Regional Analysis of the Brain Transcriptome in Mice Bred for High and Low Methamphetamine Consumption

 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Husbandry
2.2. Selective Breeding for MA Intake
2.3. Dissection of Tissue and Extraction of RNA
2.4. RNA-Seq
2.5. Network Analysis
2.6. Quantitative Reverse Transcriptase-Polymerase Chain Reaction (qRT-PCR)
2.7. Statistical Analyses
3. Results
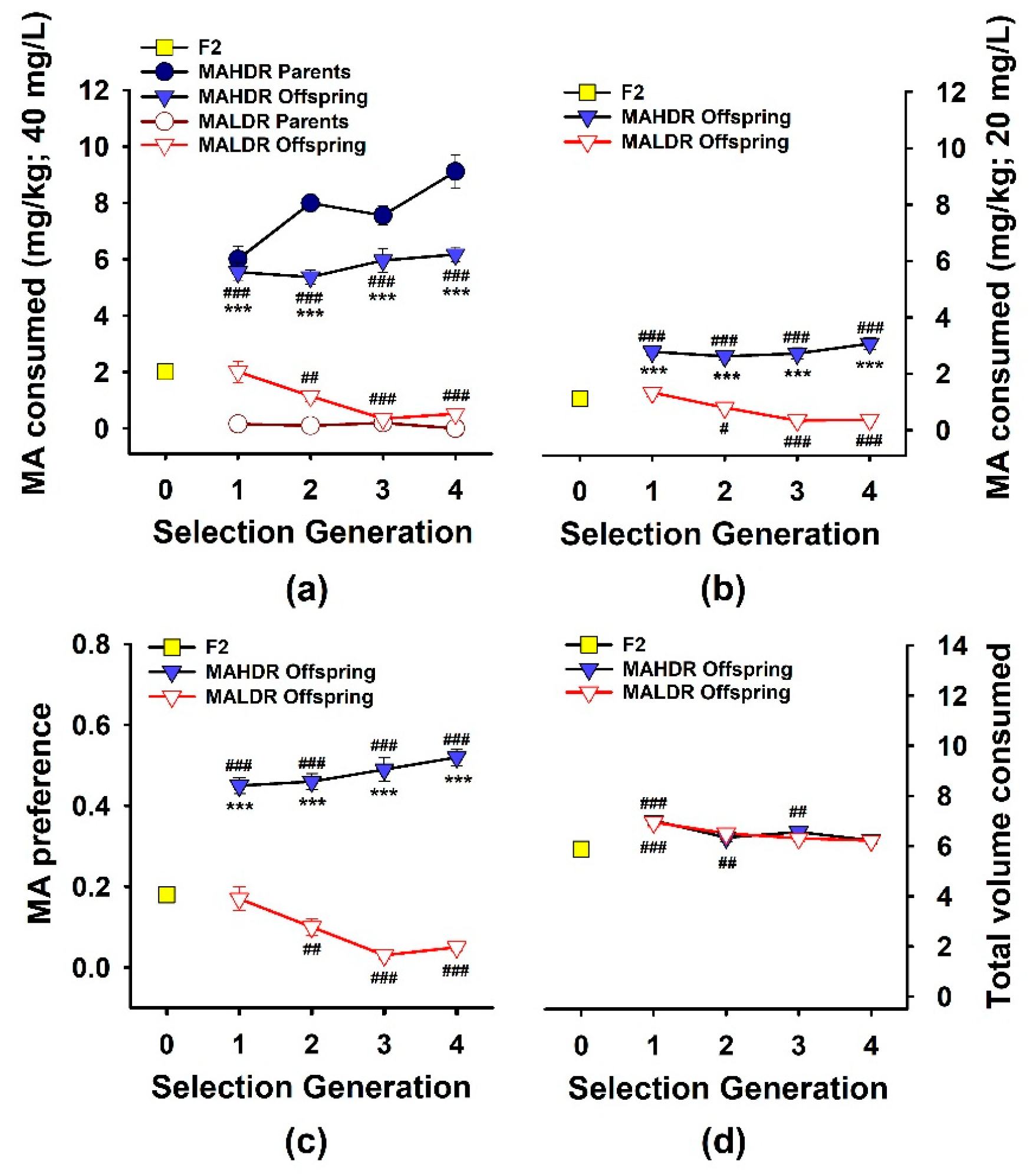
3.1. Response to Selection
3.2. Gene Expression in the VMB, PFC, and NAc
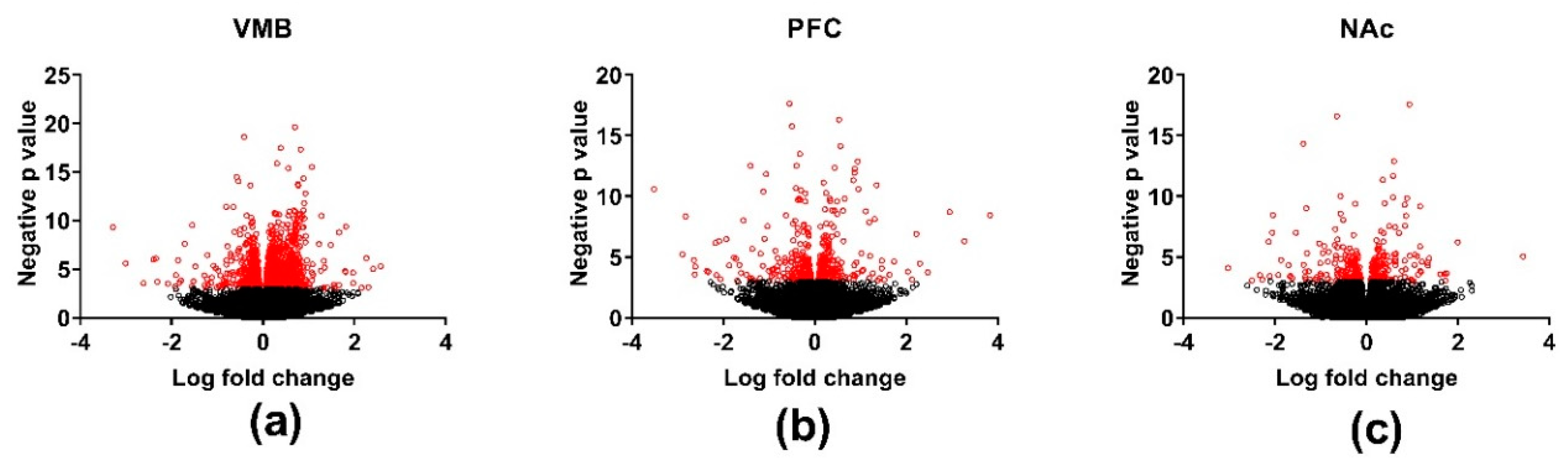
3.3. Differential Gene Expression across Brain Regions
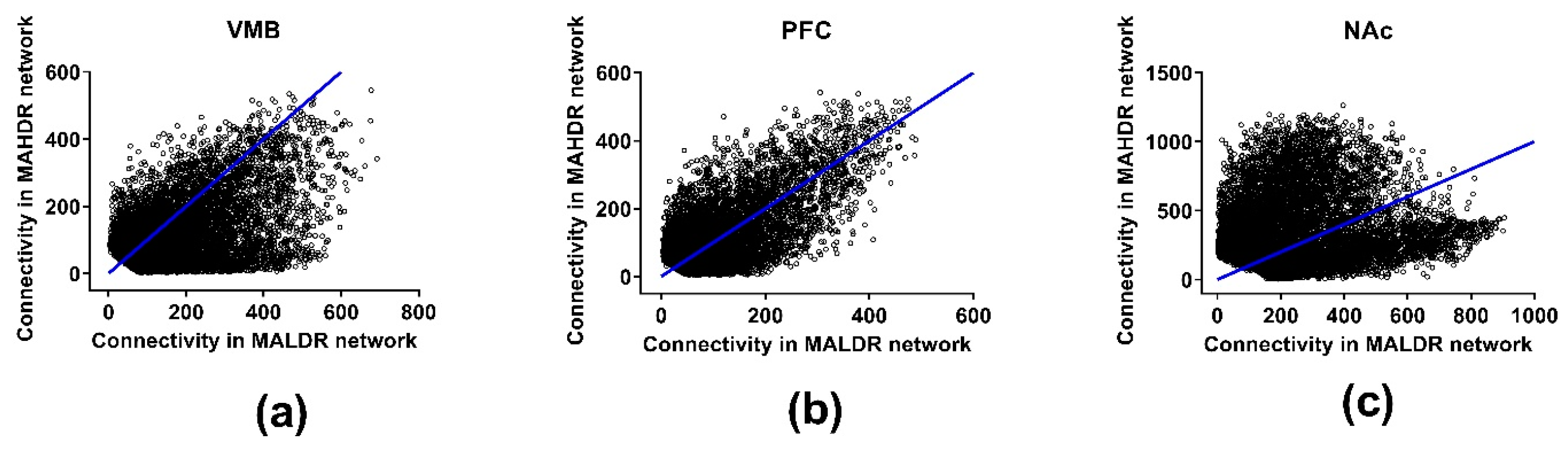
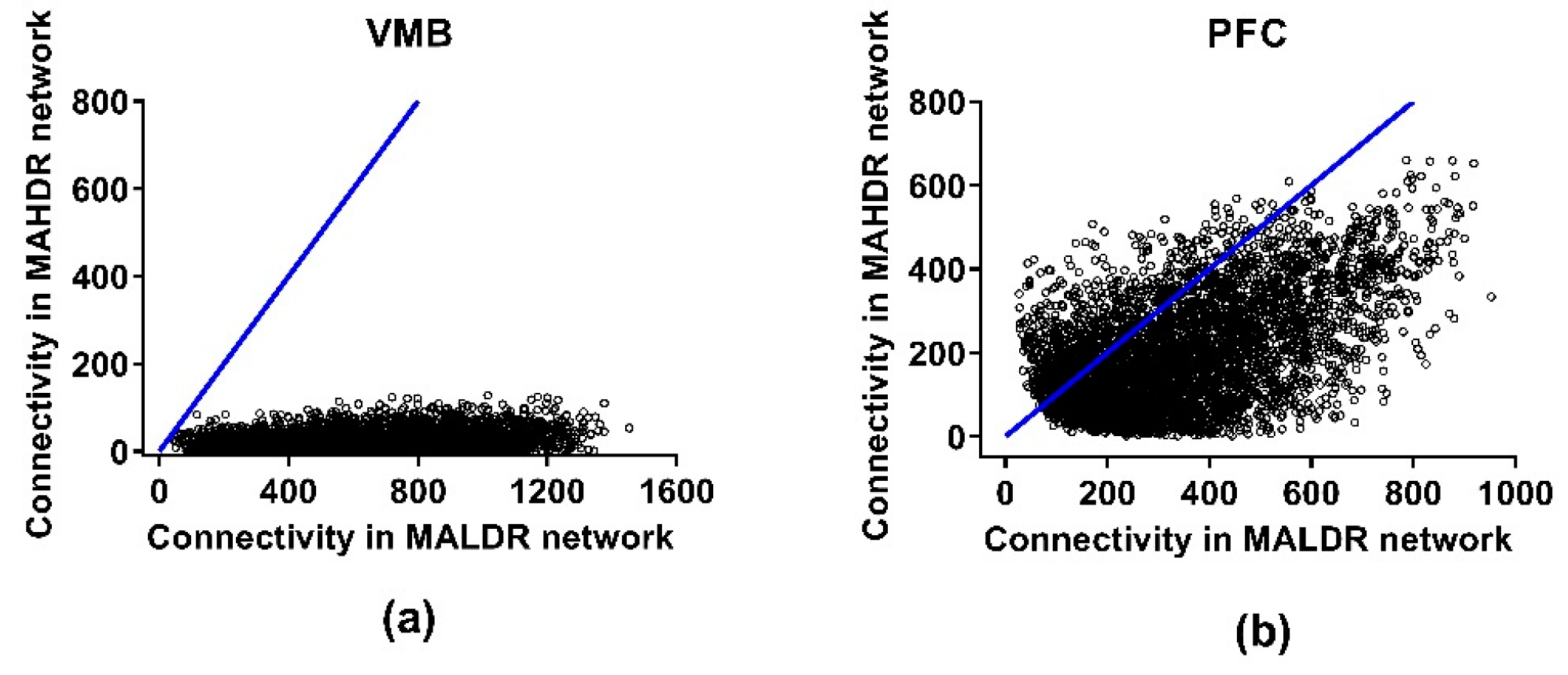
3.4. Connectivity
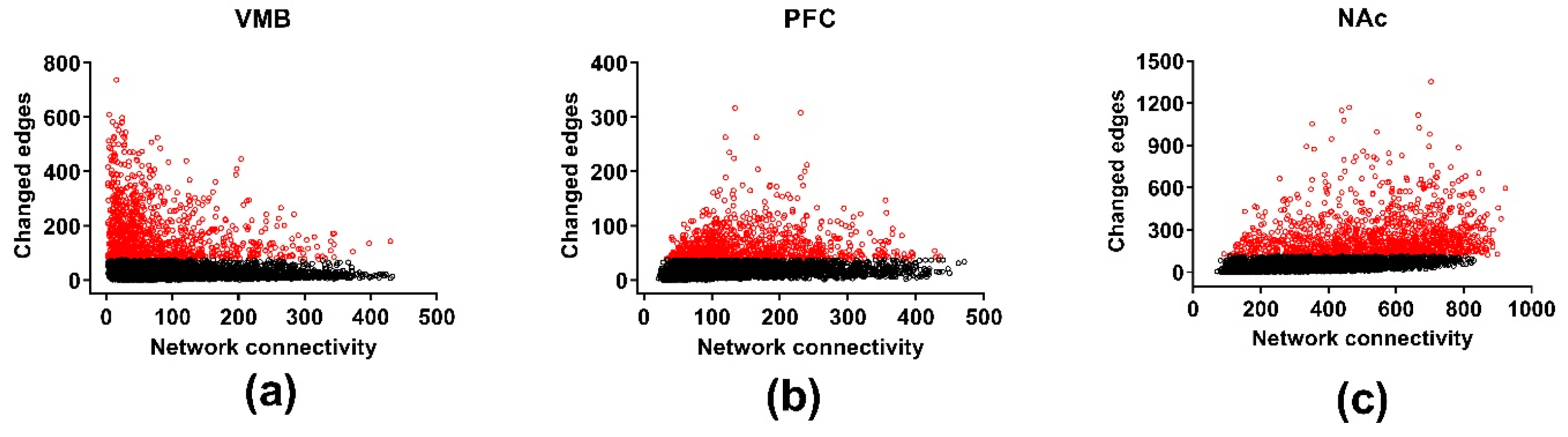
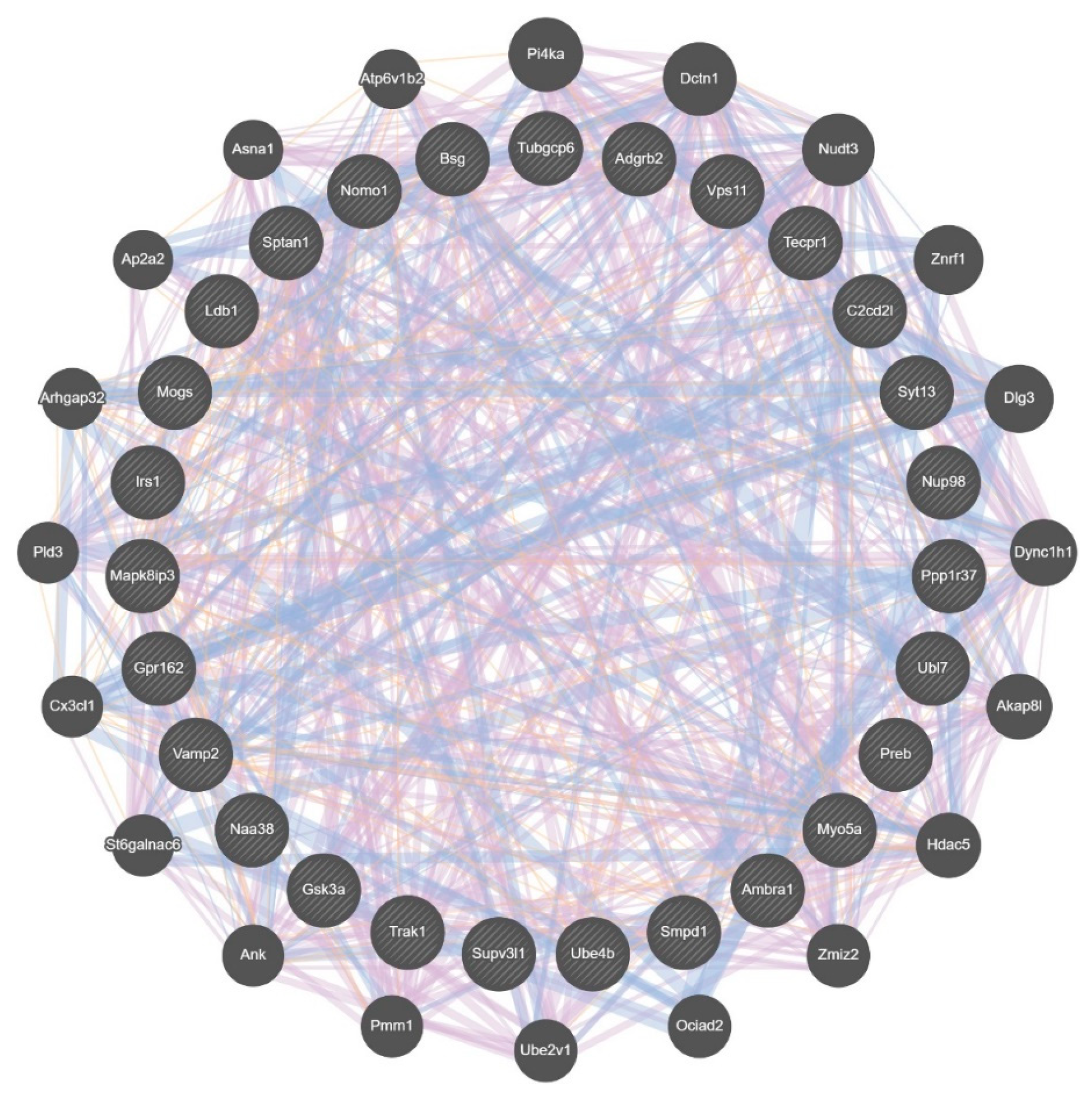
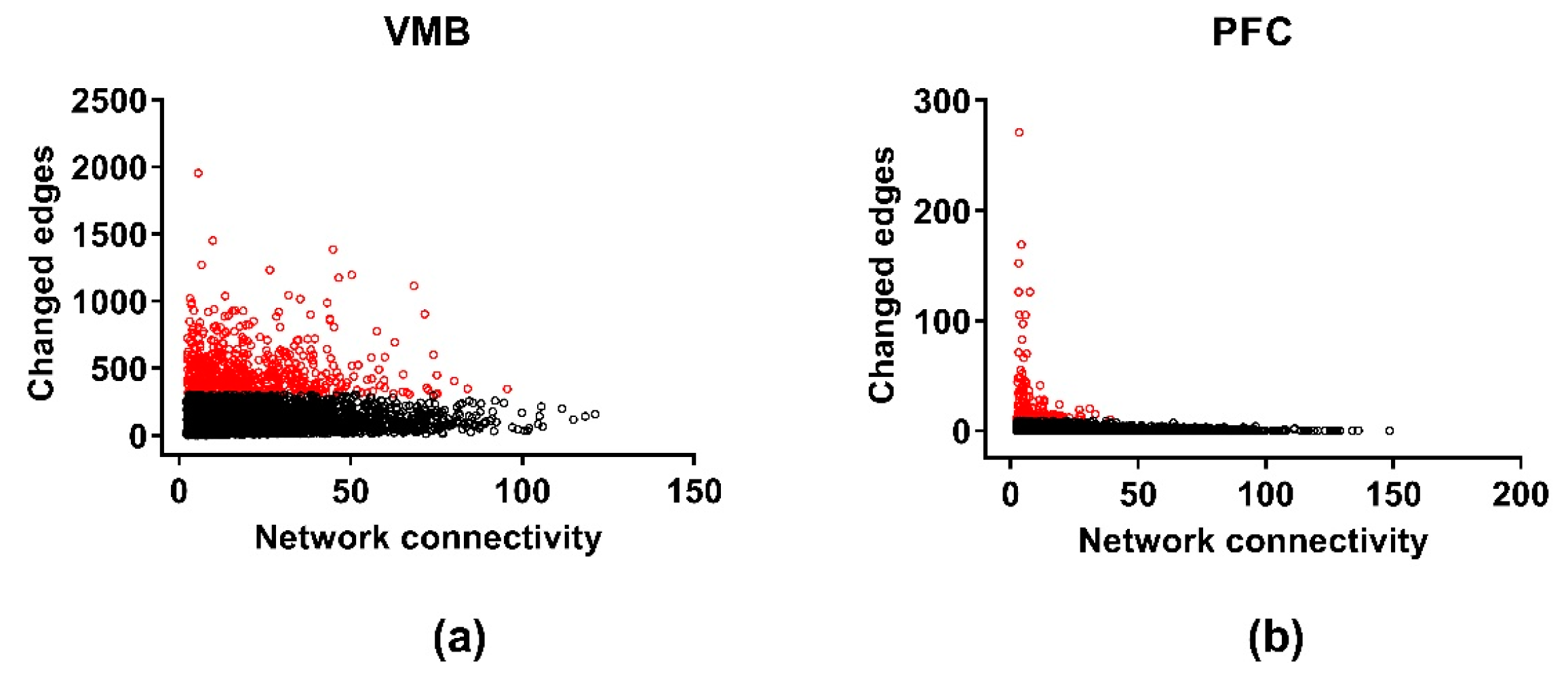
3.5. Differential Wiring across Brain Regions
3.6. Cosplicing across Brain Regions
3.7. Differential Splicing across Brain Regions
3.8. Differential Wiring of the Cospliced Genes across Brain Regions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mulligan, M.K.; Ponomarev, I.; Hitzemann, R.J.; Belknap, J.K.; Tabakoff, B.; Harris, R.A.; Crabbe, J.C.; Blednov, Y.A.; Grahame, N.J.; Phillips, T.J.; et al. Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proc. Natl. Acad. Sci. USA 2006, 103, 6368–6373. [Google Scholar] [CrossRef] [PubMed]
- Contet, C. Gene Expression Under the Influence: Transcriptional Profiling of Ethanol in the Brain. Curr. Psychopharmacol. 2012, 1, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.L.; Lopez, M.F.; Archer, K.J.; Wolen, A.R.; Becker, H.C.; Miles, M.F. Time-Course Analysis of Brain Regional Expression Network Responses to Chronic Intermittent Ethanol and Withdrawal: Implications for Mechanisms Underlying Excessive Ethanol Consumption. PLoS ONE 2016, 11, 0146257. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.B.; Zhang, L.; Kircher, D.; Wang, S.; Mayfield, R.D.; Crabbe, J.C.; Morrisett, R.A.; Harris, R.A.; Ponomarev, I. Dissecting brain networks underlying alcohol binge drinking using a systems genomics approach. Mol. Neurobiol. 2019, 56, 2791–2810. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Jayanthi, S.; McCoy, M.T.; Brannock, C.; Ladenheim, B.; Garrett, T.; Lehrmann, E.; Becker, K.G.; Cadet, J.L. Methamphetamine causes differential alterations in gene expression and patterns of histone acetylation/hypoacetylation in the rat nucleus accumbens. PLoS ONE 2012, 7, e34236. [Google Scholar] [CrossRef] [PubMed]
- Piechota, M.; Korostynski, M.; Sikora, M.; Golda, S.; Dzbek, J.; Przewlocki, R. Common transcriptional effects in the mouse striatum following chronic treatment with heroin and methamphetamine. Genes Brain Behav. 2012, 11, 404–414. [Google Scholar] [CrossRef]
- Sim, M.S.; Wu, Y.S.; Parhar, I.S.; Mohamed, Z.; Soga, T.; Pandy, V. MicroRNA expression signature of methamphetamine use and addiction in the rat nucleus accumbens. Metab. Brain Dis. 2017, 32, 1767–1783. [Google Scholar] [CrossRef]
- Peña, I.D.; Jeon, S.J.; Lee, E.; Ryu, J.H.; Shin, C.Y.; Noh, M.; Cheong, J.H. Neuronal development genes are key elements mediating the reinforcing effects of methamphetamine, amphetamine, and methylphenidate. Psychopharmacology 2013, 230, 399–413. [Google Scholar] [CrossRef]
- Cadet, J.L.; Brannock, C.; Jayanthi, S.; Krasnova, I.N. Transcriptional and epigenetic substrates of methamphetamine addiction and withdrawal: Evidence from a long-access self-administration model in the rat. Mol. Neurobiol. 2015, 51, 696–717. [Google Scholar] [CrossRef]
- Wheeler, J.M.; Reed, C.; Burkhart-Kasch, S.; Li, N.; Cunningham, C.L.; Janowsky, A.; Franken, F.H.; Hashimoto, J.G.; Scibelli, A.C.; Phillips, T.J.; et al. Genetically correlated effects of selective breeding for high and low methamphetamine consumption. Genes Brain Behav. 2009, 8, 758–771. [Google Scholar] [CrossRef]
- Belknap, J.K.; Mc Weeney, S.; Reed, C.; Burkhart-Kasch, S.; McKinnon, C.S.; Li, N.; Baba, H.; Scibelli, A.C.; Hitzemann, R.; Phillips, T.J. Genetic factors involved in risk for methamphetamine intake and sensitization. Mamm. Genome 2013, 24, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Yang, H.; Wang, L.; Liu, H.; Huo, H.; Zhang, C.; Liu, A.; Zhu, A.; Hu, J.; Lin, Y.; et al. Physiological and Transcriptome Analyses Reveal Short-Term Responses and Formation of Memory Under Drought Stress in Rice. Front. Genet. 2019, 10, 55. [Google Scholar] [CrossRef] [PubMed]
- Giannuzzi, D.; Marconato, L.; Ramy, E.; Ferraresso, S.; Scarselli, E.; Fariselli, P.; Nicosia, A.; Pegolo, S.; Leoni, G.; Laganga, P.; et al. Longitudinal transcriptomic and genetic landscape of radiotherapy response in canine melanoma. Vet. Comp. Oncol. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Warden, A.S.; Mayfield, R.D. Gene expression profiling in the human alcoholic brain. Neuropharmacology 2017, 122, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Darakjian, P.; McWeeney, S.; Colville, A.M.; Iancu, O.D.; Oberbeck, D.L.; Zheng, C.L.; Walter, N.A.R.; Harrington, C.A.; Searles, R.P.; Hitzemann, R.J. Effects of selection for ethanol preference on gene expression in the nucleus accumbens of HS-CC mice. Genes Brain Behav. 2017, 16, 462–471. [Google Scholar]
- Colville, A.M.; Iancu, O.D.; Lockwood, D.R.; Darakjian, P.; McWeeney, S.K.; Searles, R.; Zheng, C.; Hitzemann, R. Regional differences and similarities in the brain transcriptome for mice selected for ethanol preference from HS-CC founders. Front. Genet. 2018, 9, 300. [Google Scholar] [CrossRef]
- Hitzemann, R.; Bottomly, D.; Darakjian, P.; Walter, N.; Iancu, O.; Searles, R.; Wilmot, B.; McWeeney, S. Genes, behavior and next-generation RNA sequencing. Genes Brain Behav. 2013, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hitzemann, R.; Darakjian, P.; Walter, N.; Iancu, O.D.; Searles, R.; Mc Weeney, S. Introduction to Sequencing the Brain Transcriptome. Int. Rev. Neurobiol. 2014, 116, 1–19. [Google Scholar]
- Iancu, O.D.; Darakjian, P.; Kawane, S.; Bottomly, D.; Hitzemann, R.; McWeeney, S.K. Detection of Expression Quantitative Trait Loci in Complex Mouse Crosses: Impact and Alleviation of Data Quality and Complex Population Substructure. Front. Genet. 2012, 3, 157. [Google Scholar] [CrossRef]
- Iancu, O.D.; Colville, A.; Oberbeck, D.; Darakjian, P.; McWeeney, S.K.; Hitzemann, R. Cosplicing network analysis of mammalian brain RNA-Seq data utilizing WGCNA and Mantel correlations. Front. Genet. 2015, 6, 174. [Google Scholar] [CrossRef]
- Bienkowski, M.S.; Bowman, I.; Song, M.Y.; Gou, L.; Ard, T.; Cotter, K.; Zhu, M.; Benavidez, N.L.; Yamashita, S.; Abu-Jaber, J.; et al. Integration of gene expression and brain-wide connectivity reveals the multiscale organization of mouse hippocampal networks. Nat. Neurosci. 2018, 21, 1628–1643. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Kraus, W.L. From Discovery to Function: The Expanding Roles of Long Non-Coding RNAs in Physiology and Disease. Endocr. Rev. 2015, 36, 25–64. [Google Scholar] [CrossRef] [PubMed]
- National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals; The National Academies Press: Washington, DC, USA, 2011.
- Belknap, J.K.; Richards, S.P.; O’Toole, L.A.; Helms, M.L.; Phillips, T.J. Short-Term Selective Breeding as a Tool for QTL Mapping: Ethanol Preference Drinking in Mice. Behav. Genet. 1997, 27, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Shabani, S.; McKinnon, C.S.; Reed, C.; Cunningham, C.L.; Phillips, T.J. Sensitivity to Rewarding or Aversive Effects of Methamphetamine Determines Methamphetamine Intake. Genes Brain Behav. 2011, 10, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. Feature Counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A Transcriptome Database for Astrocytes, Neurons, and Oligodendrocytes: A New Resource for Understanding Brain Development and Function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef]
- Kamens, H.M.; Phillips, T.J. A role for neuronal nicotinic acetylcholine receptors in ethanol-induced stimulation, but not cocaine- or methamphetamine-induced stimulation. Psychopharmacology 2008, 196, 377–387. [Google Scholar] [CrossRef]
- Kamens, H.M.; McKinnon, C.S.; Li, N.; Helms, M.L.; Belknap, J.K.; Phillips, T.J. The α3 subunit gene of the nicotinic acetylcholine receptor is a candidate gene for ethanol stimulation. Genes Brain Behav. 2009, 8, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic. Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic. Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.A.; Verbitsky, M.; Suresh, R.; Kamens, H.M.; Reed, C.L.; Li, N.; Burkhart–Kasch, S.; McKinnon, C.S.; Belknap, J.K.; Gilliam, T.C.; et al. Gene expression differences in mice divergently selected for methamphetamine sensitivity. Mamm. Genome 2005, 16, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Eastwood, E.C.; Eshleman, A.J.; Janowsky, A.; Phillips, T.J. Verification of a genetic locus for methamphetamine intake and the impact of morphine. Mamm. Genome 2018, 29, 260–272. [Google Scholar] [CrossRef]
- Harkness, J.H.; Shi, X.; Janowsky, A.; Phillips, T.J. Trace Amine-Associated Receptor 1 Regulation of Methamphetamine Intake and Related Traits. Neuropsychopharmacology 2015, 40, 2175–2184. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Walter, N.A.R.; Harkness, J.H.; Neve, K.A.; Williams, R.W.; Lu, L.; Belknap, J.K.; Eshleman, A.J.; Phillips, T.J.; Janowsky, A. Genetic Polymorphisms Affect Mouse and Human Trace Amine-Associated Receptor 1 Function. PLoS ONE 2016, 11, 0152581. [Google Scholar] [CrossRef] [PubMed]
- Reed, C.; Baba, H.; Zhu, Z.; Erk, J.; Mootz, J.R.; Varra, N.M.; Williams, R.W.; Phillips, T.J. A spontaneous mutation in Taar1 impacts methamphetamine-related traits exclusively in DBA/2 mice from a single vendor. Front. Pharmacol. 2017, 8, 993. [Google Scholar] [CrossRef]
- Stafford, A.M.; Reed, C.; Baba, H.; Walter, N.A.R.; Mootz, J.R.K.; Williams, R.W.; Neve, K.A.; Fedorov, L.M.; Janowsky, A.; Phillips, T.J. Taar1 gene variants have a causal role in methamphetamine intake and response and interact with Oprm1. eLife 2019. submitted. [Google Scholar]
- Bottomly, D.; Walter, N.A.; Hunter, J.E.; Darakjian, P.; Kawane, S.; Buck, K.J.; Searles, R.P.; Mooney, M.; McWeeney, S.K.; Hitzemann, R. Evaluating gene expression in C57BL/6J and DBA/2J mouse striatum using RNA-Seq and microarrays. PLoS ONE 2011, 6, e17820. [Google Scholar] [CrossRef]
- Walter, N.A.; Bottomly, D.; Laderas, T.; Mooney, M.A.; Darakjian, P.; Searles, R.P.; Harrington, C.A.; McWeeney, S.K.; Hitzemann, R.; Buck, K.J. High throughput sequencing in mice: A platform comparison identifies a preponderance of cryptic SNPs. BMC Genom. 2009, 10, 379. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.-J.; Dang, D.-K.; Tran, T.-V.; Tran, H.-Q.; Jeong, J.H.; Nah, S.-Y.; Jang, C.-G.; Yamada, K.; Nabeshima, T.; Kim, H.-C. Current understanding of methamphetamine-associated dopaminergic neurodegeneration and psychotoxic behaviors. Arch. Pharmacal Res. 2017, 40, 403–428. [Google Scholar] [CrossRef] [PubMed]
- Miner, N.B.; Elmore, J.S.; Baumann, M.H.; Phillips, T.J.; Janowsky, A. Trace amine-associated receptor 1 regulation of methamphetamine-induced neurotoxicity. NeuroToxicology 2017, 63, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Conant, K.; Lim, S.T.; Randall, B.; Maguire-Zeiss, K.A. Matrix Metalloproteinase Dependent Cleavage of Cell Adhesion Molecules in the Pathogenesis of CNS Dysfunction with HIV and Methamphetamine. Curr. HIV Res. 2012, 10, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, H.; Yamada, K.; Nabeshima, T. Neuropsychotoxicity of Abused Drugs: Involvement of Matrix Metalloproteinase-2 and -9 and Tissue Inhibitor of Matrix Metalloproteinase-2 in Methamphetamine-Induced Behavioral Sensitization and Reward in Rodents. J. Pharmacol. Sci. 2008, 106, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Brown, S.; Shaikh, J.; Fishback, J.A.; Matsumoto, R.R. Relationship Between Methamphetamine Exposure and Matrix Metalloproteinase 9 Expression. NeuroReport 2008, 19, 1407–1409. [Google Scholar] [CrossRef] [PubMed]
- Conant, K.; Lonskaya, I.; Szklarczyk, A.; Krall, C.; Steiner, J.; Maguire-Zeiss, K.; Lim, S.T.; Maguire-Zeiss, K. Methamphetamine-associated cleavage of the synaptic adhesion molecule intercellular adhesion molecule-5. J. Neurochem. 2011, 118, 521–532. [Google Scholar] [CrossRef]
- Lubbers, B.R.; Smit, A.B.; Spijker, S.; Oever, M.C.V.D. Neural ECM in Addiction, Schizophrenia, and Mood Disorder; Elsevier BV: Amsterdam, The Netherlands, 2014; Volume 214, pp. 263–284. [Google Scholar]
- Lasek, A.W. Effects of Ethanol on Brain Extracellular Matrix: Implications for Alcohol Use Disorder. Alcohol. Clin. Exp. Res. 2016, 40, 2030–2042. [Google Scholar] [CrossRef]
- Smith, A.C.W.; Kupchik, Y.M.; Scofield, M.D.; Gipson, C.D.; Wiggins, A.; Thomas, C.A.; Kalivas, P.W. Synaptic plasticity mediating cocaine relapse requires matrix metalloproteinases. Nat. Neurosci. 2014, 17, 1655–1657. [Google Scholar] [CrossRef]
- Smith, A.C.; Scofield, M.D.; Kalivas, P.W. The tetrapartite synapse: Extracellular matrix remodeling contributes to corticoaccumbens plasticity underlying drug addiction. Brain Res. 2015, 1628, 29–39. [Google Scholar] [CrossRef]
- Bobadilla, A.-C.; Heinsbroek, J.A.; Gipson, C.D.; Griffin, W.C.; Fowler, C.D.; Kenny, P.J.; Kalivas, P.W. Corticostriatal Plasticity, Neuronal Ensembles and Regulation of Drug-Seeking Behavior; Elsevier BV: Amsterdam, The Netherlands, 2017; Volume 235, pp. 93–112. [Google Scholar]
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Zealy, R.W.; Fomin, M.; Davila, S.; Makowsky, D.; Thigpen, H.; McDowell, C.H.; Cummings, J.C.; Lee, E.S.; Kwon, S.-H.; Min, K.-W.; et al. Long noncoding RNA complementarity and target transcripts abundance. Biochim. et Biophys. Acta (BBA) Gene Regul. Mech. 2018, 1861, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Rubio, F.J.; Liu, Q.-R.; Li, X.; Cruz, F.C.; Leão, R.M.; Warren, B.L.; Kambhampati, S.; Babin, K.R.; McPherson, K.B.; Cimbro, R.; et al. Context-Induced Reinstatement of Methamphetamine Seeking Is Associated with Unique Molecular Alterations in Fos-Expressing Dorsolateral Striatum Neurons. J. Neurosci. 2015, 35, 5625–5639. [Google Scholar] [CrossRef] [PubMed]
- Cen, X.; Nitta, A.; Ibi, D.; Zhao, Y.; Niwa, M.; Taguchi, K.; Hamada, M.; Ito, Y.; Wang, L.; Nabeshima, T. Identification of Piccolo as a regulator of behavioral plasticity and dopamine transporter internalization. Mol. Psychiatry 2008, 13, 451–463. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Uno, K.; Nishizawa, D.; Seo, S.; Takayama, K.; Matsumura, S.; Sakai, N.; Ohi, K.; Nabeshima, T.; Hashimoto, R.; Ozaki, N.; et al. The Piccolo intronic single nucleotide polymorphism rs13438494 regulates dopamine and serotonin uptake and shows associations with dependence-like behavior in genomic association study. Curr. Mol. Med. 2015, 15, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Lominac, K.D.; Quadir, S.G.; Barrett, H.M.; McKenna, C.L.; Schwartz, L.M.; Ruiz, P.N.; Wroten, M.G.; Campbell, R.R.; Miller, B.W.; Holloway, J.J.; et al. Prefrontal glutamate correlates of methamphetamine sensitization and preference. Eur. J. Neurosci. 2016, 43, 689–702. [Google Scholar] [CrossRef]
- Szumlinski, K.K.; Lominac, K.D.; Campbell, R.R.; Cohen, M.; Fultz, E.K.; Brown, C.N.; Miller, B.W.; Quadir, S.G.; Martin, D.; Thompson, A.B.; et al. Methamphetamine addiction vulnerability: The glutamate, the bad, and the ugly. Biol. Psychiatry 2017, 81, 959–970. [Google Scholar] [CrossRef]
- Thomas, D.M.; Francescutti-Verbeem, D.M.; Liu, X.; Kuhn, D.M. Identification of differentially regulated transcripts in mouse striatum following methamphetamine treatment—An oligonucleotide microarray approach. J. Neurochem. 2004, 88, 380–393. [Google Scholar] [CrossRef]
- Bortell, N.; Basova, L.; Semenova, S.; Fox, H.S.; Ravasi, T.; Marcondes, M.C.G. Astrocyte-specific overexpressed gene signatures in response to methamphetamine exposure in vitro. J. Neuroinflammation 2017, 14, 343. [Google Scholar] [CrossRef]
- Revel, F.G.; Moreau, J.-L.; Gainetdinov, R.R.; Bradaia, A.; Sotnikova, T.D.; Mory, R.; Durkin, S.; Zbinden, K.G.; Norcross, R.; Meyer, C.A.; et al. TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proc. Natl. Acad. Sci. USA 2011, 108, 8485–8490. [Google Scholar] [CrossRef]
- Revel, F.G.; Meyer, C.A.; Bradaia, A.; Jeanneau, K.; Calcagno, E.; André, C.B.; Haenggi, M.; Miss, M.-T.; Galley, G.; Norcross, R.D.; et al. Brain-Specific Overexpression of Trace Amine-Associated Receptor 1 Alters Monoaminergic Neurotransmission and Decreases Sensitivity to Amphetamine. Neuropsychopharmacology 2012, 37, 2580–2592. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-F.; Siemian, J.N.; Seaman, R.; Zhang, Y.; Li, J.-X. Role of TAAR1 within the Subregions of the Mesocorticolimbic Dopaminergic System in Cocaine-Seeking Behavior. J. Neurosci. 2017, 37, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-F.; Li, J.-X. TAAR1 in Addiction: Looking Beyond the Tip of the Iceberg. Front. Pharmacol. 2018, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Jing, L.; Li, J.-X. Trace amine-associated receptor 1: A promising target for the treatment of psychostimulant addiction. Eur. J. Pharmacol. 2015, 761, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Just, M.A.; Keller, T.A. Converging measures of neural change at the microstructural, informational, and cortical network levels in the hippocampus during the learning of the structure of organic compounds. Brain Struct. Funct. 2019, 224, 1345–1357. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.S.; Underhill, S.M.; Stolz, D.B.; Murdoch, G.H.; Thiels, E.; Romero, G.; Amara, S.G. Amphetamine activates Rho GTPase signaling to mediate dopamine transporter internalization and acute behavioral effects of amphetamine. Proc. Natl. Acad. Sci. USA 2015, 112, E7138–E7147. [Google Scholar] [CrossRef] [PubMed]
- Koob, G.F.; Volkow, N.D. Neurocircuitry of addiction. Neuropsychopharmacology 2010, 35, 217–238. [Google Scholar] [CrossRef]
- Koob, G.F.; Volkow, N.D. Neurobiology of addiction: A neurocircuitry analysis. Lancet Psychiatry 2016, 3, 760–773. [Google Scholar] [CrossRef]
- Volkow, N.D.; Wang, G.-J.; Fowler, J.S.; Tomasi, D.; Telang, F.; Baler, R. Addiction: Decreased reward sensitivity and increased expectation sensitivity conspire to overwhelm the brain’s control circuit. BioEssays 2010, 32, 748–755. [Google Scholar] [CrossRef]
- Wise, R.A.; Bozarth, M.A. Brain mechanisms of drug reward and euphoria. Psychiatr. Med. 1985, 3, 445–460. [Google Scholar]
- Aguilar-Valles, A.; Vaissière, T.; Rumbaugh, G.; Griggs, E.M.; Mikaelsson, M.A.; Takács, I.F.; Young, E.J.; Miller, C.A. Methamphetamine-associated memory is regulated by a writer and an eraser of permissive histone methylation. Boil. Psychiatry 2014, 76, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Xie, T.; Piao, Y.; Dillon-Carter, O.; Kargul, G.J.; Lim, M.K.; Chrest, F.J.; Wersto, R.; Rowley, D.L.; Juhaszova, M.; et al. A murine dopamine neuron-specific cDNA library and microarray: Increased COX1 expression during methamphetamine neurotoxicity. Neurobiol. Dis. 2001, 8, 822–833. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bosch, P.J.; Benton, M.C.; Macartney-Coxson, D.; Kivell, B.M.; Benton, M.; Kivell, B. mRNA and microRNA analysis reveals modulation of biochemical pathways related to addiction in the ventral tegmental area of methamphetamine self-administering rats. BMC Neurosci. 2015, 16, 379. [Google Scholar] [CrossRef] [PubMed]
- Breen, M.S.; Uhlmann, A.; Nday, C.M.; Glatt, S.J.; Mitt, M.; Metsalpu, A.; Stein, D.J.; Illing, N. Candidate gene networks and blood biomarkers of methamphetamine-associated psychosis: An integrative RNA-sequencing report. Transl. Psychiatry 2016, 6, e802. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.L.; Brannock, C.; Ladenheim, B.; McCoy, M.T.; Beauvais, G. Methamphetamine preconditioning causes differential changes in striatal transcriptional responses to large doses of the drug. Dose Response 2011, 9, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.L.; Jayanthi, S.; McCoy, M.T.; Ladenheim, B.; Saint-Preux, F.; Lehrmann, E.; De, S.; Becker, K.G.; Brannock, C. Genome-wide profiling identifies a subset of methamphetamine (METH)-induced genes associated with METH-induced increased H4K5Ac binding in the rat striatum. BMC Genom. 2013, 14, 545. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.L.; Brannock, C.; Ladenheim, B.; McCoy, M.T.; Krasnova, I.N.; Lehrmann, E.; Becker, K.G.; Jayanthi, S. Enhanced Upregulation of CRH mRNA Expression in the Nucleus Accumbens of Male Rats after a Second Injection of Methamphetamine Given Thirty Days Later. PLoS ONE 2014, 9, e84665. [Google Scholar] [CrossRef]
- Choi, M.R.; Chun, J.-W.; Kwak, S.M.; Bang, S.H.; Jin, Y.-B.; Lee, Y.; Kim, H.-N.; Chang, K.-T.; Chai, Y.G.; Lee, S.-R.; et al. Effects of acute and chronic methamphetamine administration on cynomolgus monkey hippocampus structure and cellular transcriptome. Toxicol. Appl. Pharmacol. 2018, 355, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Funada, M.; Zhou, X.; Satoh, M.; Wada, K. Profiling of Methamphetamine-Induced Modifications of Gene Expression Patterns in the Mouse Brain. Ann. N. Y. Acad. Sci. 2004, 1025, 76–83. [Google Scholar] [CrossRef]
- Jayanthi, S.; McCoy, M.T.; Ladenheim, B.; Cadet, J.L. Methamphetamine Causes Coordinate Regulation of Src, Cas, Crk, and the Jun N-Terminal Kinase-Jun Pathway. Mol. Pharmacol. 2002, 61, 1124–1131. [Google Scholar] [CrossRef]
- Krasnova, I.N.; Chiflikyan, M.; Justinova, Z.; McCoy, M.T.; Ladenheim, B.; Jayanthi, S.; Quintero, C.; Brannock, C.; Barnes, C.; Adair, J.E.; et al. CREB Phosphorylation Regulates Striatal Transcriptional Responses in the Self-Administration Model of Methamphetamine Addiction in the Rat. Neurobiol. Dis. 2013, 58, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Krasnova, I.N.; Gerra, M.C.; Walther, D.; Jayanthi, S.; Ladenheim, B.; McCoy, M.T.; Brannock, C.; Cadet, J.L. Compulsive methamphetamine taking in the presence of punishment is associated with increased oxytocin expression in the nucleus accumbens of rats. Sci. Rep. 2017, 7, 8331. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, Y.; Kubota, Y.; Ito, C. Serial Analysis of Gene Expression in Methamphetamine- and Phencyclidine-Treated Rodent Cerebral Cortices: Are There Common Mechanisms? Ann. New York Acad. Sci. 2004, 1025, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Piechota, M.; Korostynski, M.; Solecki, W.; Gieryk, A.; Slezak, M.; Bilecki, W.; Ziolkowska, B.; Kostrzewa, E.; Cymerman, I.; Swiech, L.; et al. The dissection of transcriptional modules regulated by various drugs of abuse in the mouse striatum. Genome Boil. 2010, 11, R48. [Google Scholar] [CrossRef] [PubMed]
- Sodersten, E.; Toskas, K.; Rraklli, V.; Tiklová, K.; Björklund, Å.K.; Ringnér, M.; Perlmann, T.; Holmberg, J. A comprehensive map coupling histone modifications with gene regulation in adult dopaminergic and serotonergic neurons. Nat. Commun. 2018, 9, 1226. [Google Scholar] [CrossRef] [PubMed]
- Torres, O.V.; McCoy, M.T.; Ladenheim, B.; Jayanthi, S.; Brannock, C.; Tulloch, I.; Krasnova, I.N.; Cadet, J.L. CAMKII-conditional deletion of histone deacetylase 2 potentiates acute methamphetamine-induced expression of immediate early genes in the mouse nucleus accumbens. Sci. Rep. 2015, 5, 13396. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Tong, L.; Barrett, T.; Yuan, J.; Hatzidimitriou, G.; McCann, U.D.; Becker, K.G.; Donovan, D.M.; Ricaurte, G.A. Changes in Gene Expression Linked to Methamphetamine-Induced Dopaminergic Neurotoxicity. J. Neurosci. 2002, 22, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Xiong, K.; Long, L.; Zhang, X.; Qu, H.; Deng, H.; Ding, Y.; Cai, J.; Wang, S.; Wang, M.; Liao, L.; et al. Overview of long non-coding RNA and mRNA expression in response to methamphetamine treatment in vitro. Toxicol. In Vitro 2017, 44, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Imai, K.; Takamatsu, Y.; Kamegaya, E.; Kishida, M.; Hagino, Y.; Hara, Y.; Shimada, K.; Yamamoto, T.; Sora, I.; et al. Methamphetamine modulation of gene expression in the brain: Analysis using customized cDNA microarray system with the mouse homologues of KIAA genes. Mol. Brain Res. 2005, 137, 40–46. [Google Scholar] [CrossRef]
- Yang, M.H.; Jung, M.-S.; Lee, M.J.; Yoo, K.H.; Yook, Y.J.; Park, E.Y.; Choi, S.H.; Suh, Y.J.; Kim, K.-W.; Park, J.H. Gene expression profiling of the rewarding effect caused by methamphetamine in the mesolimbic dopamine system. Mol. Cells 2008, 26, 121–130. [Google Scholar]
- Yazdani, N.; Parker, C.C.; Shen, Y.; Reed, E.R.; Guido, M.A.; Kole, L.A.; Kirkpatrick, S.L.; Lim, J.E.; Sokoloff, G.; Cheng, R.; et al. Hnrnph1 is a quantitative trait gene for methamphetamine sensitivity. PLoS Genet. 2015, 11, e1005713. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, N.; Shen, Y.; Johnson, W.E.; Bryant, C.D. Striatal transcriptome analysis of a congenic mouse line (chromosome 11: 50–60Mb) exhibiting reduced methamphetamine sensitivity. Genom. Data 2016, 8, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhu, J.; Liu, Y.; Chen, Y.; Li, Y.; Huang, L.; Chen, S.; Li, T.; Dang, Y.; Chen, T. Methamphetamine induces alterations in the long non-coding RNAs expression profile in the nucleus accumbens of the mouse. BMC Neurosci. 2015, 16, 18. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Li, J.; Dong, N.; Guan, F.; Liu, Y.; Ma, D.; Goh, E.L.K.; Chen, T. mRNA changes in nucleus accumbens related to methamphetamine addiction in mice. Sci. Rep. 2016, 6, 36993. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhu, J.; Liu, Y.; Chen, Y.; Li, Y.; Chen, S.; Li, T.; Dang, Y.; Chen, T. Chronic methamphetamine regulates the expression of MicroRNAs and putative target genes in the nucleus accumbens of mice. J. Neurosci. Res. 2015, 93, 1600–1610. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Th | Drd1 | Drd2 | Drd5 | Slc6a3 | Comt | |
|---|---|---|---|---|---|---|
| VMB | 19235 | 135 | 3760 | 128 | 31070 | 3229 |
| PFC | 70 | 267 | 54 | 154 | ND | 2817 |
| NAc | 534 | 13086 | 6049 | 354 | ND | 5950 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hitzemann, R.; Iancu, O.D.; Reed, C.; Baba, H.; Lockwood, D.R.; Phillips, T.J. Regional Analysis of the Brain Transcriptome in Mice Bred for High and Low Methamphetamine Consumption. Brain Sci. 2019, 9, 155. https://doi.org/10.3390/brainsci9070155
Hitzemann R, Iancu OD, Reed C, Baba H, Lockwood DR, Phillips TJ. Regional Analysis of the Brain Transcriptome in Mice Bred for High and Low Methamphetamine Consumption. Brain Sciences. 2019; 9(7):155. https://doi.org/10.3390/brainsci9070155
Chicago/Turabian StyleHitzemann, Robert, Ovidiu D. Iancu, Cheryl Reed, Harue Baba, Denesa R. Lockwood, and Tamara J. Phillips. 2019. "Regional Analysis of the Brain Transcriptome in Mice Bred for High and Low Methamphetamine Consumption" Brain Sciences 9, no. 7: 155. https://doi.org/10.3390/brainsci9070155
APA StyleHitzemann, R., Iancu, O. D., Reed, C., Baba, H., Lockwood, D. R., & Phillips, T. J. (2019). Regional Analysis of the Brain Transcriptome in Mice Bred for High and Low Methamphetamine Consumption. Brain Sciences, 9(7), 155. https://doi.org/10.3390/brainsci9070155