An Overview of Experimental and Clinical Spinal Cord Findings in Alzheimer’s Disease

{kind=link}
{kind=link}
Abstract
1. Introduction
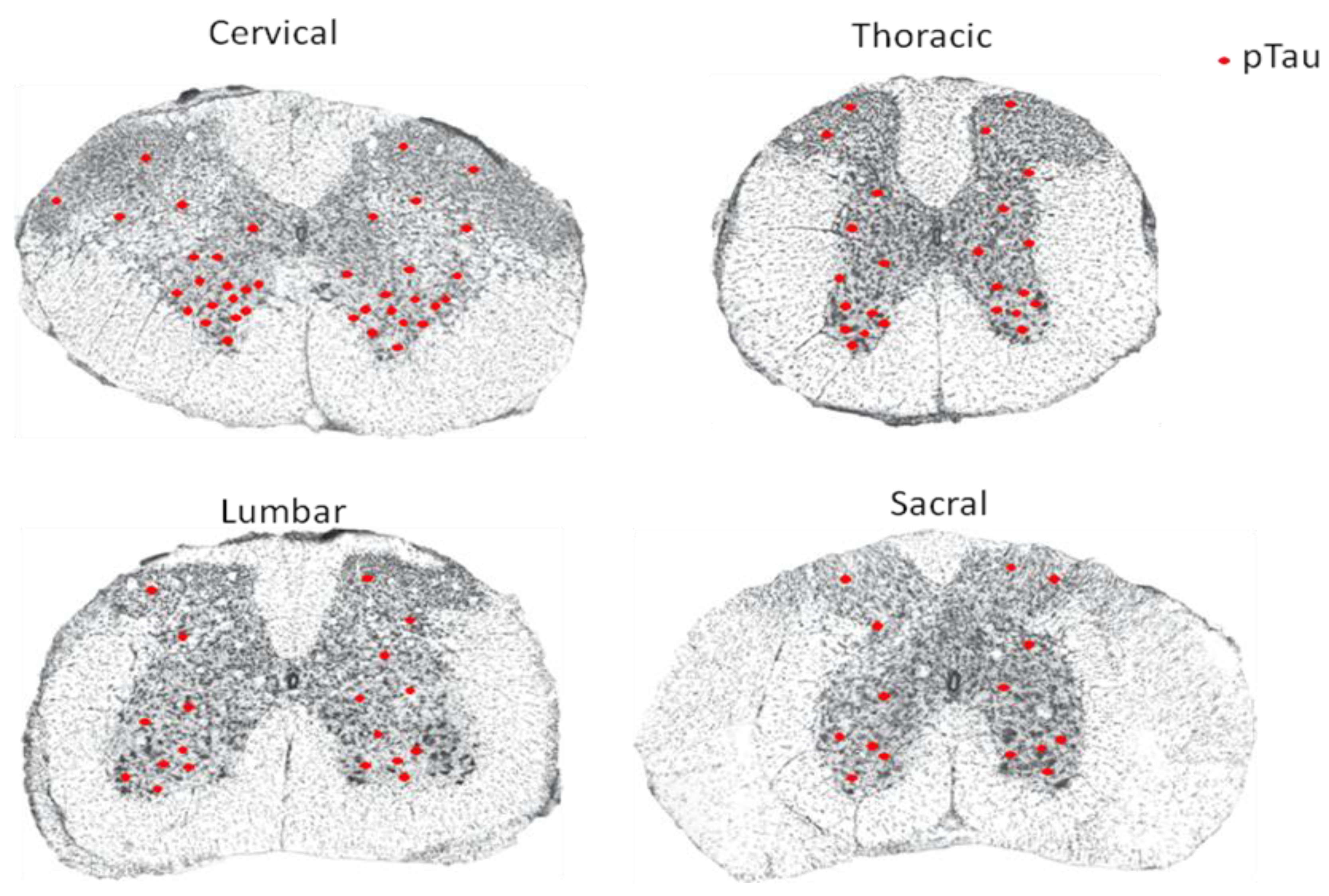
2. Abnormal Phosphorylation of Tau in the Spinal Cord of AD Patients and Animal Models
3. Deposition of β-Amyloid Protein (Aβ) in the Spinal Cord of AD Patients and Animal Models
4. The Role of Inflammation in the Spinal Cord of AD Patients and Animal Models
5. Other Pathological Changes in the Spinal Cord of AD Patients
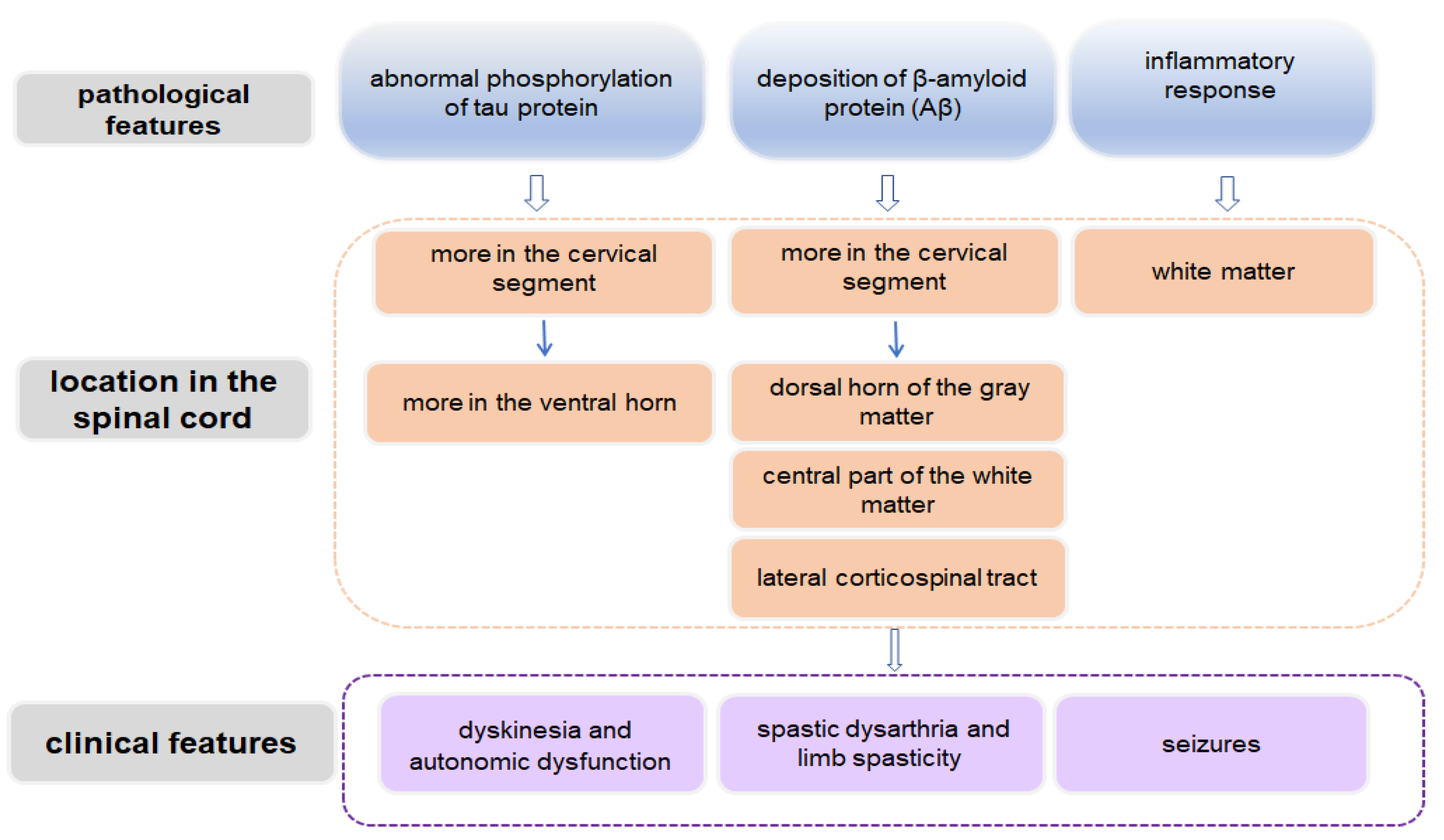
6. Summary and Prospective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, Y.H.; Ji, Y. Development of alzheimer’s disease in the world. Chin. J. Modern Neurol. Dis. 2015, 15, 507–511, [Article in Chinese]. [Google Scholar]
- 2014 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2014, 10, e47–e92. [CrossRef]
- Masoodi, T.A.; Al Shammari, S.A.; Al-Muammar, M.N.; Alhamdan, A.A.; Talluri, V.R. Exploration of deleterious single nucleotide polymorphisms in late-onset Alzheimer disease susceptibility genes. Gene 2013, 512, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Galante, D.; Corsaro, A.; Florio, T.; Vella, S.; Pagano, A.; Sbrana, F.; Vassalli, M.; Perico, A.; D’Arrigo, C. Differential Toxicity, Conformation and Morphology of Typical Initial Aggregation States of Abeta1-42 and Abetapy3-42 Beta-Amyloids. Int. J. Biochem. Cell Biol. 2012, 44, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Mohsenzadegan, M.; Mirshafiey, A. The immunopathogenic role of reactive oxygen species in Alzheimer disease. Iran. J. Allergy Asthma Immunol. 2012, 11, 203–216. [Google Scholar] [PubMed]
- Azizi, G.; Mirshafiey, A. The potential role of proinflammatory and antiinflammatory cytokines in Alzheimer disease pathogenesis. Immunopharmacol. Immunotoxicol. 2012, 34, 881–895. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, E.; Pavlovic, A.; Jovanovic, Z.; Veselinovic, N.; Despotovic, I.; Stojkovic, T.; Sternic, N.; Kostic, V. Vascular Risk Factors in Alzheimer’s Disease - Preliminary Report. J. Neurol. Sci. 2012, 322, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.Y.; Jagust, W.J.; Initiative, F.T.A.D.N. Vascular burden and Alzheimer disease pathologic progression. Neurol. 2012, 79, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Raudino, F. Involvement of the Spinal Cord in the Alzheimer’s Disease: A Literature Review. Arch. Neurosci. 2016, 3, e33834. [Google Scholar] [CrossRef]
- Girgis, S.I.; Yates, C.M.; Fink, G.; MacIntyre, I. Calcitonin gene-related peptide and calcitonin immunoreactivity in brain and spinal cord in Alzheimer-type dementia. J. Neurol. Sci. 1990, 99, 69–74. [Google Scholar] [CrossRef]
- Yeh, T.S.; Ho, Y.C.; Hsu, C.L.; Pan, S.L. Spinal cord injury and Alzheimer’s disease risk: A population-based, retrospective cohort study. Spinal Cord 2018, 56, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Cao, M.X.; Li, Y.M.; Zhang, Q.Y.; Shao, H.R.; Dong, X.D. The Exploration between Tau Protein and Related Molecules in Alzheimer’s Disease. Med. Res. Educ. 2014, 31, 72–75, 87. [Google Scholar]
- Botha, H.; Mantyh, W.G.; Graff-Radford, J.; Machulda, M.M.; Przybelski, S.A.; Wiste, H.J.; Senjem, M.L.; Parisi, J.E.; Petersen, R.C.; Murray, M.E.; et al. Tau-negative amnestic dementia masquerading as Alzheimer disease dementia. Neurology 2018, 90, e940–e946. [Google Scholar] [CrossRef] [PubMed]
- Ikezu, T.; Chen, C.; DeLeo, A.M.; Zeldich, E.; Fallin, M.D.; Kanaan, N.M.; Lunetta, K.L.; Abraham, C.R.; Logue, M.W.; Farrer, L.A. Tau Phosphorylation is Impacted by Rare AKAP9 Mutations Associated with Alzheimer Disease in African Americans. J. Neuroimmune Pharmacol. 2018, 13, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Tiernan, C.T.; Mufson, E.J.; Kanaan, N.M.; E Counts, S. Tau Oligomer Pathology in Nucleus Basalis Neurons During the Progression of Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2018, 77, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M. On the Distribution of Senile Changes in the Spinal Cord. Psychiatry Clin. Neurosci. 1978, 32, 249–251. [Google Scholar] [CrossRef]
- Saito, Y.; Murayama, S. Expression of tau immunoreactivity in the spinal motor neurons of Alzheimer’s disease. Neurology 2000, 55, 1727–1729. [Google Scholar] [CrossRef] [PubMed]
- Dugger, B.N.; Hidalgo, J.A.; Chiarolanza, G.; Mariner, M.; Henry-Watson, J.; Sue, L.I.; Beach, T.G. The Distribution of Phosphorylated Tau in Spinal Cords of Alzheimer’s Disease and Non-Demented Individuals. J. Alzheimers Dis. 2013, 34, 529–536. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, L.; Liu, J.; Gui, Q.; Guo, Y.; Hu, Y.; Zhang, H. Histopathological and immunohistochemical study of spinal cord tissues in neurodegenerative diseases. Chin. J. Pathol. 2015, 44, 587–593. [Google Scholar]
- Parkkinen, L.; Soininen, H.; Alafuzoff, I. Regional distribution of alpha-synuclein pathology in unimpaired aging and Alzheimer disease. J. Neuropathol. Exp. Neurol. 2003, 62, 363–367. [Google Scholar] [CrossRef]
- Leroy, K.; Bretteville, A.; Schindowski, K.; Gilissen, E.; Authelet, M.; De Decker, R.; Yilmaz, Z.; Buée, L.; Brion, J.-P. Early Axonopathy Preceding Neurofibrillary Tangles in Mutant Tau Transgenic Mice. Am. J. Pathol. 2007, 171, 976–992. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.J.; Wang, L.N.; Zhu, M.W.; Zhang, H.H.; Hu, Y.Z.; Han, Z.T.; Li, J.M.; Wang, D.X. Expression of tau-related protein in spinal cord of patients with Alzheimer’s disease. Chin. J. Pathol. 2011, 40, 161–164, [Article in Chinese]. [Google Scholar]
- Guo, Y.; Wang, L.; Zhu, M.; Zhang, H.; Hu, Y.; Han, Z.; Liu, J.; Zhao, W.; Wang, D. Detection of Hyperphosphorylated Tau Protein and Alpha-Synuclein in Spinal Cord of Patients with Alzheimer’s Disease. Neuropsychiatr. Dis. Treat. 2016, 12, 445–452, [Article in Chinese]. [Google Scholar] [PubMed]
- Schmidt, M.L.; Zhukareva, V.; Perl, D.P.; Sheridan, S.K.; Schuck, T.; Lee, V.M.-Y.; Trojanowski, J.Q. Spinal Cord Neurofibrillary Pathology in Alzheimer Disease and Guam Parkinsonism-Dementia Complex. J. Neuropathol. Exp. Neurol. 2001, 60, 1075–1086. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Watson, C.; Paxinos, G.; Kayalioglu, G.; Heise, C. Atlas of the Mouse Spinal Cord; Elsevier Ltd.: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef]
- Arriagada, P.V.; Marzloff, K.; Hyman, B.T. Distribution of Alzheimer-Type Pathologic Changes in Nondemented Elderly Individuals Matches the Pattern in Alzheimer’s Disease. Neurology 1992, 42, 1681–1688. [Google Scholar] [CrossRef]
- A Carson, J.; Turner, A.J. Beta-amyloid catabolism: Roles for neprilysin (NEP) and other metallopeptidases? J. Neurochem. 2002, 81, 1–8. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Beckett, C.; Belyaev, N.D.; Turner, A.J. Are Amyloid-Degrading Enzymes Viable Therapeutic Targets in Alzheimer’s Disease? J. Neurochem. 2012, 120, 167–185. [Google Scholar] [CrossRef]
- Fisk, L.; Nalivaeva, N.N.; Boyle, J.P.; Peers, C.S.; Turner, A.J. Effects of Hypoxia and Oxidative Stress on Expression of Neprilysin in Human Neuroblastoma Cells and Rat Cortical Neurones and Astrocytes. Neurochem. Res. 2007, 32, 1741–1748. [Google Scholar] [CrossRef]
- Dubrovskaia, N.M.; Nalivaeva, N.N.; A Plesneva, S.; A Feponova, A.; Turner, A.J.; A Zhuravin, I. [Changes in the activity of amyloid-degrading metallopeptidases leads to disruption of memory in rats]. Журнал высшей нервнoй деятельнoсти им И П Павлoва 2009, 59, 630–638. [Google Scholar] [CrossRef]
- Bird, S.M.; Sohrabi, H.R.; Sutton, T.A.; Weinborn, M.; Rainey-Smith, S.R.; Brown, B.; Patterson, L.; Taddei, K.; Gupta, V.; Carruthers, M.; et al. Cerebral Amyloid-Beta Accumulation and Deposition Following Traumatic Brain Injury--a Narrative Review and Meta-Analysis of Animal Studies. Neurosci. Biobehav. Rev. 2016, 64, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Su, H.; Zhang, Y.; Chau, W.H.; Ng, C.T.; Song, Y.Q.; Huang, J.D.; Wu, W.; Lin, Z.X. Amyloid Pathology in Spinal Cord of the Transgenic Alzheimer’s Disease Mice Is Correlated to the Corticospinal Tract Pathway. J. Alzheimers Dis. 2013, 35, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Li, L.X.; Zhang, S.F.; Zhang, X.; Le, W.D. Autophagic Changes in Brain and Spinal Cord of Mice with Alzheimer’s Disease. J. Shanghai Jiaotong Univ. 2012, 32, 536–542, [Article in Chinese]. [Google Scholar]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The Autophagy-Related Protein Beclin 1 Shows Reduced Expression in Early Alzheimer Disease and Regulates Amyloid Beta Accumulation in Mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [PubMed]
- Wirths, O.; Weis, J.; Szczygielski, J.; Multhaup, G.; Bayer, T.A. Axonopathy in an APP/PS1 transgenic mouse model of Alzheimer’s disease. Acta Neuropathol. 2006, 111, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, S.; Trawicka, A.; Jenneckens, C.; Bayer, T.A.; Wirths, O. Motor Deficits, Neuron Loss, and Reduced Anxiety Coinciding with Axonal Degeneration and Intraneuronal Abeta Aggregation in the 5xfad Mouse Model of Alzheimer’s Disease. Neurobiol. Aging 2012, 33, e29–e40. [Google Scholar] [CrossRef]
- Chu, T.-H.; Cummins, K.; Sparling, J.S.; Tsutsui, S.; Brideau, C.; Nilsson, K.P.R.; Joseph, J.T.; Stys, P.K. Axonal and myelinic pathology in 5xFAD Alzheimer’s mouse spinal cord. PLoS ONE 2017, 12, e0188218. [Google Scholar] [CrossRef]
- Seo, J.-S.; Leem, Y.-H.; Kim, S.-W.; Lee, K.-W.; Han, P.-L. Severe Motor Neuron Degeneration in the Spinal Cord of the Tg2576 Mouse Model of Alzheimer Disease. J. Alzheimer’s Dis. 2010, 21, 263–276. [Google Scholar] [CrossRef]
- Verkkoniemi, A.; Kalimo, H.; Paetau, A.; Somer, M.; Iwatsubo, T.; Hardy, J.; Haltia, M. Variant Alzheimer Disease With Spastic paraparesis: Neuropathological phenotype. J. Neuropathol. Exp. Neurol. 2001, 60, 483–492. [Google Scholar] [CrossRef]
- Rudzinski, L.A.; Fletcher, R.M.; Dickson, D.W.; Crook, R.; Hutton, M.L.; Adamson, J.; Graff-Radford, N.R. Early Onset Alzheimer’s Disease with Spastic Paraparesis, Dysarthria and Seizures and N135S Mutation in PSEN1. Alzheimer Dis. Assoc. Disord. 2008, 22, 299–307. [Google Scholar] [CrossRef]
- Ogomori, K.; Kitamoto, T.; Tateishi, J.; Sato, Y.; Suetsugu, M.; Abe, M. Beta-Protein Amyloid Is Widely Distributed in the Central Nervous System of Patients with Alzheimer’s Disease. Am. J. Pathol. 1989, 134, 243–251. [Google Scholar] [PubMed]
- Finnie, G.S.; Gunnarsson, R.; Manavis, J.; Blumbergs, P.C.; Mander, K.A.; Edwards, S.; Van den Heuvel, C.; Finnie, J.W. Characterization of an ‘Amyloid Only’ Transgenic (B6c3-Tg(Appswe,Psen1de9)85dbo/Mmjax) Mouse Model of Alzheimer’s Disease. J. Comp. Pathol. 2017, 156, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Lucin, K.M.; Wyss-Coray, T. Immune Activation in Brain Aging and Neurodegeneration: Too Much or Too Little? Neuron 2009, 64, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J. Microglia and Neuroprotection: Implications for Alzheimer’s Disease. Brain Res. Rev. 2005, 48, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Schwartz, J.B.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann. Neurol. 2018, 83, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Klyubin, I.; Cuello, A.C.; Rowan, M.J. Nlrp3-Dependent Synaptic Plasticity Deficit in an Alzheimer’s Disease Amyloidosis Model in Vivo. Neurobiol. Dis. 2018, 114, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Torika, N.; Asraf, K.; Apte, R.N.; Fleisher-Berkovich, S. Candesartan Ameliorates Brain Inflammation Associated with Alzheimer’s Disease. CNS Neurosci. Ther. 2018, 24, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Breyhan, H.; Marcello, A.; Cotel, M.C.; Bruck, W.; Bayer, T.A. Inflammatory Changes Are Tightly Associated with Neurodegeneration in the Brain and Spinal Cord of the App/Ps1ki Mouse Model of Alzheimer’s Disease. Neurobiol. Aging 2010, 31, 747–757. [Google Scholar] [CrossRef]
- Guo, Y.J.; Wang, L.N.; Zhu, M.W.; Zhang, H.H.; Hu, Y.Z.; Han, Z.T.; Zhao, W.Q.; Wang, D.X. Expression of A-Synuclein in the Spinal Cord of Alzheimer’s Disease. Chin. J. Contemp. Neurol. Neurosurg. 2010, 10, 483–487. [Google Scholar]
- Saito, Y.; Kawashima, A.; Ruberu, N.N.; Fujiwara, H.; Koyama, S.; Sawabe, M.; Arai, T.; Nagura, H.; Yamanouchi, H.; Hasegawa, M.; et al. Accumulation of phosphorylated alpha-synuclein in aging human brain. J. Neuropathol. Exp. Neurol. 2003, 62, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Bloch, A.; Probst, A.; Bissig, H.; Adams, H.; Tolnay, M. alpha-Synuclein pathology of the spinal and peripheral autonomic nervous system in neurologically unimpaired elderly subjects. Neuropathol. Appl. Neurobiol. 2006, 32, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; White Iii, C.L.; Akiyama, H.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; et al. Arizona Parkinson’s Disease Consortium. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010, 119, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.M.; Dammer, E.B.; Diner, I.; Yi, H.; Seyfried, N.T.; Gearing, M.; Glass, J.D.; Montine, T.J.; Levey, A.I.; Lah, J.J. Aggregates of Small Nuclear Ribonucleic Acids (snRNAs) in Alzheimer’s Disease. Brain Pathol. 2014, 24, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Dore, V.; Burnham, S.C.; Masters, C.L.; Rowe, C.C. Imaging Tau and Amyloid-Beta Proteinopathies in Alzheimer Disease and Other Conditions. Nat. Rev. Neurol. 2018, 14, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Djogo, N.; Jakovcevski, I.; Muller, C.; Lee, H.J.; Xu, J.C.; Jakovcevski, M.; Kugler, S.; Loers, G.; Schachner, M. Adhesion Molecule L1 Binds to Amyloid Beta and Reduces Alzheimer’s Disease Pathology in Mice. Neurobiol. Dis. 2013, 56, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, G.S.; A Banks, R.; Kim, B.; Changolkar, L.; Riddle, D.M.; Leight, S.N.; Irwin, D.J.; Trojanowski, J.Q.; Lee, V.M.Y. Detection of Alzheimer Disease (AD)-Specific Tau Pathology in AD and NonAD Tauopathies by Immunohistochemistry With Novel Conformation-Selective Tau Antibodies. J. Neuropathol. Exp. Neurol. 2018, 77, 216–228. [Google Scholar] [CrossRef]
- Quiroz, Y.T.; Sperling, R.A.; Norton, D.J.; Baena, A.; Arboleda-Velásquez, J.F.; Cosio, D.; Schultz, A.; LaPoint, M.; Guzman-Velez, E.; Miller, J.B.; et al. Association Between Amyloid and Tau Accumulation in Young Adults With Autosomal Dominant Alzheimer Disease. JAMA Neurol. 2018, 75, 548–556. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Q.; Zhao, W.-J.; Ou, G.-Y.; Xue, W.-K. An Overview of Experimental and Clinical Spinal Cord Findings in Alzheimer’s Disease. Brain Sci. 2019, 9, 168. https://doi.org/10.3390/brainsci9070168
Xie Q, Zhao W-J, Ou G-Y, Xue W-K. An Overview of Experimental and Clinical Spinal Cord Findings in Alzheimer’s Disease. Brain Sciences. 2019; 9(7):168. https://doi.org/10.3390/brainsci9070168
Chicago/Turabian StyleXie, Qing, Wei-Jiang Zhao, Guan-Yong Ou, and Wei-Kang Xue. 2019. "An Overview of Experimental and Clinical Spinal Cord Findings in Alzheimer’s Disease" Brain Sciences 9, no. 7: 168. https://doi.org/10.3390/brainsci9070168
APA StyleXie, Q., Zhao, W.-J., Ou, G.-Y., & Xue, W.-K. (2019). An Overview of Experimental and Clinical Spinal Cord Findings in Alzheimer’s Disease. Brain Sciences, 9(7), 168. https://doi.org/10.3390/brainsci9070168