New Design of the Electrophoretic Part of CLARITY Technology for Confocal Light Microscopy of Rat and Human Brains

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
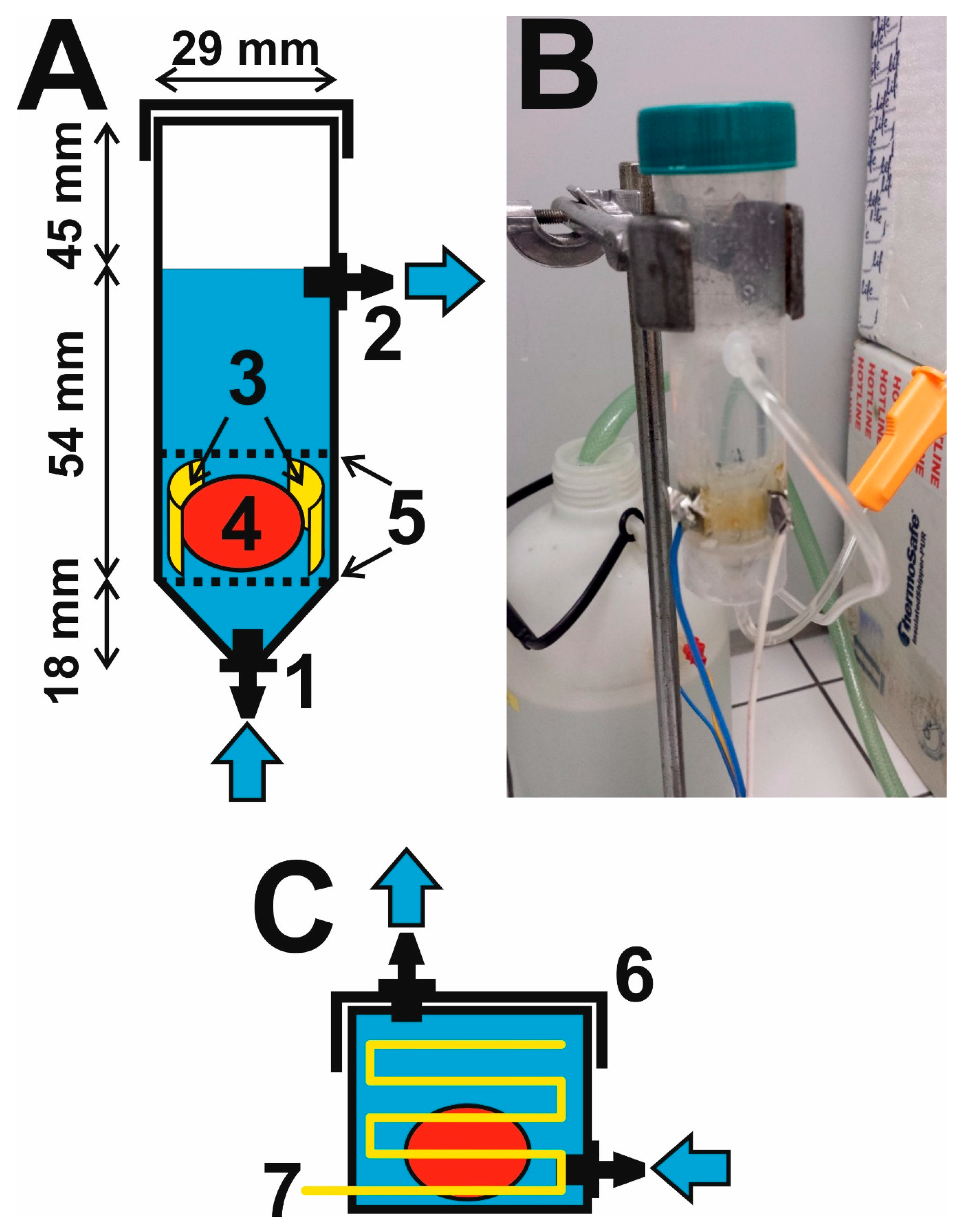
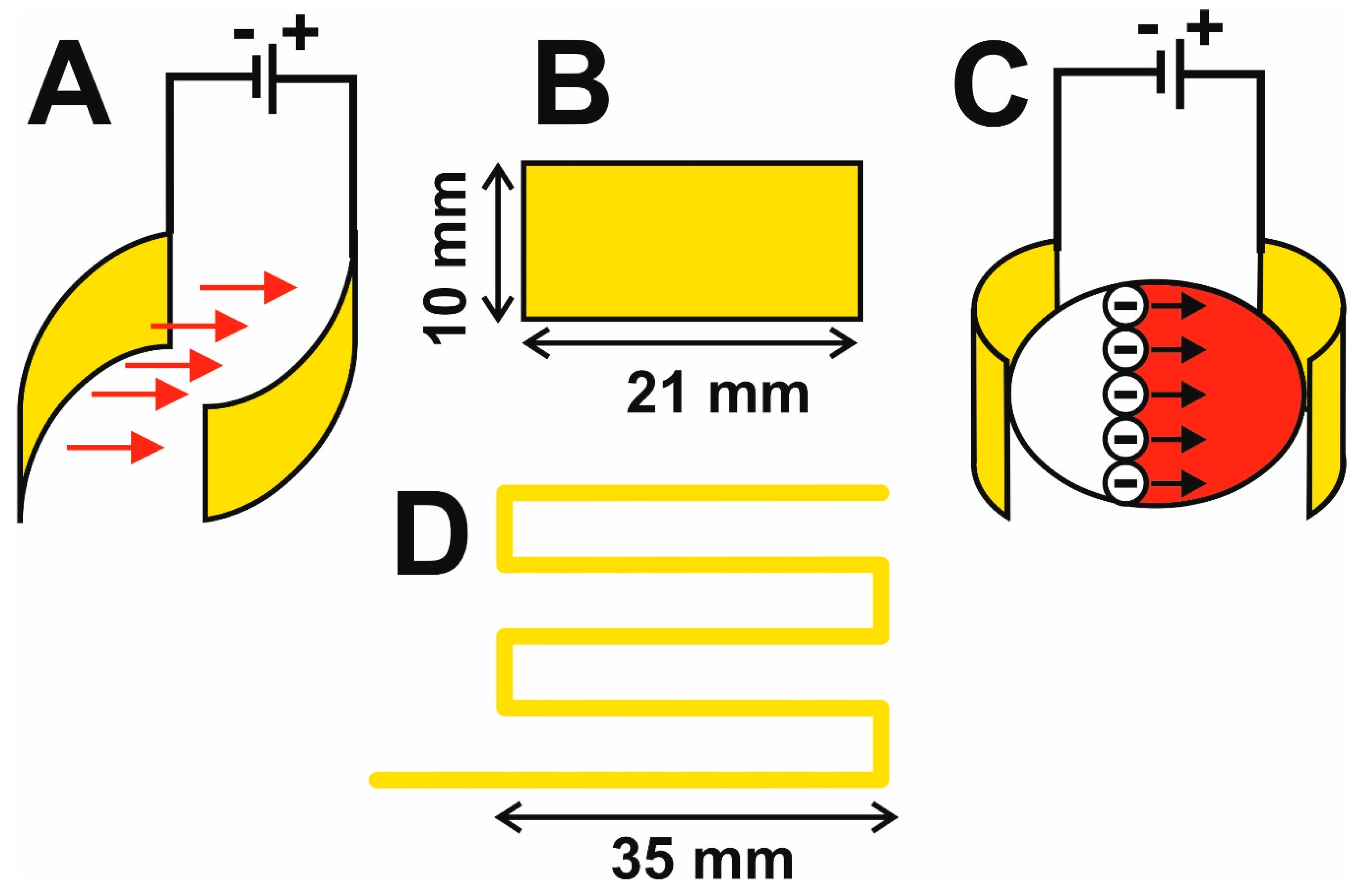
2.1. Construction of a Flow-Through Electrophoretic Cell
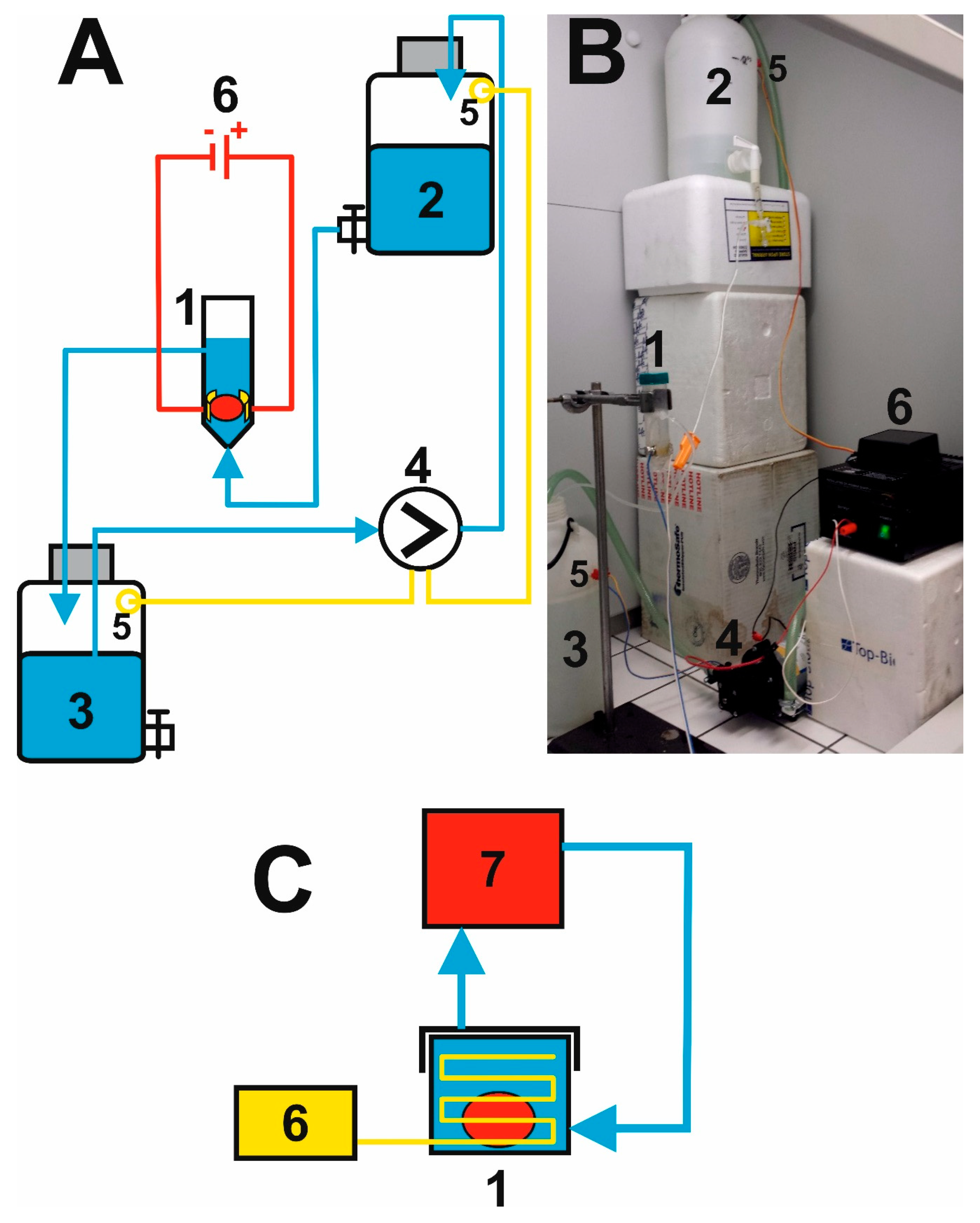
2.2. Putting Together the Electrophoretic Washing Equipment
2.3. Tissue Samples, Fixation, and Preparation
2.4. Preparation of Solutions for CLARITY
2.4.1. Components for Hydrogel Preparation
2.4.2. Penetration of Brain Tissue by the Hydrogel Solution
2.4.3. Hydrogel Tissue Embedding
2.4.4. Preparation of the Clearing Solution
2.5. Brain Tissue Staining and Confocal Microscopy
2.5.1. Brain Tissue Staining for Confocal Microscopy
2.5.2. Confocal Microscopy Procedure
2.6. Ethical and Legal Statement
3. Results and Discussion
3.1. Innovation of the Electrophoretic Washing Cell
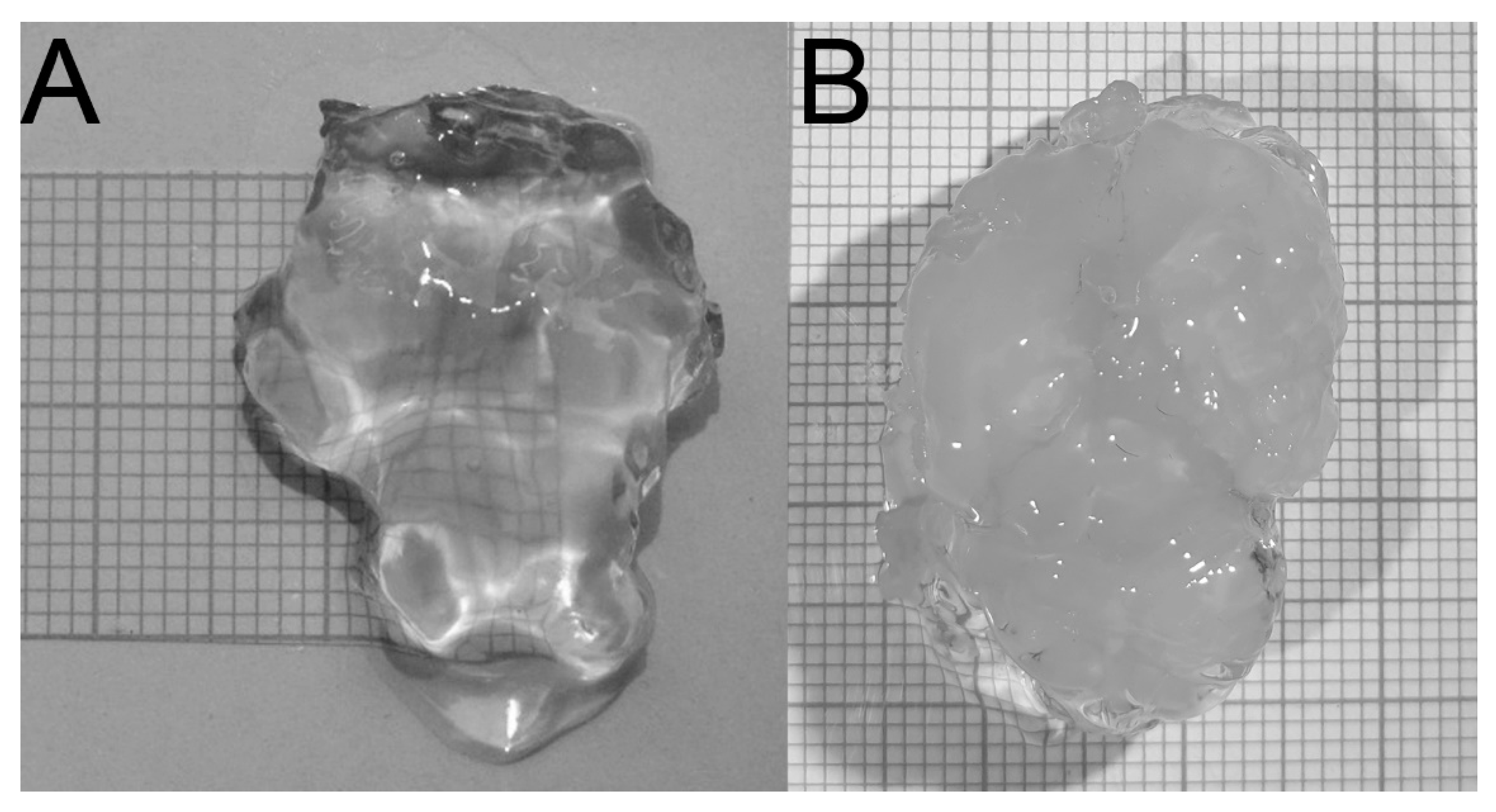
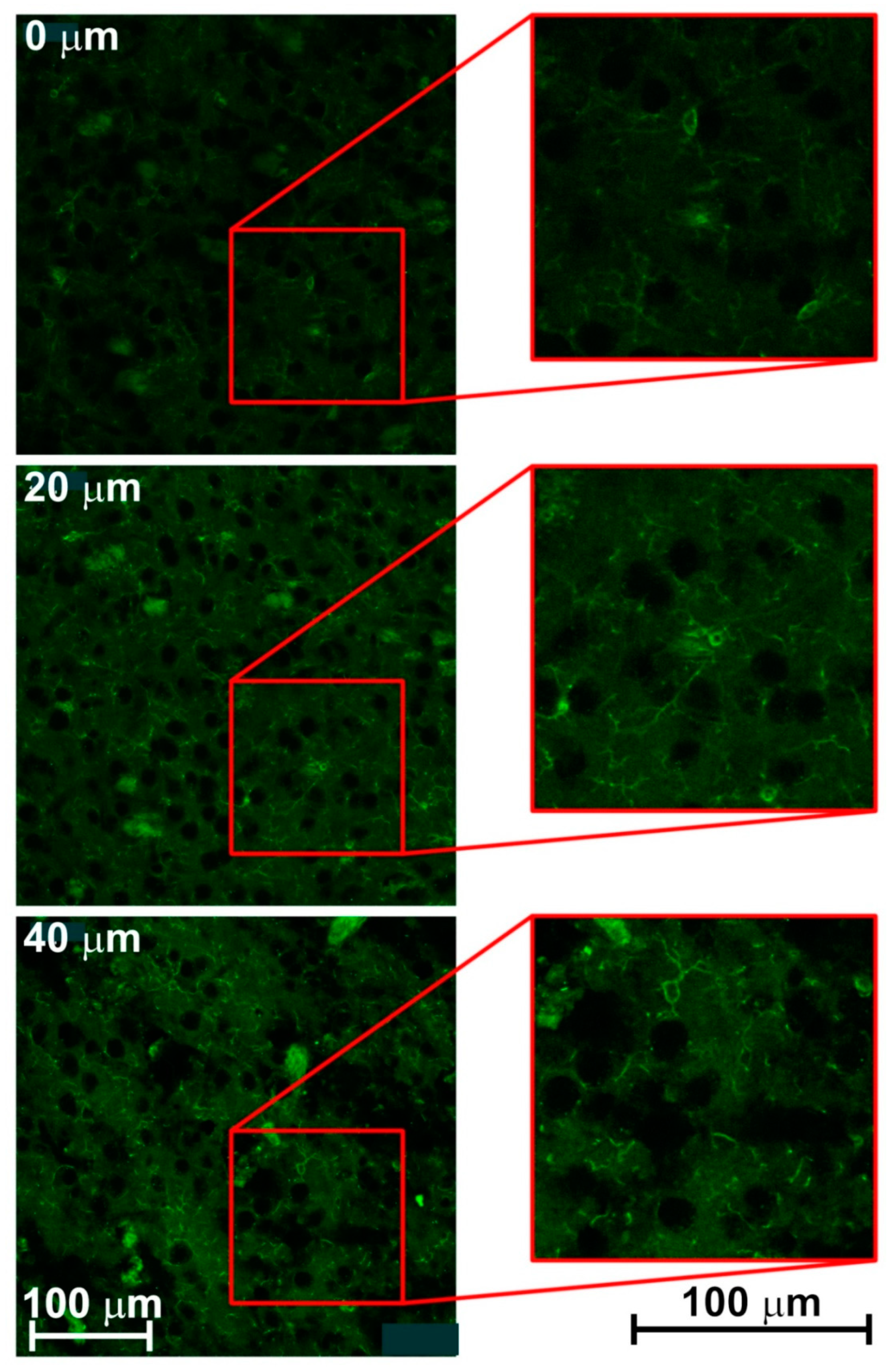
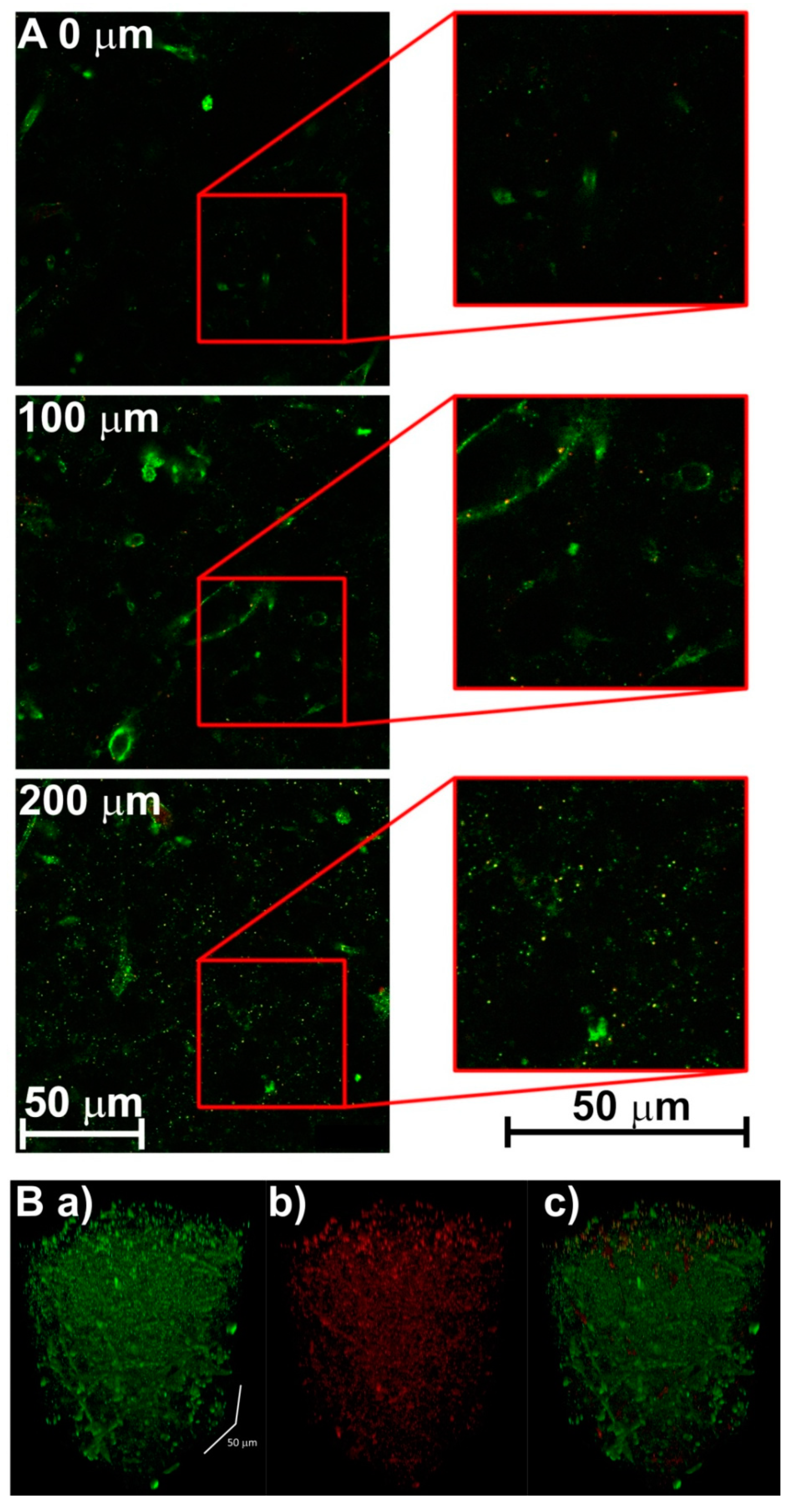
3.2. Creating 3-D Images of Brain Tissue
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chung, K.; Wallace, J.; Kim, S.Y.; Kalyanasundaram, S.; Andalman, A.S.; Davidson, T.J.; Mirzabekov, J.J.; Zalocusky, K.A.; Mattis, J.; Denisin, A.K.; et al. Structural and molecular interrogation of intact biological systems. Nature 2013, 497, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.; Deisseroth, K. Clarity for mapping the nervous system. Nat. Methods 2013, 10, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Tomer, R.; Ye, L.; Hsueh, B.; Deisseroth, K. Advanced clarity for rapid and high-resolution imaging of intact tissues. Nat. Protoc. 2014, 9, 1682–1697. [Google Scholar] [CrossRef] [PubMed]
- Epp, J.R.; Niibori, Y.; Hsiang, H.L.; Mercaldo, V.; Deisseroth, K.; Josselyn, S.A.; Frankland, P.W. Optimization of clarity for clearing whole-brain and other intact organs. eNeuro 2015, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.K.L.; Hurry, M.E.D.; Ng, O.T.W.; DeFelice, J.; Lai, H.M.; Pearce, R.K.B.; Wong, G.T.C.; Chang, R.C.C.; Gentleman, S.M. Bringing clarity to the human brain: Visualization of lewy pathology in three dimensions. Neuropathol. Appl. Neurobiol. 2016, 42, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Cho, J.H.; Murray, E.; Bakh, N.; Choi, H.; Ohn, K.; Ruelas, L.; Hubbert, A.; McCue, M.; Vassallo, S.L.; et al. Stochastic electrotransport selectively enhances the transport of highly electromobile molecules. Proc. Natl. Acad. Sci. USA 2015, 112, E6274–E6283. [Google Scholar] [CrossRef] [PubMed]
- Poguzhelskaya, E.; Artamonov, D.; Bolshakova, A.; Vlasova, O.; Bezprozvanny, I. Simplified method to perform clarity imaging. Mol. Neurodegener. 2014, 9, 1–5. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Phillips, J.; Laude, A.; Lightowlers, R.; Morris, C.M.; Turnbull, D.M.; Lax, N.Z. Development of passive clarity and immunofluorescent labelling of multiple proteins in human cerebellum: Understanding mechanisms of neurodegeneration in mitochondrial disease. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.X.; Asano, S.M.; Upadhyayula, S.; Pisarev, I.; Milkie, D.E.; Liu, T.L.; Singh, V.; Graves, A.; Huynh, G.H.; Zhao, Y.X.; et al. Cortical column and whole-brain imaging with molecular contrast and nanoscale resolution. Science 2019, 363, 8302. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Lee, M.; Seo, J.M.; Park, H.S.; Cho, Y.E. Optimization of the optical transparency of rodent tissues by modified pact-based passive clearing. Exp. Mol. Med. 2016, 48, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Unnersjo-Jess, D.; Scott, L.; Blom, H.; Brismar, H. Super-resolution stimulated emission depletion imaging of slit diaphragm proteins in optically cleared kidney tissue. Kidney Int. 2016, 89, 243–247. [Google Scholar] [CrossRef] [PubMed]
- McCollum, L.A.; McCullumsmith, R.E.; Roberts, R.C. Tyrosine hydroxylase localization in the nucleus accumbens in schizophrenia. Brain Struct. Funct. 2016, 221, 4451–4458. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Tanaka, K.; Suzuki, S.; Dembo, T.; Fukuuchi, Y. Enhanced expression of iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke 2001, 32, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial activation in traumatic brain injury. Front. Aging Neurosci. 2017, 9, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.K.; Ji, K.; Min, K.; Joe, E.H. Brain inflammation and microglia: Facts and misconceptions. Exp. Neurobiol. 2013, 22, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Choi, J.; Jo, Y.; Kim, J.Y.; Jang, Y.J.; Lee, H.M.; Kim, S.Y.; Lee, H.J.; Cho, K.; Jung, N.; et al. Act-presto: Rapid and consistent tissue clearing and labeling method for 3-dimensional (3d) imaging. Sci. Rep. 2016, 6, 1–13. [Google Scholar]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zach, P.; Mrzílková, J.; Pala, J.; Uttl, L.; Kútna, V.; Musil, V.; Sommerová, B.; Tůma, P. New Design of the Electrophoretic Part of CLARITY Technology for Confocal Light Microscopy of Rat and Human Brains. Brain Sci. 2019, 9, 218. https://doi.org/10.3390/brainsci9090218
Zach P, Mrzílková J, Pala J, Uttl L, Kútna V, Musil V, Sommerová B, Tůma P. New Design of the Electrophoretic Part of CLARITY Technology for Confocal Light Microscopy of Rat and Human Brains. Brain Sciences. 2019; 9(9):218. https://doi.org/10.3390/brainsci9090218
Chicago/Turabian StyleZach, Petr, Jana Mrzílková, Jan Pala, Libor Uttl, Viera Kútna, Vladimír Musil, Blanka Sommerová, and Petr Tůma. 2019. "New Design of the Electrophoretic Part of CLARITY Technology for Confocal Light Microscopy of Rat and Human Brains" Brain Sciences 9, no. 9: 218. https://doi.org/10.3390/brainsci9090218
APA StyleZach, P., Mrzílková, J., Pala, J., Uttl, L., Kútna, V., Musil, V., Sommerová, B., & Tůma, P. (2019). New Design of the Electrophoretic Part of CLARITY Technology for Confocal Light Microscopy of Rat and Human Brains. Brain Sciences, 9(9), 218. https://doi.org/10.3390/brainsci9090218