/10.3390/antiox10010010
Chicago/Turabian StyleAnastasiadi, Rozalia-Maria, Federico Berti, Silvia Colomban, Claudio Tavagnacco, Luciano Navarini, and Marina Resmini. 2021. "Simultaneous Quantification of Antioxidants Paraxanthine and Caffeine in Human Saliva by Electrochemical Sensing for CYP1A2 Phenotyping" Antioxidants 10, no. 1: 10. https://doi.org/10.3390/antiox10010010
APA StyleAnastasiadi, R.-M., Berti, F., Colomban, S., Tavagnacco, C., Navarini, L., & Resmini, M. (2021). Simultaneous Quantification of Antioxidants Paraxanthine and Caffeine in Human Saliva by Electrochemical Sensing for CYP1A2 Phenotyping. Antioxidants, 10(1), 10. https://doi.org/10.3390/antiox10010010