ABCA7 and Pathogenic Pathways of Alzheimer’s Disease


{kind=link}
{kind=link}
Abstract
1. Introduction
2. ABCA7 Gene Variants and Alzheimer’s Disease
3. Biochemical and Functional Features of ABCA7
3.1. ABCA7 Structure
3.2. ABCA7 Expression and Organ/Tissue Distribution
3.3. ABCA7 and Lipid Metabolism
3.4. ABCA7 and Phagocytosis
4. ABCA7 and Alzheimer Disease-Related Phenotypes
4.1. ABCA7, Neurobehaviors, and Aβ Pathology in Mouse Models
4.2. ABCA7 and Microglial Aβ Clearance
4.3. ABCA7 and APP Processing
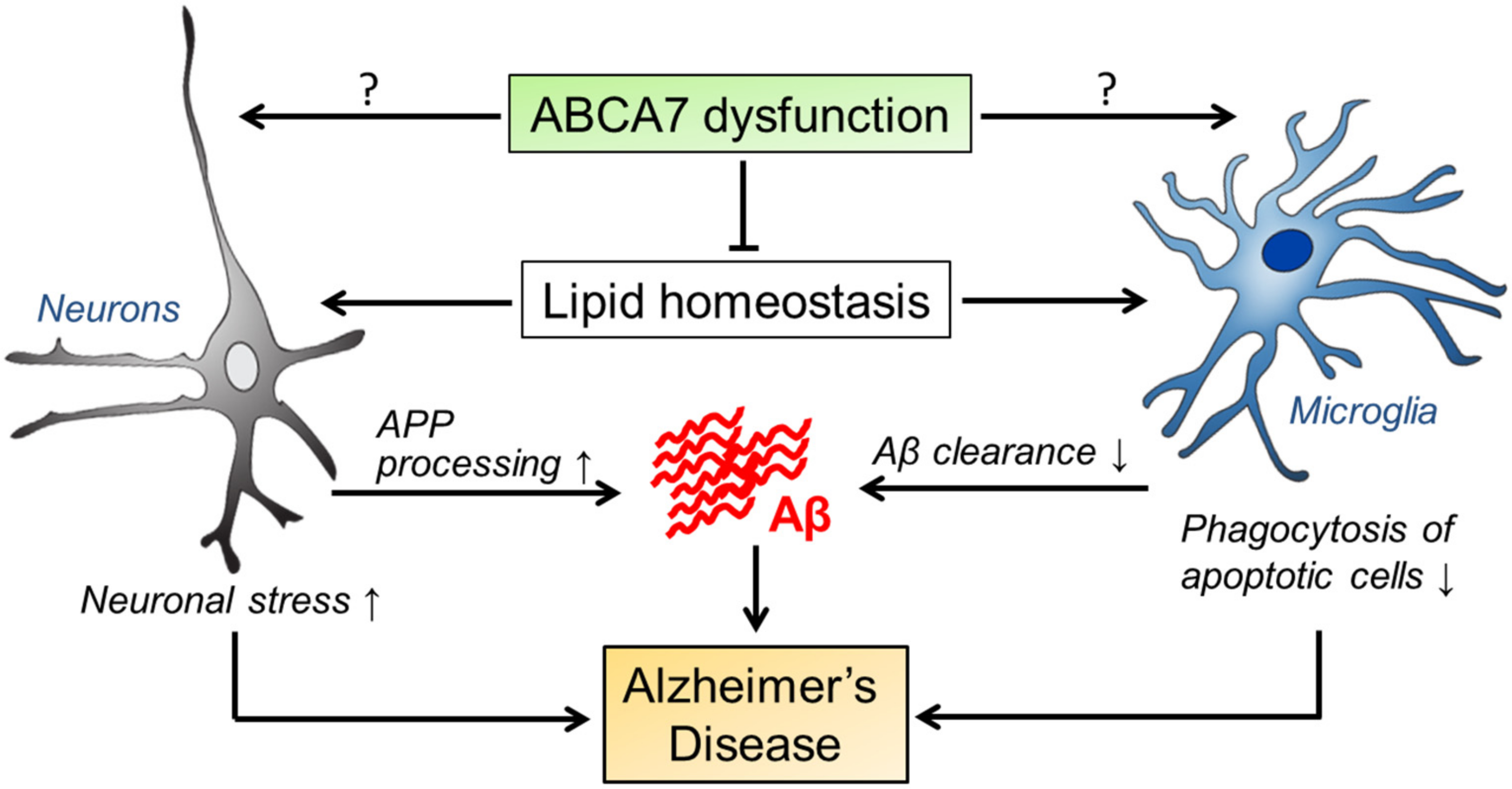
5. Summary and Perspective
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ABC | ATP binding cassette |
| ABCA7 | ATP-binding cassette transporter A7 |
| AD | Alzheimer’s disease |
| apoA | apolipoprotein A |
| apoE | apolipoprotein E |
| APP | amyloid precursor protein |
| Aβ | amyloid-β |
| BACE1 | β-site amyloid precursor protein cleaving enzyme 1 |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| GWAS | genome-wide association study |
| HMG-CoA | β-hydroxy β-methylglutaryl-CoA |
| HEK | human embryonic kidney |
| KO | knockout |
| LDLR | low density lipoprotein receptor |
| LPC | lysophosphatidyl choline |
| LXR | liver-X-receptor |
| MAF | minor allele frequency |
| MCI | mild cognitive impairment |
| NBD | nucleotide binding domain |
| OR | odds ratio |
| PC | phosphatidylcholine |
| PS | phosphatidylserine |
| PTC | premature termination codon |
| ROS | reactive oxygen species |
| RXR | retinoid-X-receptor |
| SNP | single nucleotide polymorphism |
| SREBP | sterol-responsive/regulatory element binding protein |
References
- Alzheimer’s Association. 2017 alzheimer’s disease facts and figures. Alzheimer’s Dement. 2017, 13, 325–373. [Google Scholar]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2016 alzheimer’s disease facts and figures. Alzheimer’s Dement. 2016, 12, 459–509. [Google Scholar]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Aisen, P.S.; De Strooper, B.; Fox, N.C.; Lemere, C.A.; Ringman, J.M.; Salloway, S.; Sperling, R.A.; Windisch, M.; Xiong, C. Autosomal-dominant alzheimer’s disease: A review and proposal for the prevention of alzheimer’s disease. Alzheimer’s Res. Ther. 2011, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Weisgraber, K.H.; Huang, D.Y.; Dong, L.M.; Salvesen, G.S.; Pericak-Vance, M.; Schmechel, D.; Saunders, A.M.; Goldgaber, D.; Roses, A.D. Binding of human apolipoprotein E to synthetic amyloid beta peptide: Isoform-specific effects and implications for late-onset alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 8098–8102. [Google Scholar] [CrossRef] [PubMed]
- Bu, G. Apolipoprotein E and its receptors in alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Xu, H.; Bu, G. APOE and Aβ in alzheimer’s disease: Accidental encounters or partners? Neuron 2014, 81, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, R.L. Genome-wide association studies, alzheimer disease, and understudied populations. JAMA 2013, 309, 1527–1528. [Google Scholar] [CrossRef] [PubMed]
- Loy, C.T.; Schofield, P.R.; Turner, A.M.; Kwok, J.B. Genetics of dementia. Lancet 2014, 383, 828–840. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006312. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and alzheimer disease. A meta-analysis. APOE and alzheimer disease meta analysis consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Risch, N.J.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C., Jr.; Rimmler, J.B.; Locke, P.A.; Conneally, P.M.; Schmader, K.E.; et al. Protective effect of apolipoprotein E type 2 allele for late onset alzheimer disease. Nat. Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Efthymiou, A.G.; Goate, A.M. Late onset alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, L.M.; Goukasian, N.; Porat, S.; Hwang, K.S.; Eastman, J.A.; Hurtz, S.; Wang, B.; Vang, N.; Sears, R.; Klein, E.; et al. Common variants in ABCA7 and MS4A6A are associated with cortical and hippocampal atrophy. Neurobiol. Aging 2016, 39, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Carrasquillo, M.M.; Crook, J.E.; Pedraza, O.; Thomas, C.S.; Pankratz, V.S.; Allen, M.; Nguyen, T.; Malphrus, K.G.; Ma, L.; Bisceglio, G.D.; et al. Late-onset alzheimer’s risk variants in memory decline, incident mild cognitive impairment, and alzheimer’s disease. Neurobiol. Aging 2015, 36, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Logue, M.W.; Schu, M.; Vardarajan, B.N.; Buros, J.; Green, R.C.; Go, R.C.; Griffith, P.; Obisesan, T.O.; Shatz, R.; Borenstein, A.; et al. A comprehensive genetic association study of alzheimer disease in African Americans. Arch. Neurol. 2011, 68, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Jun, G.; Naj, A.; Rajbhandary, R.; Vardarajan, B.N.; Wang, L.S.; Valladares, O.; Lin, C.F.; Larson, E.B.; Graff-Radford, N.R.; et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4,and the risk of late-onset alzheimer disease in African Americans. JAMA 2013, 309, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Cukier, H.N.; Kunkle, B.W.; Vardarajan, B.N.; Rolati, S.; Hamilton-Nelson, K.L.; Kohli, M.A.; Whitehead, P.L.; Dombroski, B.A.; Van Booven, D.; Lang, R.; et al. ABCA7 frameshift deletion associated with alzheimer disease in African Americans. Neurol. Genet 2016, 2, e79. [Google Scholar] [CrossRef] [PubMed]
- Cuyvers, E.; De Roeck, A.; Van den Bossche, T.; Van Cauwenberghe, C.; Bettens, K.; Vermeulen, S.; Mattheijssens, M.; Peeters, K.; Engelborghs, S.; Vandenbulcke, M.; et al. Mutations in ABCA7 in a Belgian cohort of alzheimer’s disease patients: A targeted resequencing study. Lancet Neurol. 2015, 14, 814–822. [Google Scholar] [CrossRef]
- N’Songo, A.; Carrasquillo, M.M.; Wang, X.; Burgess, J.D.; Nguyen, T.; Asmann, Y.W.; Serie, D.J.; Younkin, S.G.; Allen, M.; Pedraza, O.; et al. African American exome sequencing identifies potential risk variants at alzheimer disease loci. Neurol. Genet. 2017, 3, e141. [Google Scholar] [CrossRef] [PubMed]
- Sassi, C.; Nalls, M.A.; Ridge, P.G.; Gibbs, J.R.; Ding, J.; Lupton, M.K.; Troakes, C.; Lunnon, K.; Al-Sarraj, S.; Brown, K.S.; et al. ABCA7 p.G215s as potential protective factor for alzheimer’s disease. Neurobiol. Aging 2016, 46, 235.e1–235.e9. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.; Stefansson, H.; Jonsson, T.; Johannsdottir, H.; Ingason, A.; Helgason, H.; Sulem, P.; Magnusson, O.T.; Gudjonsson, S.A.; Unnsteinsdottir, U.; et al. Loss-of-function variants in ABCA7 confer risk of alzheimer’s disease. Nat. Genet. 2015, 47, 445–447. [Google Scholar] [CrossRef] [PubMed]
- Bellenguez, C.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Le Guennec, K.; Nicolas, G.; Chauhan, G.; Wallon, D.; Rousseau, S.; Richard, A.C.; et al. Contribution to alzheimer’s disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol. Aging 2017, 59, 220.e1–220.e9. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Lincoln, S.J.; Corda, M.; Watzlawik, J.O.; Carrasquillo, M.M.; Reddy, J.S.; Burgess, J.D.; Nguyen, T.; Malphrus, K.; Petersen, R.C.; et al. ABCA7 loss-of-function variants, expression, and neurologic disease risk. Neurol. Genet. 2017, 3, e126. [Google Scholar] [CrossRef] [PubMed]
- Le Guennec, K.; Nicolas, G.; Quenez, O.; Charbonnier, C.; Wallon, D.; Bellenguez, C.; Grenier-Boley, B.; Rousseau, S.; Richard, A.C.; Rovelet-Lecrux, A.; et al. ABCA7 rare variants and alzheimer disease risk. Neurology 2016, 86, 2134–2137. [Google Scholar] [CrossRef] [PubMed]
- Del-Aguila, J.L.; Fernandez, M.V.; Jimenez, J.; Black, K.; Ma, S.; Deming, Y.; Carrell, D.; Saef, B.; Alzheimer’s Disease Neuroimaging Initiative; Howells, B.; et al. Role of ABCA7 loss-of-function variant in alzheimer’s disease: A replication study in European-Americans. Alzheimer’s Res. Ther. 2015, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- De Roeck, A.; Van den Bossche, T.; van der Zee, J.; Verheijen, J.; De Coster, W.; Van Dongen, J.; Dillen, L.; Baradaran-Heravi, Y.; Heeman, B.; Sanchez-Valle, R.; et al. Deleterious ABCA7 mutations and transcript rescue mechanisms in early onset alzheimer’s disease. Acta Neuropathol. 2017, 134, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Vardarajan, B.N.; Ghani, M.; Kahn, A.; Sheikh, S.; Sato, C.; Barral, S.; Lee, J.H.; Cheng, R.; Reitz, C.; Lantigua, R.; et al. Rare coding mutations identified by sequencing of alzheimer disease genome-wide association studies loci. Ann. Neurol. 2015, 78, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, J.B.; Fardo, D.W.; Estus, S. ABCA7 expression is associated with alzheimer’s disease polymorphism and disease status. Neurosci. Lett. 2013, 556, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F. ABC transporters: From microorganisms to man. Annu. Rev. Cell Biol. 1992, 8, 67–113. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Allikmets, R. Evolution of ATP-binding cassette transporter genes. Curr. Opin. Genet. Dev. 1995, 5, 779–785. [Google Scholar] [CrossRef]
- Kaminski, W.E.; Orso, E.; Diederich, W.; Klucken, J.; Drobnik, W.; Schmitz, G. Identification of a novel human sterol-sensitive ATP-binding cassette transporter (ABCA7). Biochem. Biophys. Res. Commun. 2000, 273, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Bjork, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef] [PubMed]
- Abe-Dohmae, S.; Ikeda, Y.; Matsuo, M.; Hayashi, M.; Okuhira, K.; Ueda, K.; Yokoyama, S. Human ABCA7 supports apolipoprotein-mediated release of cellular cholesterol and phospholipid to generate high density lipoprotein. J. Biol. Chem. 2004, 279, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, M.L.; Morris, A.L.; Rhee, J.S.; Andersson, L.P.; Mendez, A.J.; Freeman, M.W. Naturally occurring mutations in the largest extracellular loops of abca1 can disrupt its direct interaction with apolipoprotein AI. J. Biol. Chem. 2002, 277, 33178–33187. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Silver, D.L.; Costet, P.; Tall, A.R. Specific binding of ApoA-I, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1. J. Biol. Chem. 2000, 275, 33053–33058. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Lan, D.; Gerbod-Giannone, M.; Linsel-Nitschke, P.; Jehle, A.W.; Chen, W.; Martinez, L.O.; Tall, A.R. ATP-binding cassette transporter A7 (ABCA7) binds apolipoprotein A-I and mediates cellular phospholipid but not cholesterol efflux. J. Biol. Chem. 2003, 278, 42906–42912. [Google Scholar] [CrossRef] [PubMed]
- Oram, J.F. Molecular basis of cholesterol homeostasis: Lessons from tangier disease and ABCA1. Trends Mol. Med. 2002, 8, 168–173. [Google Scholar] [CrossRef]
- Denis, M.; Bissonnette, R.; Haidar, B.; Krimbou, L.; Bouvier, M.; Genest, J. Expression, regulation, and activity of ABCA1 in human cell lines. Mol. Genet. Metab. 2003, 78, 265–274. [Google Scholar] [CrossRef]
- Iwamoto, N.; Abe-Dohmae, S.; Sato, R.; Yokoyama, S. ABCA7 expression is regulated by cellular cholesterol through the SREBP2 pathway and associated with phagocytosis. J. Lipid Res. 2006, 47, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Sato, R.; Inoue, J.; Kawabe, Y.; Kodama, T.; Takano, T.; Maeda, M. Sterol-dependent transcriptional regulation of sterol regulatory element-binding protein-2. J. Biol. Chem. 1996, 271, 26461–26464. [Google Scholar] [CrossRef] [PubMed]
- Broccardo, C.; Osorio, J.; Luciani, M.F.; Schriml, L.M.; Prades, C.; Shulenin, S.; Arnould, I.; Naudin, L.; Lafargue, C.; Rosier, M.; et al. Comparative analysis of the promoter structure and genomic organization of the human and mouse ABCA7 gene encoding a novel ABCA transporter. Cytogenet. Cell Genet. 2001, 92, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Shoji, A.; Kubo, Y.; Nada, S.; Yamaguchi, A. Cloning of rat ABCA7 and its preferential expression in platelets. Biochem. Biophys. Res. Commun. 2003, 304, 777–782. [Google Scholar] [CrossRef]
- Kim, W.S.; Guillemin, G.J.; Glaros, E.N.; Lim, C.K.; Garner, B. Quantitation of ATP-binding cassette subfamily-A transporter gene expression in primary human brain cells. Neuroreport 2006, 17, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Fitzgerald, M.L.; Kang, K.W.; Okuhira, K.; Bell, S.A.; Manning, J.J.; Koehn, S.L.; Lu, N.F.; Moore, K.J.; Freeman, M.W. ABCA7 null mice retain normal macrophage phosphatidyleholine and cholesterol efflux activity despite alterations in adipose mass and serum cholesterol levels. J. Biol. Chem. 2005, 280, 3989–3995. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Abe-Dohmae, S.; Munehira, Y.; Aoki, R.; Kawamoto, S.; Furuya, A.; Shitara, K.; Amachi, T.; Kioka, N.; Matsuo, M.; et al. Posttranscriptional regulation of human abca7 and its function for the ApoA-I-dependent lipid release. Biochem. Biophys. Res. Commun. 2003, 311, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Sakae, N.; Liu, C.C.; Shinohara, M.; Frisch-Daiello, J.; Ma, L.; Yamazaki, Y.; Tachibana, M.; Younkin, L.; Kurti, A.; Carrasquillo, M.M.; et al. ABCA7 deficiency accelerates amyloid-beta generation and alzheimer’s neuronal pathology. J. Neurosci. 2016, 36, 3848–3859. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Abe-Dohmae, S.; Okazaki, M.; Ueda, K.; Yokoyama, S. Heterogeneity of high density lipoprotein generated by ABCA1 and ABCA7. J. Lipid Res. 2005, 46, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Linsel-Nitschke, P.; Jehle, A.W.; Shan, J.; Cao, G.Q.; Bacic, D.; Lan, D.B.; Wang, N.; Tall, A.R. Potential role of ABCA7 in cellular lipid efflux to ApoA-I. J. Lipid Res. 2005, 46, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, M.; Toda, Y.; Manucat, N.B.; Akatsu, H.; Fukumoto, M.; Kono, N.; Arai, H.; Kioka, N.; Ueda, K. Lysophosphatidylcholine export by human ABCA7. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids. 2017, 1862, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Kielar, D.; Kaminski, W.E.; Liebisch, G.; Piehler, A.; Wenzel, J.J.; Mohle, C.; Heimerl, S.; Langmann, T.; Friedrich, S.O.; Bottcher, A.; et al. Adenosine triphosphate binding cassette (ABC) transporters are expressed and regulated during terminal keratinocyte differentiation: A potential role for ABCA7 in epidermal lipid reorganization. J. Investig. Dermatol. 2003, 121, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Nowyhed, H.N.; Chandra, S.; Kiosses, W.; Marcovecchio, P.; Andary, F.; Zhao, M.; Fitzgerald, M.L.; Kronenberg, M.; Hedrick, C.C. ATP binding cassette transporter ABCA7 regulates NKT cell development and function by controlling cd1d expression and lipid raft content. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Henson, P.M.; Bratton, D.L.; Fadok, V.A. Apoptotic cell removal. Curr. Biol. 2001, 11, R795–R805. [Google Scholar] [CrossRef]
- Wu, Y.C.; Horvitz, H.R. The C. Elegans cell corpse engulfment gene ced-7 encodes a protein similar to ABC transporters. Cell 1998, 93, 951–960. [Google Scholar] [CrossRef]
- Li, H.Y.; Karl, T.; Garner, B. Understanding the function of ABCA7 in alzheimer’s disease. Biochem. Soc. Trans. 2015, 43, 920–923. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Abe-Dohmae, S.; Iwamoto, N.; Fitzgerald, M.L.; Yokoyama, S. Helical apolipoproteins of high-density lipoprotein enhance phagocytosis by stabilizing ATP-binding cassette transporter A7. J. Lipid Res. 2010, 51, 2591–2599. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Abe-Dohmae, S.; Iwamoto, N.; Yokoyama, S. Roles of ATP-binding cassette transporter A7 in cholesterol homeostasis and host defense system. J. Atheroscler. Thromb. 2011, 18, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Jehle, A.W.; Gardai, S.J.; Li, S.; Linsel-Nitschke, P.; Morimoto, K.; Janssen, W.J.; Vandivier, R.W.; Wang, N.; Greenberg, S.; Dale, B.M.; et al. ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J. Cell Biol. 2006, 174, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Logge, W.; Cheng, D.; Chesworth, R.; Bhatia, S.; Garner, B.; Kim, W.S.; Karl, T. Role of ABCA7 in mouse behaviours relevant to neurodegenerative diseases. PLoS ONE 2012, 7, e45959. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Karl, T.; Garner, B. ABCA7 deletion does not affect adult neurogenesis in the mouse. Biosci. Rep. 2016, 36, e00308. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Li, H.; Ruberu, K.; Chan, S.; Elliott, D.A.; Low, J.K.; Cheng, D.; Karl, T.; Garner, B. Deletion of ABCA7 increases cerebral amyloid-beta accumulation in the j20 mouse model of alzheimer’s disease. J. Neurosci. 2013, 33, 4387–4394. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Abe-Dohmae, S.; Yokoyama, S.; St George-Hyslop, P.; Fraser, P.E. ATP-binding cassette transporter A7 (ABCA7) loss of function alters alzheimer amyloid processing. J. Biol. Chem. 2015, 290, 24152–24165. [Google Scholar] [CrossRef] [PubMed]
- Mosher, K.I.; Wyss-Coray, T. Microglial dysfunction in brain aging and alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, S.; Jiang, Q.; Lee, C.Y.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Landreth, G.E. Microglia mediate the clearance of soluble abeta through fluid phase macropinocytosis. J. Neurosci. 2009, 29, 4252–4262. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Hsiao, J.H.; Paxinos, G.; Halliday, G.M.; Kim, W.S. ABCA7 mediates phagocytic clearance of amyloid-beta in the brain. J. Alzheimer’s Dis. 2016, 54, 569–584. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-beta clearance in alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of app. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Kim, W.S.; Kwok, J.B.; Hill, A.F.; Cappai, R.; Rye, K.A.; Garner, B. ATP-binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. J. Neurochem. 2008, 106, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Rahmanto, A.S.; Kamili, A.; Rye, K.A.; Guillemin, G.J.; Gelissen, I.C.; Jessup, W.; Hill, A.F.; Garner, B. Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein E discs and suppression of amyloid-beta peptide generation. J. Biol. Chem. 2007, 282, 2851–2861. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Guglielmotto, M.; Medana, C.; Catalano, M.G.; Cutrupi, S.; Borghi, R.; Tamagno, E.; Boccuzzi, G.; Aragno, M. Dysregulation of SREBP2 induces BACE1 expression. Neurobiol. Dis. 2011, 44, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Bodovitz, S.; Klein, W.L. Cholesterol modulates alpha-secretase cleavage of amyloid precursor protein. J. Biol. Chem. 1996, 271, 4436–4440. [Google Scholar] [CrossRef] [PubMed]
- Tun, H.; Marlow, L.; Pinnix, I.; Kinsey, R.; Sambamurti, K. Lipid rafts play an important role in a beta biogenesis by regulating the beta-secretase pathway. J. Mol. Neurosci. 2002, 19, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, K.S.; Cheng, H.; Kim, S.H.; Chen, Y.; Barnes, N.Y.; Parent, A.T.; Sisodia, S.S.; Thinakaran, G. Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J. Biol. Chem. 2005, 280, 25892–25900. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yuan, Y.; Hu, B.; Wu, L. Study on lentivirus-mediated ABCA7 improves neurocognitive function and related mechanisms in the C57BL/6 mouse model of alzheimer’s disease. J. Mol. Neurosci. 2017, 61, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Volmar, C.H.; Salah-Uddin, H.; Janczura, K.J.; Halley, P.; Lambert, G.; Wodrich, A.; Manoah, S.; Patel, N.H.; Sartor, G.C.; Mehta, N.; et al. M344 promotes nonamyloidogenic amyloid precursor protein processing while normalizing alzheimer’s disease genes and improving memory. Proc. Natl. Acad. Sci. USA 2017, 114, E9135–E9144. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aikawa, T.; Holm, M.-L.; Kanekiyo, T. ABCA7 and Pathogenic Pathways of Alzheimer’s Disease. Brain Sci. 2018, 8, 27. https://doi.org/10.3390/brainsci8020027
Aikawa T, Holm M-L, Kanekiyo T. ABCA7 and Pathogenic Pathways of Alzheimer’s Disease. Brain Sciences. 2018; 8(2):27. https://doi.org/10.3390/brainsci8020027
Chicago/Turabian StyleAikawa, Tomonori, Marie-Louise Holm, and Takahisa Kanekiyo. 2018. "ABCA7 and Pathogenic Pathways of Alzheimer’s Disease" Brain Sciences 8, no. 2: 27. https://doi.org/10.3390/brainsci8020027
APA StyleAikawa, T., Holm, M.-L., & Kanekiyo, T. (2018). ABCA7 and Pathogenic Pathways of Alzheimer’s Disease. Brain Sciences, 8(2), 27. https://doi.org/10.3390/brainsci8020027