1. Introduction
Hereditary spastic paraplegias (HSPs) are a group of neurodegenerative disorders characterized by weakness and spasticity of the lower extremities. HSPs are classified clinically, or as to the inheritance pattern, or causative genes. According to clinical information, HSPs are classified into a pure form, which presents leg spasticity accompanied by bladder disturbance and disturbance of vibration sense, and a complicated form, which presents leg spasticity and a lot of complications such as ataxia, a thin corpus callosum, chorioretinal dystrophy, mental retardation, extrapyramidal signs, and peripheral neuropathy [
1]. As to the mode of inheritance, HSPs are classified into autosomal dominant, autosomal recessive, X-linked, and mitochondrial inheritance. Complicated forms tend to exhibit autosomal recessive rather than autosomal dominant inheritance. To date, the causative genes or gene loci of HSPs, from SPG1 to SPG79, have been reported, and other hereditary diseases including Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS), Chediak-Higashi syndrome, adrenoleukodystrophy, dystonia 4, and others, also exhibit leg spasticity as their one of the symptoms.
There are estimated to be 3–10 HSP patients per 100,000 individuals in Europe [
2,
3]. On the other hand, in Japan, there are estimated to be 0.2 cases per 100,000 individuals [
4]. However, it has not been understood what subtypes of HSP exist in Japan. Furthermore, there is no causative treatment for HSPs so far. Therefore, the Ministry of Health, Labor and Welfare in Japan formed the Research Committee for Ataxic Disease to clarify the molecular epidemiology and pathogeneses of ataxic diseases including HSPs, and to identify targets of treatment. The Japan Spastic Paraplegia Research Consortium (JASPAC) is one of the groups of the Research Committee for Ataxic Disease. Here, we describe the JASPAC and its findings in detail.
2. Patients and Methods
The JASPAC was established in 2006. Its purpose was to collect genome resources for HSPs, to conduct comprehensive mutation analysis, to clarify the molecular epidemiologies of HSPs, and to clarify the molecular pathogeneses of HSPs. We collected HSP families using the family history or clinical information. Our HSP families include probands having affected parents or children, probands having an affected brother or sister, probands having parents of consanguineous marriage, and probands showing a complicated form with normal parents. According to our criteria, we have already recruited 714 patients. Written informed consent was obtained from all participants. We collected neurological examination results through our survey form.
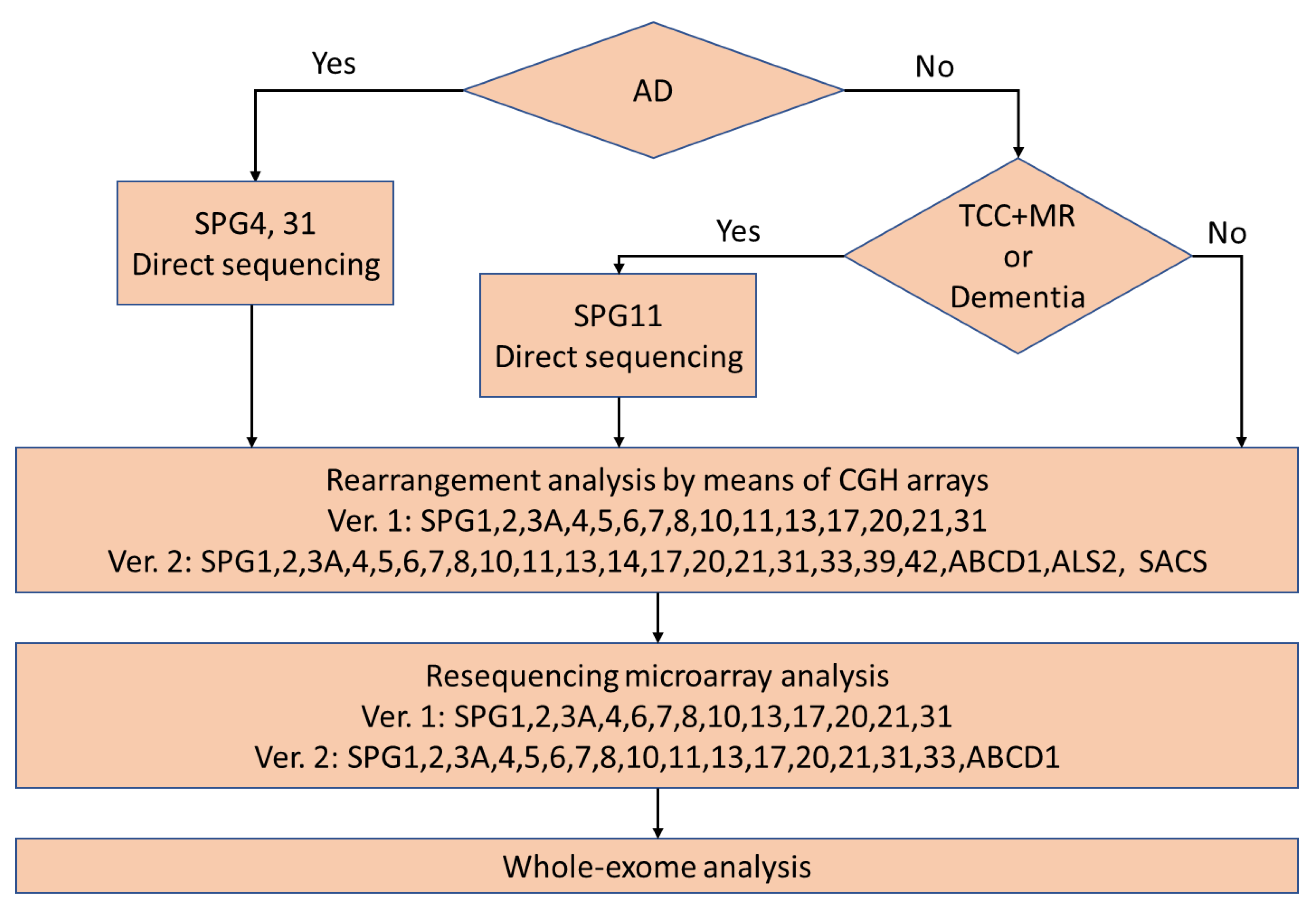
Our previous flow chart for the comprehensive mutation analysis is presented in
Figure 1. Patients determined to be autosomal dominant were subjected to direct sequencing of SPG4 and SPG31 at first. Patients with dementia or a thin corpus callosum and mental retardation were subjected to direct sequencing of SPG11 at first. Then, other patients and patients with no mutation of SPG4, SPG31, or SPG11 were subjected to rearrangement analysis by means of comparative genomic hybridization. The next analysis was resequencing microarray analysis. Finally, whole-exome analysis was performed. Exon capture was performed using a SureSelect All Exon V4+UTRs Kit and a V5+UTRs or V6+UTRs Kit followed by massively parallel sequencing using an Illumina Hiseq 2000 (100bp paired end). We aligned the exome data with a Burrows-Wheeler Aligner and extracted single nucleotide variations using Sequence Alignment/Map (SAM) tools. We checked for causative genes of neurodegenerative diseases. Furthermore, we compared the variants and their frequencies in an in-house control database consisting of 1261 subjects registered in Exome Aggregation Consortium (ExAC). We removed variants with a minor allele frequency of >0.2%, considering the disease frequency of HSP in Japan. Currently, we are performing whole-exome analysis before comparative genomic hybridization and resequencing microarray analysis. To date, we have completed the analysis in 383 patients.
3. Results
We found 224 patients (58%) with variants for 45 known HSP or neurodegenerative genes. These patients are listed in
Table 1. The most frequent HSP was SPG4 (100 patients, 26%) among all HSP patients, followed by SPG31 (14 patients, 3.7%), SPG3A (13 patients, 3.4%), SPG11 (10 patients, 2.6%), and SPG10 (six patients, 1.6%). Other HSP genotypes, including SPG55, were found in five patients or less [
5]. We found 25 causative genes in each one HSP family. Furthermore, we identified a lot of variants of other genes associated with different diseases, including
LYST [
6],
SACS,
ABCD1,
TUBB4A,
ALS2,
KCNA2,
ABHD12,
ANO10,
CLCN1,
CSF1R,
GALC,
GFAP,
KCND3,
MECP2,
OPA1,
PLA2G6,
POLR3A,
PSEN1,
RNASEH1,
SLC25A15,
SYNE1,
TYROBP,
VPS13C, and
WDR45. These genes were reported as the cause of spasticity as one of the symptoms. Therefore, we classified these patients as complicated HSP patients accordingly.
We present clinical information on major pure form HSPs such as SPG4, SPG3A, SPG31, and SPG10 in
Table 2. The average onset ages were 30.6, 14.3, 20.7 and 26.7 years old, respectively. Some of the patients exhibited ataxia, 9.0%, 7.7%, 7.0% and 16.7%, respectively. Neuropathy was also found in 31.0%, 0%, 29.0% and 33.3%, respectively.
4. Discussion
Our comprehensive mutation analysis revealed causative gene mutations in 224 HSP patients (58%). According to earlier reports [
7,
8], exome analysis revealed causative variants in 36% and 46% of their case series. Our HSP patients were collected carefully, which meant patients have to have a clear family history of autosomal dominant inheritance, severe phenotype or a family history that indicated autosomal recessive inheritance. This might have led to the high percentage of genetic diagnosis.
This study further expands the earlier molecular analyses of HSPs in Japan [
7]. SPG4 is the most frequent cause of HSPs in Japan, as it is worldwide. It is the most frequent in 26% of the HSP families, which is similar to the earlier reported frequency [
8]. Only three other HSPs, i.e., SPG31, SPG3A and SPG11, were found in over ten patients. HSPs with less than ten patients were caused by 44 genes. This indicates that HSPs are extremely heterogeneous diseases.
Furthermore, 24 genes, which comprise half the causative genes identified in this study, are not “SPG” genes, including the causative genes of Parkinson’s disease, amyotrophic lateral sclerosis, adrenoleukodystrophy, and others. These hereditary neurodegenerative disorders sometimes reveal upper motor neuron impairment. Furthermore, hereditary neurodegenerative diseases often involve several impaired systems, including the pyramidal tracts. Therefore, it is necessary to analyze genes comprehensively. JASPAC is succeeding in performing comprehensive gene analysis of HSP patients.
Our other purpose is to clarify the natural histories of HSPs and molecular pathogeneses of HSPs. To clarify the natural histories, we are tracking collected patients prospectively. Furthermore, we are now trying to find new causative genes and elucidate the molecular mechanisms underlying HSPs.
Author Contributions
Conceptualization, S.T. and Y.T.; Methodology, H.I. and S.T.; Formal Analysis, K.K. and H.I.; Resources, K.K. and Y.T.; Data Curation, K.K. and H.I.; Writing-Original Draft Preparation, K.K.; Writing-Review & Editing, H.I., S.T. and Y.T.; Project Administration, K.K., H.I., S.T. and Y.T.
Funding
This work was supported by Grants-in-Aid from the Research Committee for Ataxic Disease (Y.T.), the Ministry of Health, Labor and Welfare, Japan, JSPS KAKENHI Grant Numbers JP17K17772 (K.K.) and JP18K07495 (Y.T.) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan, and grants for AMED under Grant Number JP 17ek0109078 (Y.T.).
Acknowledgments
We thank all participants and cooperating doctors in the Japan Spastic Paraplegia Research Consortium.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fink, J.K. Hereditary spastic paraplegia: Clinical principles and genetic advances. Semin. Neurol. 2014, 34, 293–305. [Google Scholar] [CrossRef] [PubMed]
- McMonagle, P.; Webb, S.; Hutchinson, M. The prevalence of “pure” autosomal dominant hereditary spastic paraparesis in the island of Ireland. J. Neurol. Neurosurg. Psychiatry 2002, 72, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Coutinho, P.; Pinheiro, C.D.; Neves, J.M.; Serrano, P. Hereditary ataxias and spastic paraplegias: Methodological aspects of a prevalence study in Portugal. J. Clin. Epidemiol. 1997, 50, 1377–1384. [Google Scholar] [CrossRef]
- Hirayama, K.; Takayanagi, T.; Nakamura, R.; Yanagisawa, N.; Hattori, T.; Kita, K.; Yanagimoto, S.; Fujita, M.; Nagaoka, M.; Satomura, Y.; et al. Spinocerebellar degenerations in Japan: A nationwide epidemiological and clinical study. Acta Neuro. Scand. Suppl. 1994, 153, 1–22. [Google Scholar] [CrossRef]
- Shimazaki, H.; Takiyama, Y.; Ishiura, H.; Sakai, C.; Matsushima, Y.; Hatakeyama, H.; Honda, J.; Sakoe, K.; Naoi, T.; Namekawa, M.; et al. A homozygous mutation of C12orf65 causes spastic paraplegia with optic atrophy and neuropathy. J. Med. Genet. 2012, 49, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, H.; Honda, J.; Naoi, T.; Namekawa, M.; Nakano, I.; Yazaki, M.; Nakamura, K.; Yoshida, K.; Ikeda, S.; Ishiura, H.; et al. Autosomal-recessive complicated spastic paraplegia with a novel lysosomal trafficking regulator gene mutation. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1024–1028. [Google Scholar] [CrossRef] [PubMed]
- Ishiura, H.; Takahashi, Y.; Hayashi, T.; Saito, K.; Furuya, H.; Watanabe, M.; Murata, M.; Suzuki, M.; Sugiura, A.; Sawai, S.; et al. Molecular epidemiology and clinical spectrum of hereditary spastic paraplegia in the Japanese population based on comprehensive mutational analyses. J. Hum. Genet. 2014, 59, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Schüle, R.; Wiethoff, S.; Martus, P.; Karle, K.N.; Otto, S.; Klebe, S.; Klimpe, S.; Gallenmüller, C.; Kurzwelly, D.; Henkel, D.; et al. Hereditary spastic paraplegia: Clinicogenetic lessons from 608 patients. Ann. Neurol. 2016, 79, 646–658. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).


{kind=link}