
Phenotypic Discordance in Siblings with Identical Compound Heterozygous PARK2 Mutations


{kind=link}
{kind=link}
Abstract
:1. Introduction
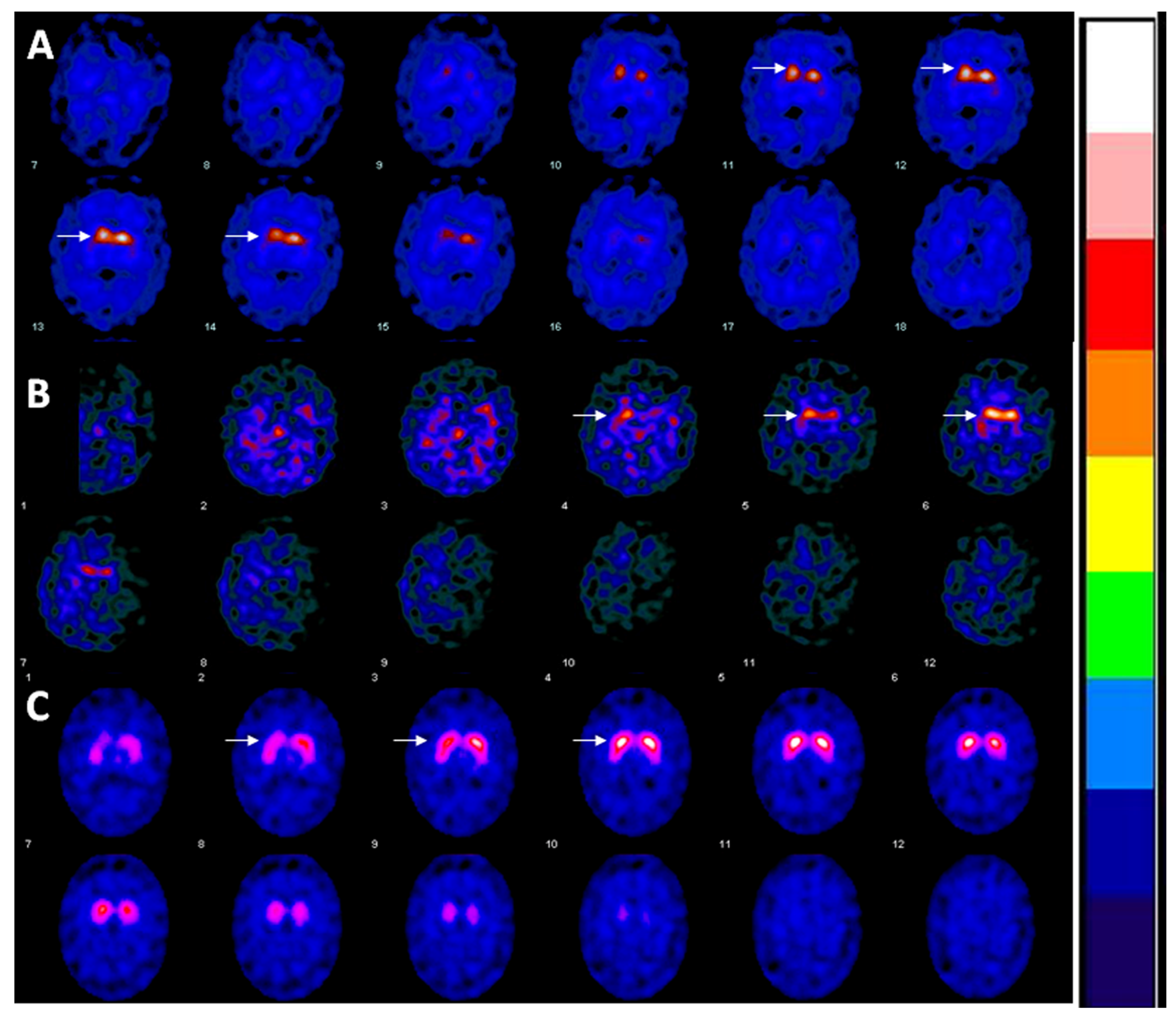
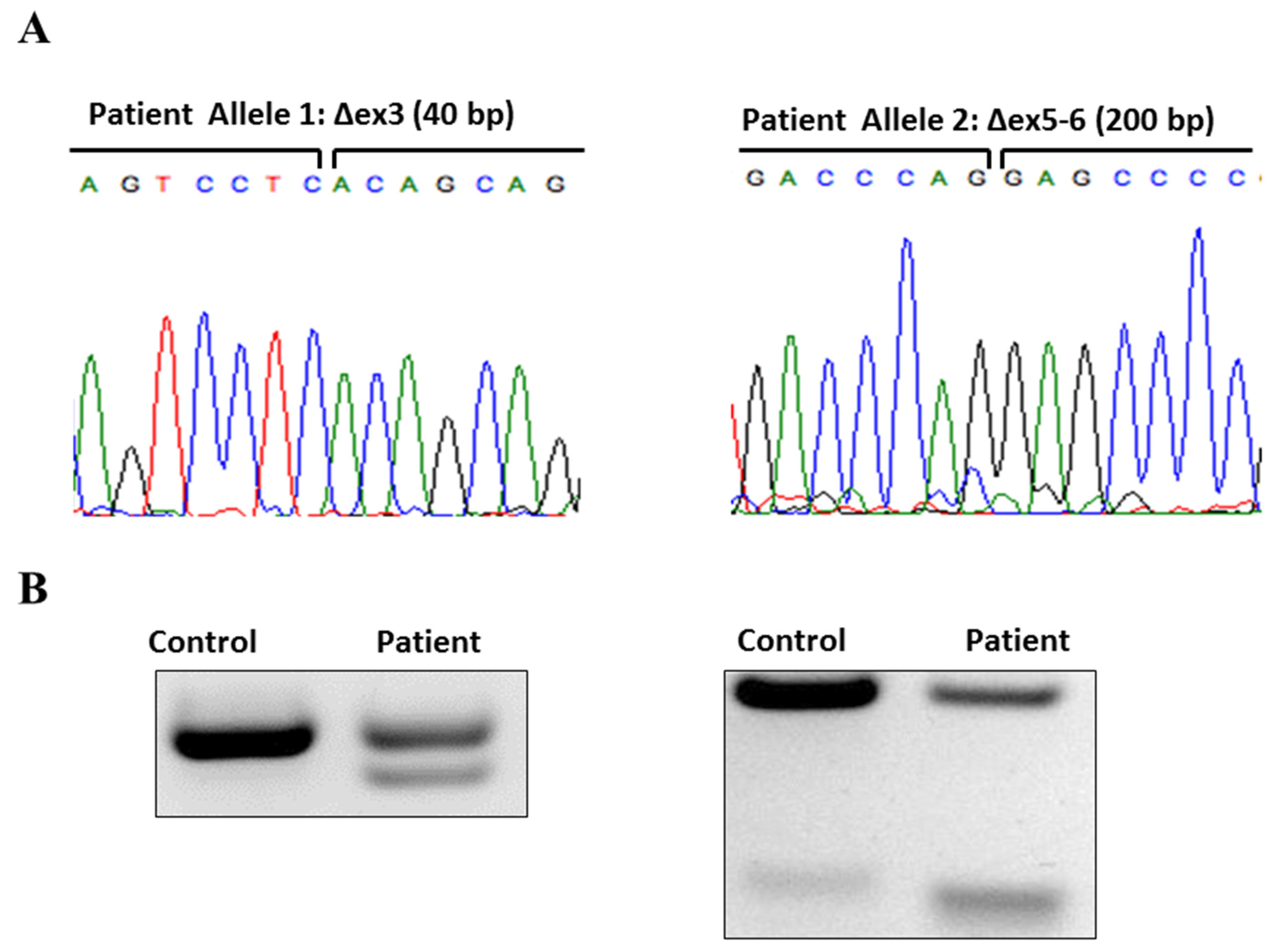
2. Case
3. Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Puschmann, A. Monogenic Parkinson’s disease and parkinsonism: Clinical phenotypes and frequencies of known mutations. Park. Relat. Disord. 2013, 19, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [PubMed]
- Doherty, K.M.; Silveira-Moriyama, L.; Parkkinen, L.; Healy, D.G.; Farrell, M.; Mencacci, N.E.; Ahmed, Z.; Brett, F.M.; Hardy, J.; Quinn, N.; et al. Parkin disease: A clinicopathologic entity? JAMA Neurol. 2013, 70, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Periquet, M.; Latouche, M.; Lohmann, E.; Rawal, N.; De Michele, G.; Ricard, S.; Teive, H.; Fraix, V.; Vidailhet, M.; Nicholl, D.; et al. Parkin mutations are frequent in patients with isolated early-onset parkinsonism. Brain 2003, 126, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Lucking, C.B.; Durr, A.; Bonifati, V.; Vaughan, J.; De Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denèfle, P.; Wood, N.W.; et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N. Engl. J. Med. 2000, 342, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Pramstaller, P.P.; Kis, B.; Page, C.C.; Kann, M.; Leung, J.; Woodward, H.; Castellan, C.C.; Scherer, M.; Vieregge, P.; et al. Parkin deletions in a family with adult-onset, tremor-dominant parkinsonism: Expanding the phenotype. Ann. Neurol. 2000, 48, 65–71. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 2010, 25, S32–S39. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef] [PubMed]
- Periquet, M.; Lucking, C.; Vaughan, J.; Bonifati, V.; Durr, A.; De Michele, G.; Horstink, M.; Farrer, M.; Illarioshkin, S.N.; Pollak, P.; et al. Origin of the mutations in the parkin gene in Europe: Exon rearrangements are independent recurrent events, whereas point mutations may result from Founder effects. Am. J. Hum. Genet. 2001, 68, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Abbas, N.; Lucking, C.B.; Ricard, S.; Durr, A.; Bonifati, V.; De Michele, G.; Bouley, S.; Vaughan, J.R.; Gasser, T.; Marconi, R.; et al. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson’s Disease. Hum. Mol. Genet. 1999, 8, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Nichols, W.C.; Pankratz, N.; Uniacke, S.K.; Pauciulo, M.W.; Halter, C.; Rudolph, A.; Conneally, P.; Foroud, T. Linkage stratification and mutation analysis at the Parkin locus identifies mutation positive Parkinson’s disease families. J. Med. Genet. 2002, 39, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Varrone, A.; Pellecchia, M.T.; Amboni, M.; Sansone, V.; Salvatore, E.; Ghezzi, D.; Garavaglia, B.; Brice, A.; Brunetti, A.; Bonavita, V.; et al. Imaging of dopaminergic dysfunction with [123I]FP-CIT SPECT in early-onset parkin disease. Neurology 2004, 63, 2097–2103. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.L.; Brooks, D.J.; Pavese, N.; Sweeney, M.G.; Wood, N.W.; Lees, A.J.; Piccini, P. Progression of nigrostriatal dysfunction in a parkin kindred: An [18F]dopa PET and clinical study. Brain 2002, 125, 2248–2256. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.L.; Graham, E.; Critchley, P.; Schrag, A.E.; Wood, N.W.; Lees, A.J.; Bhatia, K.P.; Quinn, N. Parkin disease: A phenotypic study of a large case series. Brain 2003, 126, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Le, W.D.; Hunter, C.B.; Ondo, W.G.; Guo, Y.; Xie, W.J.; Jankovic, J. Heterogeneous phenotype in a family with compound heterozygous parkin gene mutations. Arch. Neurol. 2006, 63, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Aboud, A.A.; Tidball, A.M.; Kumar, K.K.; Neely, M.D.; Han, B.; Ess, K.C.; Hong, C.C.; Erikson, K.M.; Hedera, P.; Bowman, A.B. PARK2 patient neuroprogenitors show increased mitochondrial sensitivity to copper. Neurobiol. Dis. 2015, 73, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Thobois, S.; Ribeiro, M.J.; Lohmann, E.; Durr, A.; Pollak, P.; Rascol, O.; Guillouet, S.; Chapoy, E.; Costes, N.; Agid, Y.; et al. Young-onset Parkinson disease with and without parkin gene mutations: A fluorodopa F 18 positron emission tomography study. Arch. Neurol. 2003, 60, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Scherfler, C.; Khan, N.L.; Pavese, N.; Eunson, L.; Graham, E.; Lees, A.J.; Quinn, N.P.; Wood, N.W.; Brooks, D.J.; Piccini, P.P. Striatal and cortical pre- and postsynaptic dopaminergic dysfunction in sporadic parkin-linked parkinsonism. Brain 2004, 127, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Jeon, B.S.; Jeong, J.M.; Park, S.S.; Kim, J.M.; Chang, Y.S.; Song, H.C.; Kim, K.M.; Yoon, K.Y.; Lee, M.C.; Lee, S.B. Dopamine transporter density measured by [123I]beta-CIT single-photon emission computed tomography is normal in dopa-responsive dystonia. Ann. Neurol. 1998, 43, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.L.; Scherfler, C.; Graham, E.; Bhatia, K.P.; Quinn, N.; Lees, A.J.; Brooks, D.J.; Wood, N.W.; Piccini, P. Dopaminergic dysfunction in unrelated, asymptomatic carriers of a single parkin mutation. Neurology 2005, 64, 134–136. [Google Scholar] [CrossRef] [PubMed]
- Koentjoro, B.; Park, J.S.; Ha, A.D.; Sue, C.M. Phenotypic variability of parkin mutations in single kindred. Mov. Disord. 2012, 27, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Latourelle, J.C.; Wooten, G.F.; Lew, M.F.; Klein, C.; Shill, H.A.; et al. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: The GenePD study. Arch. Neurol. 2006, 63, 826–832. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isaacs, D.; Claassen, D.; Bowman, A.B.; Hedera, P. Phenotypic Discordance in Siblings with Identical Compound Heterozygous PARK2 Mutations. Brain Sci. 2017, 7, 71. https://doi.org/10.3390/brainsci7070071
Isaacs D, Claassen D, Bowman AB, Hedera P. Phenotypic Discordance in Siblings with Identical Compound Heterozygous PARK2 Mutations. Brain Sciences. 2017; 7(7):71. https://doi.org/10.3390/brainsci7070071
Chicago/Turabian StyleIsaacs, David, Daniel Claassen, Aaron B. Bowman, and Peter Hedera. 2017. "Phenotypic Discordance in Siblings with Identical Compound Heterozygous PARK2 Mutations" Brain Sciences 7, no. 7: 71. https://doi.org/10.3390/brainsci7070071