
In Vitro, Ex Vivo and In Vivo Techniques to Study Neuronal Migration in the Developing Cerebral Cortex

 , , , and
, , , and 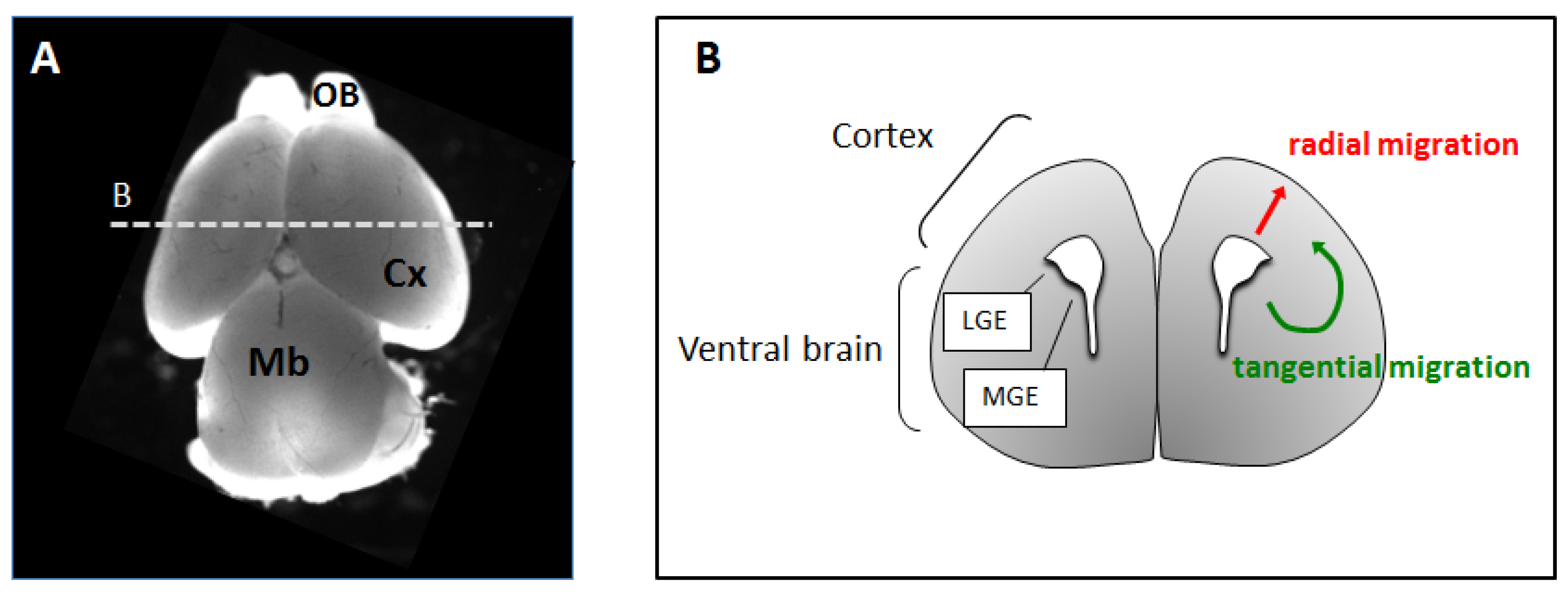{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Neuronal Migration in the Developing Cerebral Cortex
2.1. Radial Migration
2.2. Tangential Migration
3. How to Study Neuronal Migration in the Cerebral Cortex
3.1. In Vitro Assays
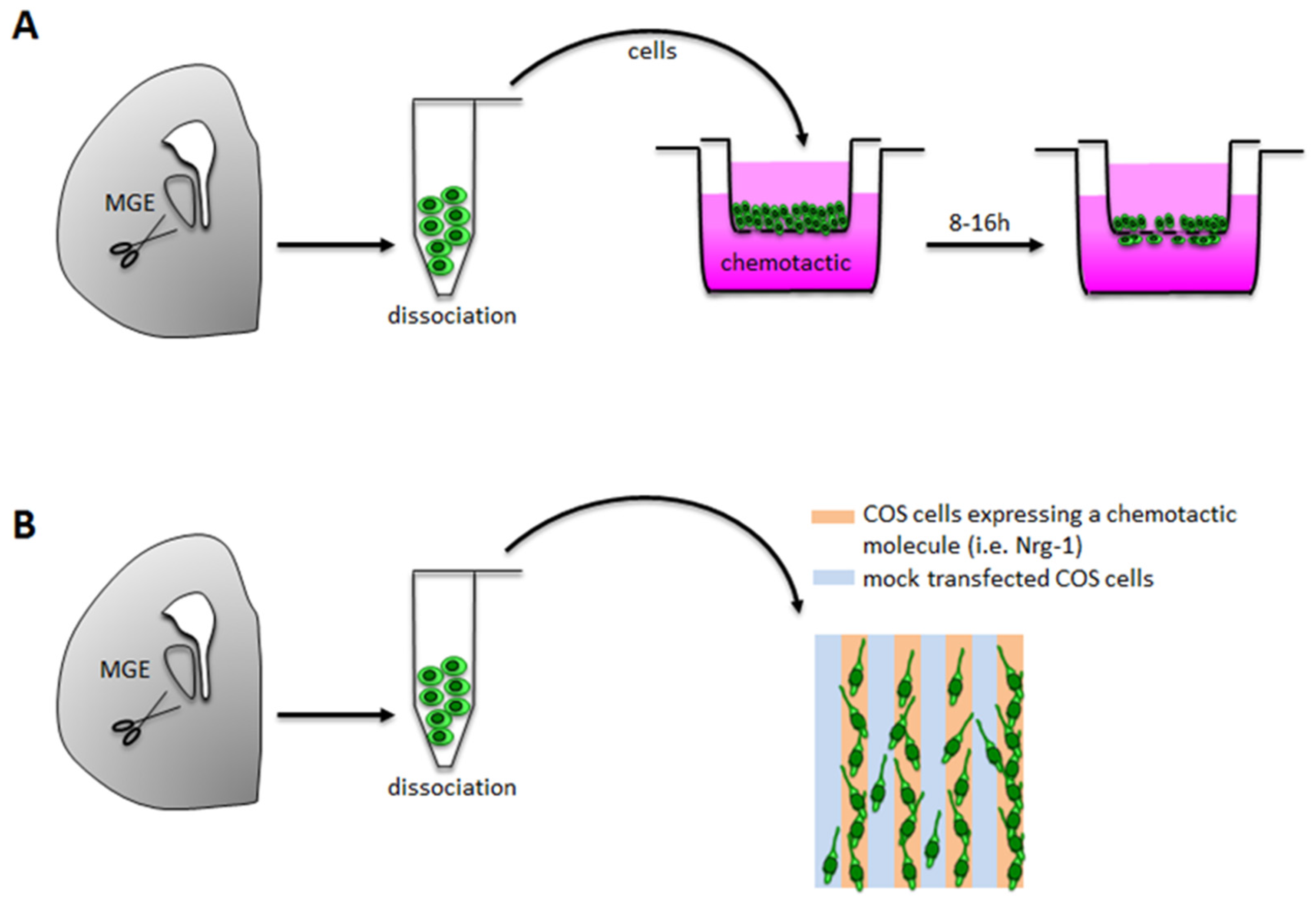
3.1.1. Boyden Chamber/Transwell Assays
Technical Details
Applications
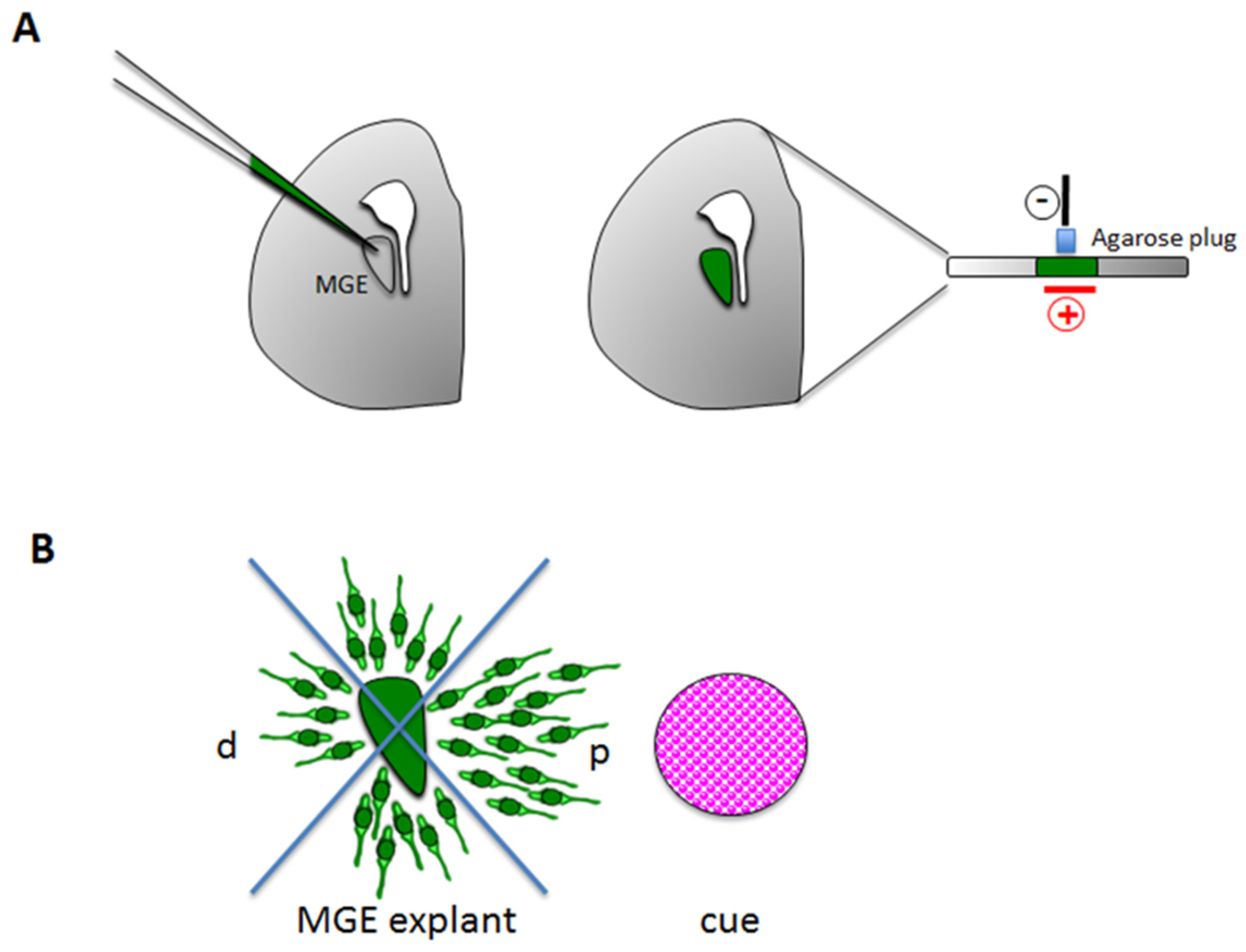
3.1.2. Stripe Choice Assay
3.2. Ex Vivo Assays
3.2.1. Organotypic Slices
Technical Details
Applications
3.2.2. Ex Vivo Explants
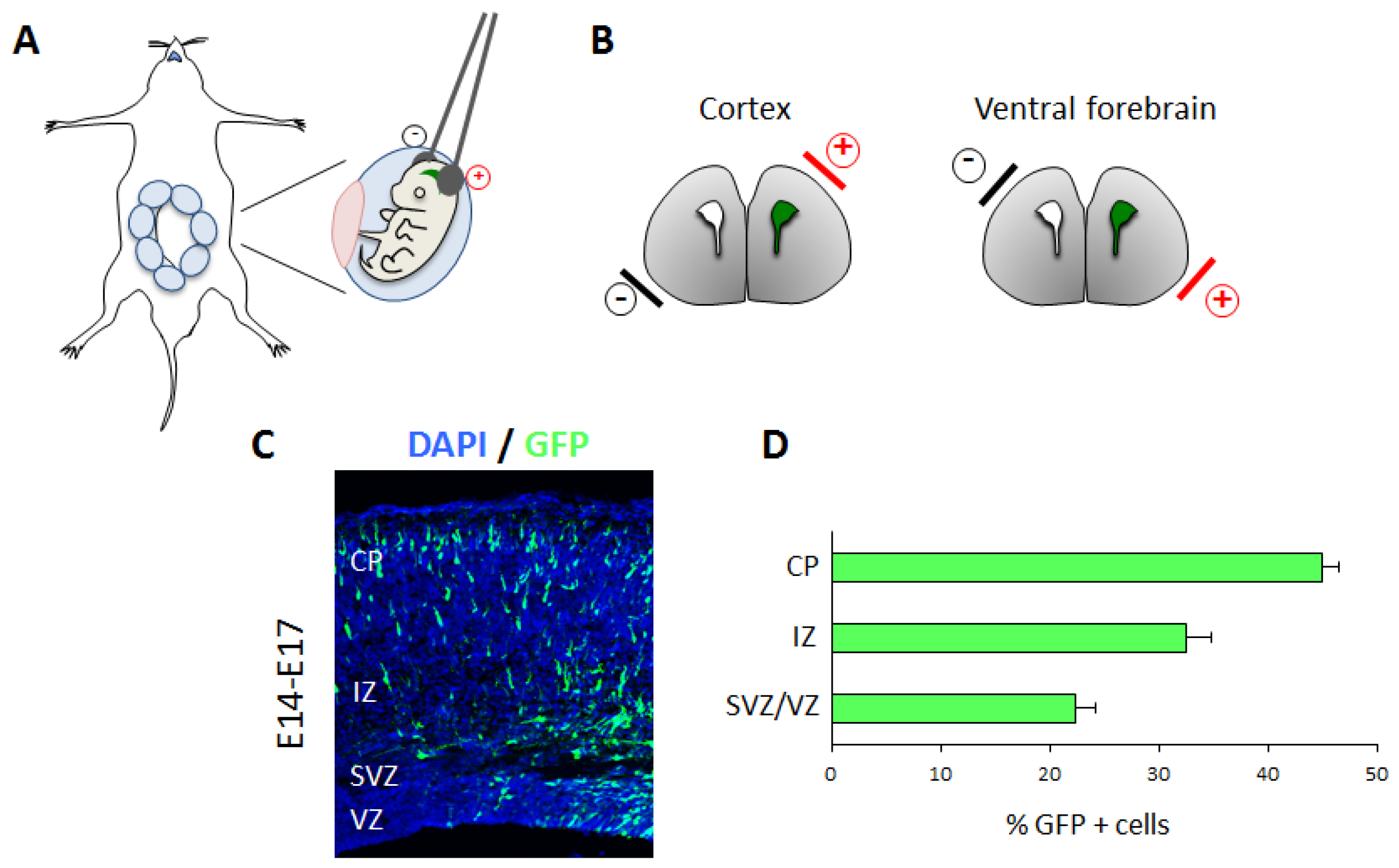
3.3. In Utero Electroporation to Study In Vivo Cortical Neuronal Migration
3.3.1. Technical Details
3.3.2. Applications
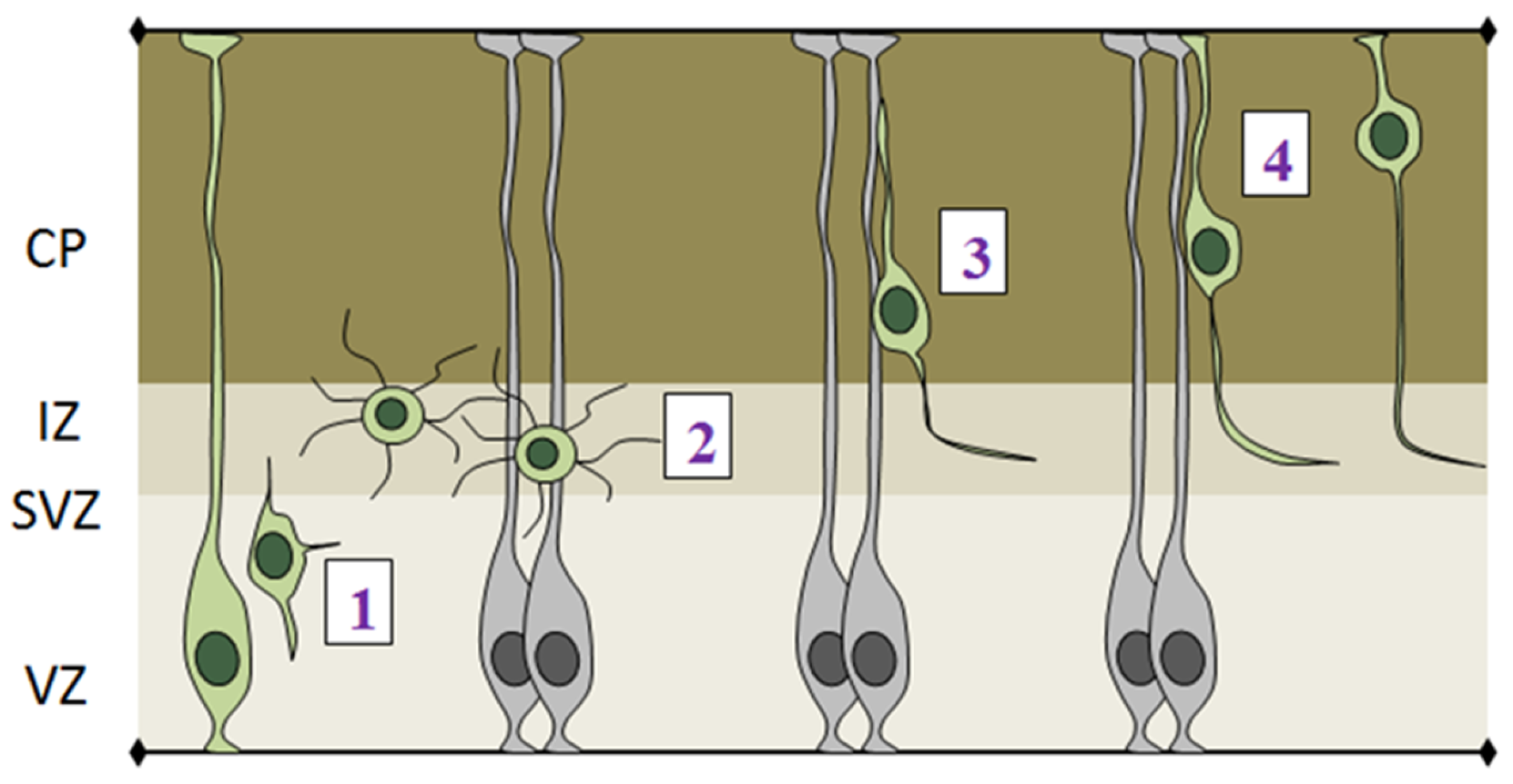
- (1)
- The initial step of radial migration consists in the detachment of the apical process from the VZ surface, which is accompanied by a loss of bi-polarity by RGCs (Figure 5-phase 1). Deregulation of apical detachment disrupts the integrity of the neural epithelium and results in neuronal migration disorders as shown for example by perturbing FilaminA and β1 integrin function [70,71].
- (2)
- Post-mitotic cells leave the proliferative VZ and SVZ and reach the IZ, where they acquire a multipolar shape, which is characterized by multiple protrusions that dynamically extend and retract from the cell body (Figure 5-phase 2). Several genes have been shown to play important roles in exiting the IZ, since their knock down causes accumulation of multipolar cells in this region. Interestingly, the majority of these genes are involved in cytoskeletal remodeling via the actin or microtubule networks, such as ENA/VASP homology proteins (EVH), Dcx, Lis and Rnd2 [72,73,74].
- (3)
- After leaving the IZ, cells acquire a bipolar shape, with a leading process facing the pial surface and a trailing process facing the VZ. The leading process of migrating neurons embraces the radial glia filament and uses it as a guide for migration. Bipolar cells migrating in the CP undergo a mode of radial migration called locomotion (Figure 5-phase 3). This is characterized by repetitive cycles of leading process extension, forward displacement of the nucleus and partial retraction of the trailing process. These events require strict coordination between centrosomal and nuclear movements, as shown by several studies that use fluorescently labeled centrin to track centrosome in in vivo migrating cells [75,76].
- (4)
4. Advantages and Limitations of the Techniques
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Evsyukova, I.; Plestant, C.; Anton, E.S. Integrative mechanisms of oriented neuronal migration in the developing brain. Annu. Rev. Cell Dev. Biol. 2013, 29, 299–353. [Google Scholar] [CrossRef] [PubMed]
- Valiente, M.; Marin, O. Neuronal migration mechanisms in development and disease. Curr. Opin. Neurobiol. 2010, 20, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Marin, O.; Rubenstein, J.L. Cell migration in the forebrain. Annu. Rev. Neurosci. 2003, 26, 441–483. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, B.J.; Arlotta, P.; Menezes, J.R.; Macklis, J.D. Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 2007, 8, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Greig, L.C.; Woodworth, M.B.; Galazo, M.J.; Padmanabhan, H.; Macklis, J.D. Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci. 2013, 14, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Kriegstein, A.; Parnavelas, J.G. Progress in corticogenesis. Cereb. Cortex 2006, 16, i1–i2. [Google Scholar] [CrossRef] [PubMed]
- Lui, J.H.; Hansen, D.V.; Kriegstein, A.R. Development and evolution of the human neocortex. Cell 2011, 146, 18–36. [Google Scholar] [CrossRef] [PubMed]
- Marin, O.; Muller, U. Lineage origins of gabaergic versus glutamatergic neurons in the neocortex. Curr. Opin. Neurobiol. 2014, 26, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Nadarajah, B.; Parnavelas, J.G. Modes of neuronal migration in the developing cerebral cortex. Nat. Rev. Neurosci. 2002, 3, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Rubin, A.N.; Alfonsi, F.; Humphreys, M.P.; Choi, C.K.; Rocha, S.F.; Kessaris, N. The germinal zones of the basal ganglia but not the septum generate gabaergic interneurons for the cortex. J. Neurosci. 2010, 30, 12050–12062. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.; Pierani, A. Tangential migration of glutamatergic neurons and cortical patterning during development: Lessons from cajal-retzius cells. Dev. Neurobiol. 2016, 76, 847–881. [Google Scholar] [CrossRef] [PubMed]
- Francis, F.; Meyer, G.; Fallet-Bianco, C.; Moreno, S.; Kappeler, C.; Socorro, A.C.; Tuy, F.P.; Beldjord, C.; Chelly, J. Human disorders of cortical development: From past to present. Eur. J. Neurosci. 2006, 23, 877–893. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, R.; Parrini, E. Neuronal migration disorders. Neurobiol. Dis. 2010, 38, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, J.G. Classical lissencephaly and double cortex (subcortical band heterotopia): Lis1 and doublecortin. Curr. Opin. Neurol. 2000, 13, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Marin, O. Developmental timing and critical windows for the treatment of psychiatric disorders. Nat. Med. 2016, 22, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Gotz, M.; Huttner, W.B. The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Anthony, T.E.; Klein, C.; Fishell, G.; Heintz, N. Radial glia serve as neuronal progenitors in all regions of the central nervous system. Neuron 2004, 41, 881–890. [Google Scholar] [CrossRef]
- Miyoshi, G.; Hjerling-Leffler, J.; Karayannis, T.; Sousa, V.H.; Butt, S.J.; Battiste, J.; Johnson, J.E.; Machold, R.P.; Fishell, G. Genetic fate mapping reveals that the caudal ganglionic eminence produces a large and diverse population of superficial cortical interneurons. J. Neurosci. 2010, 30, 1582–1594. [Google Scholar] [CrossRef] [PubMed]
- Gelman, D.M.; Martini, F.J.; Nobrega-Pereira, S.; Pierani, A.; Kessaris, N.; Marin, O. The embryonic preoptic area is a novel source of cortical gabaergic interneurons. J. Neurosci. 2009, 29, 9380–9389. [Google Scholar] [CrossRef] [PubMed]
- Ang, E.S., Jr.; Haydar, T.F.; Gluncic, V.; Rakic, P. Four-dimensional migratory coordinates of gabaergic interneurons in the developing mouse cortex. J. Neurosci. 2003, 23, 5805–5815. [Google Scholar] [PubMed]
- Tanaka, D.; Nakaya, Y.; Yanagawa, Y.; Obata, K.; Murakami, F. Multimodal tangential migration of neocortical GABAergic neurons independent of GPI-anchored proteins. Development 2003, 130, 5803–5813. [Google Scholar] [CrossRef] [PubMed]
- Polleux, F.; Whitford, K.L.; Dijkhuizen, P.A.; Vitalis, T.; Ghosh, A. Control of cortical interneuron migration by neurotrophins and PI3-kinase signaling. Development 2002, 129, 3147–3160. [Google Scholar] [PubMed]
- Lodato, S.; Rouaux, C.; Quast, K.B.; Jantrachotechatchawan, C.; Studer, M.; Hensch, T.K.; Arlotta, P. Excitatory projection neuron subtypes control the distribution of local inhibitory interneurons in the cerebral cortex. Neuron 2011, 69, 763–779. [Google Scholar] [CrossRef] [PubMed]
- Kwan, K.Y.; Sestan, N.; Anton, E.S. Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development 2012, 139, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, G.; Ciceri, G.; Marin, O. Integration of gabaergic interneurons into cortical cell assemblies: Lessons from embryos and adults. Neuron 2013, 79, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Z.; Yuan, Y.H.; Zhang, Y.; Wang, X.F.; Chu, S.F.; Han, N.; Chen, N.H. Chemokine-like factor 1 promotes the migration of rat primary cortical neurons by the induction of actin polymerization. Neuroreport 2014, 25, 1221–1226. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, J.F.; Mione, M.C.; Pappas, I.S.; Parnavelas, J.G. Basic fibroblast growth factor prolongs the proliferation of rat cortical progenitor cells in vitro without altering their cell cycle parameters. Cerebral cortex 1997, 7, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Rakic, S.; Kanatani, S.; Hunt, D.; Faux, C.; Cariboni, A.; Chiara, F.; Khan, S.; Wansbury, O.; Howard, B.; Nakajima, K.; et al. Cdk5 phosphorylation of ErbB4 is required for tangential migration of cortical interneurons. Cereb. Cortex 2015, 25, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Miranda, L.R.; Cariboni, A.; Faux, C.; Ruhrberg, C.; Cho, J.H.; Cloutier, J.F.; Eickholt, B.J.; Parnavelas, J.G.; Andrews, W.D. Robo1 regulates semaphorin signaling to guide the migration of cortical interneurons through the ventral forebrain. J. Neurosci. 2011, 31, 6174–6187. [Google Scholar] [CrossRef] [PubMed]
- Flames, N.; Long, J.E.; Garratt, A.N.; Fischer, T.M.; Gassmann, M.; Birchmeier, C.; Lai, C.; Rubenstein, J.L.; Marin, O. Short- and long-range attraction of cortical gabaergic interneurons by neuregulin-1. Neuron 2004, 44, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Giesing, M.; Neumann, G.; Egge, H.; Zilliken, F. Lipid metabolism of developing central nervous tissues in organotypic cultures. I. Lipid distribution and fatty acid profiles of the medium for rat brain cortex in vitro. Nutr. Metab. 1975, 19, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Tobet, S.A.; Hanna, I.K.; Schwarting, G.A. Migration of neurons containing gonadotropin releasing hormone (GNRH) in slices from embryonic nasal compartment and forebrain. Brain Res. Dev. Brain Res. 1996, 97, 287–292. [Google Scholar] [CrossRef]
- Gahwiler, B.H. Organotypic monolayer cultures of nervous tissue. J. Neurosci. Methods 1981, 4, 329–342. [Google Scholar] [CrossRef]
- Gahwiler, B.H.; Capogna, M.; Debanne, D.; McKinney, R.A.; Thompson, S.M. Organotypic slice cultures: A technique has come of age. Trends Neurosci. 1997, 20, 471–477. [Google Scholar] [CrossRef]
- Gahwiler, B.H.; Thompson, S.M.; Muller, D. Preparation and maintenance of organotypic slice cultures of cns tissue. Curr. Protoc. Neurosci. 2001. [Google Scholar] [CrossRef]
- Stoppini, L.; Buchs, P.A.; Muller, D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 1991, 37, 173–182. [Google Scholar] [CrossRef]
- Humpel, C. Organotypic brain slice cultures: A review. Neuroscience 2015, 305, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Tielens, S.; Godin, J.D.; Nguyen, L. Real-time recordings of migrating cortical neurons from gfp and cre recombinase expressing mice. Curr. Protoc. Neurosci. 2016. [Google Scholar] [CrossRef]
- Anderson, S.A.; Eisenstat, D.D.; Shi, L.; Rubenstein, J.L. Interneuron migration from basal forebrain to neocortex: Dependence on dlx genes. Science 1997, 278, 474–476. [Google Scholar] [CrossRef] [PubMed]
- Crandall, J.E.; Goodman, T.; McCarthy, D.M.; Duester, G.; Bhide, P.G.; Drager, U.C.; McCaffery, P. Retinoic acid influences neuronal migration from the ganglionic eminence to the cerebral cortex. J. Neurochem. 2011, 119, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Lavdas, A.A.; Grigoriou, M.; Pachnis, V.; Parnavelas, J.G. The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J. Neurosci. 1999, 19, 7881–7888. [Google Scholar] [PubMed]
- Stenman, J.; Toresson, H.; Campbell, K. Identification of two distinct progenitor populations in the lateral ganglionic eminence: Implications for striatal and olfactory bulb neurogenesis. J. Neurosci. 2003, 23, 167–174. [Google Scholar] [PubMed]
- Tamamaki, N.; Yanagawa, Y.; Tomioka, R.; Miyazaki, J.; Obata, K.; Kaneko, T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the gad67-gfp knock-in mouse. J. Comp. Neurol. 2003, 467, 60–79. [Google Scholar] [CrossRef] [PubMed]
- Bellion, A.; Baudoin, J.P.; Alvarez, C.; Bornens, M.; Metin, C. Nucleokinesis in tangentially migrating neurons comprises two alternating phases: Forward migration of the golgi/centrosome associated with centrosome splitting and myosin contraction at the rear. J. Neurosci. 2005, 25, 5691–5699. [Google Scholar] [CrossRef] [PubMed]
- Metin, C.; Vallee, R.B.; Rakic, P.; Bhide, P.G. Modes and mishaps of neuronal migration in the mammalian brain. J. Neurosci. 2008, 28, 11746–11752. [Google Scholar] [CrossRef] [PubMed]
- Filipovic, R.; Kumar, S.S.; Bahr, B.A.; Loturco, J. Slice culture method for studying migration of neuronal progenitor cells derived from human embryonic stem cells (hesc). Curr. Protoc. Stem Cell Biol. 2014. [Google Scholar] [CrossRef]
- Andrews, W.D.; Zito, A.; Memi, F.; Jones, G.; Tamamaki, N.; Parnavelas, J.G. Limk2 mediates semaphorin signalling in cortical interneurons migrating through the subpallium. Biol. Open 2013, 2, 277–282. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alifragis, P.; Liapi, A.; Parnavelas, J.G. Lhx6 regulates the migration of cortical interneurons from the ventral telencephalon but does not specify their gaba phenotype. J. Neurosci. 2004, 24, 5643–5648. [Google Scholar] [CrossRef] [PubMed]
- Friocourt, G.; Liu, J.S.; Antypa, M.; Rakic, S.; Walsh, C.A.; Parnavelas, J.G. Both doublecortin and doublecortin-like kinase play a role in cortical interneuron migration. J. Neurosci. 2007, 27, 3875–3883. [Google Scholar] [CrossRef] [PubMed]
- Marin, O.; Plump, A.S.; Flames, N.; Sanchez-Camacho, C.; Tessier-Lavigne, M.; Rubenstein, J.L. Directional guidance of interneuron migration to the cerebral cortex relies on subcortical Slit1/2-independent repulsion and cortical attraction. Development 2003, 130, 1889–1901. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, V.; Stappers, E.; Vandesande, B.; Dimidschstein, J.; Kroes, R.; Francis, A.; Conidi, A.; Lesage, F.; Dries, R.; Cazzola, S.; et al. Directed migration of cortical interneurons depends on the cell-autonomous action of SIP1. Neuron 2013, 77, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Godin, J.D.; Thomas, N.; Laguesse, S.; Malinouskaya, L.; Close, P.; Malaise, O.; Purnelle, A.; Raineteau, O.; Campbell, K.; Fero, M.; et al. P27(kip1) is a microtubule-associated protein that promotes microtubule polymerization during neuron migration. Dev. Cell 2012, 23, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Nery, F.C.; da Hora, C.C.; Yaqub, U.; Zhang, X.; McCarthy, D.M.; Bhide, P.G.; Irimia, D.; Breakefield, X.O. New methods for investigation of neuronal migration in embryonic brain explants. J. Neurosci. Methods 2015, 239, 80–84. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.M.; Zhang, X.; Darnell, S.B.; Sangrey, G.R.; Yanagawa, Y.; Sadri-Vakili, G.; Bhide, P.G. Cocaine alters BDNF expression and neuronal migration in the embryonic mouse forebrain. J. Neurosci. 2011, 31, 13400–13411. [Google Scholar] [CrossRef] [PubMed]
- Lysko, D.E.; Putt, M.; Golden, J.A. Sdf1 regulates leading process branching and speed of migrating interneurons. J. Neurosci. 2011, 31, 1739–1745. [Google Scholar] [CrossRef] [PubMed]
- Lysko, D.E.; Putt, M.; Golden, J.A. Sdf1 reduces interneuron leading process branching through dual regulation of actin and microtubules. J. Neurosci. 2014, 34, 4941–4962. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, G.; Sanchez-Alcaniz, J.A.; Osorio, C.; Valiente, M.; Garcia-Frigola, C.; Marin, O. Neuregulin 3 mediates cortical plate invasion and laminar allocation of gabaergic interneurons. Cell Rep. 2017, 18, 1157–1170. [Google Scholar] [CrossRef] [PubMed]
- LoTurco, J.; Manent, J.B.; Sidiqi, F. New and improved tools for in utero electroporation studies of developing cerebral cortex. Cereb. Cortex 2009, 19, i120–i125. [Google Scholar] [CrossRef] [PubMed]
- Angevine, J.B., Jr.; Sidman, R.L. Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature 1961, 192, 766–768. [Google Scholar] [CrossRef] [PubMed]
- Rakic, P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J. Comp. Neurol. 1972, 145, 61–83. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, T.; Mizutani, Y.; Ohmori, Y.; Okumura, J. Comparison of three nonviral transfection methods for foreign gene expression in early chicken embryos in ovo. Biochem. Biophys. Res. Commun. 1997, 230, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi-Shimogori, T.; Grove, E.A. Neocortex patterning by the secreted signaling molecule FGF8. Science 2001, 294, 1071–1074. [Google Scholar] [CrossRef] [PubMed]
- Tabata, H.; Nakajima, K. Efficient in utero gene transfer system to the developing mouse brain using electroporation: Visualization of neuronal migration in the developing cortex. Neuroscience 2001, 103, 865–872. [Google Scholar] [CrossRef]
- Saito, T.; Nakatsuji, N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev. Biol. 2001, 240, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Borrell, V.; Yoshimura, Y.; Callaway, E.M. Targeted gene delivery to telencephalic inhibitory neurons by directional in utero electroporation. J. Neurosci. Methods 2005, 143, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Pacary, E.; Guillemot, F. Cerebral cortex electroporation to study projection neuron migration. Curr. Protoc. Neurosci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pacary, E.; Guillemot, F. In utero electroporation to study mouse brain development. Methods Mol. Biol. 2014, 1082, 285–293. [Google Scholar] [PubMed]
- Kriegstein, A.; Alvarez-Buylla, A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, N.A.; Dailey, M.E.; Smith, S.J.; McConnell, S.K. Diverse migratory pathways in the developing cerebral cortex. Science 1992, 258, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Yoneda, T.; Hatanaka, Y.; Kubota, C.; Murakami, F.; Sato, M. Filamin a-interacting protein (FILIP) regulates cortical cell migration out of the ventricular zone. Nat. Cell Biol. 2002, 4, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Loulier, K.; Lathia, J.D.; Marthiens, V.; Relucio, J.; Mughal, M.R.; Tang, S.C.; Coksaygan, T.; Hall, P.E.; Chigurupati, S.; Patton, B.; et al. Beta1 integrin maintains integrity of the embryonic neocortical stem cell niche. PLoS Biol. 2009, 7, e1000176. [Google Scholar] [CrossRef] [PubMed]
- Heng, J.I.; Nguyen, L.; Castro, D.S.; Zimmer, C.; Wildner, H.; Armant, O.; Skowronska-Krawczyk, D.; Bedogni, F.; Matter, J.M.; Hevner, R.; et al. Neurogenin 2 controls cortical neuron migration through regulation of RND2. Nature 2008, 455, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Ramos, R.L.; Paramasivam, M.; Siddiqi, F.; Ackman, J.B.; LoTurco, J.J. The role of dcx and lis1 in migration through the lateral cortical stream of developing forebrain. Dev. Neurosci. 2008, 30, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, A.V.; Rubinson, D.A.; Dent, E.W.; Edward van Veen, J.; Leslie, J.D.; Zhang, J.; Mebane, L.M.; Philippar, U.; Pinheiro, E.M.; Burds, A.A.; et al. ENA/VASP is required for neuritogenesis in the developing cortex. Neuron 2007, 56, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Pacary, E.; Heng, J.; Azzarelli, R.; Riou, P.; Castro, D.; Lebel-Potter, M.; Parras, C.; Bell, D.M.; Ridley, A.J.; Parsons, M.; et al. Proneural transcription factors regulate different steps of cortical neuron migration through rnd-mediated inhibition of RHOA signaling. Neuron 2011, 69, 1069–1084. [Google Scholar] [CrossRef] [PubMed]
- Imai, J.H.; Wang, X.; Shi, S.H. Kaede-centrin1 labeling of mother and daughter centrosomes in mammalian neocortical neural progenitors. Curr. Protoc. Stem Cell Biol. 2010. [Google Scholar] [CrossRef]
- Nadarajah, B.; Alifragis, P.; Wong, R.O.; Parnavelas, J.G. Neuronal migration in the developing cerebral cortex: Observations based on real-time imaging. Cereb. Cortex 2003, 13, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Yokota, Y.; Gashghaei, H.T.; Han, C.; Watson, H.; Campbell, K.J.; Anton, E.S. Radial glial dependent and independent dynamics of interneuronal migration in the developing cerebral cortex. PLoS ONE 2007, 2, e794. [Google Scholar] [CrossRef] [PubMed]
- Martini, F.J.; Valiente, M.; Lopez Bendito, G.; Szabo, G.; Moya, F.; Valdeolmillos, M.; Marin, O. Biased selection of leading process branches mediates chemotaxis during tangential neuronal migration. Development 2009, 136, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Metin, C.; Baudoin, J.P.; Rakic, S.; Parnavelas, J.G. Cell and molecular mechanisms involved in the migration of cortical interneurons. Eur. J. Neurosci. 2006, 23, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Cepko, C.L. Controlled expression of transgenes introduced by in vivo electroporation. Proc. Natl. Acad. Sci. USA 2007, 104, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Reiner, O.; Coquelle, F.M.; Peter, B.; Levy, T.; Kaplan, A.; Sapir, T.; Orr, I.; Barkai, N.; Eichele, G.; Bergmann, S. The evolving doublecortin (DCX) superfamily. BMC Genom. 2006, 7, 188. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Ramos, R.L.; Ackman, J.B.; Thomas, A.M.; Lee, R.V.; LoTurco, J.J. Rnai reveals doublecortin is required for radial migration in rat neocortex. Nat. Neurosci. 2003, 6, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Sapir, T.; Sapoznik, S.; Levy, T.; Finkelshtein, D.; Shmueli, A.; Timm, T.; Mandelkow, E.M.; Reiner, O. Accurate balance of the polarity kinase mark2/par-1 is required for proper cortical neuronal migration. J. Neurosci. 2008, 28, 5710–5720. [Google Scholar] [CrossRef] [PubMed]
- Kappeler, C.; Saillour, Y.; Baudoin, J.P.; Tuy, F.P.; Alvarez, C.; Houbron, C.; Gaspar, P.; Hamard, G.; Chelly, J.; Metin, C.; et al. Branching and nucleokinesis defects in migrating interneurons derived from doublecortin knockout mice. Hum. Mol. Genet. 2006, 15, 1387–1400. [Google Scholar] [CrossRef] [PubMed]
- Corbo, J.C.; Deuel, T.A.; Long, J.M.; LaPorte, P.; Tsai, E.; Wynshaw-Boris, A.; Walsh, C.A. Doublecortin is required in mice for lamination of the hippocampus but not the neocortex. J. Neurosci. 2002, 22, 7548–7557. [Google Scholar] [PubMed]
- Broix, L.; Jagline, H.; E, L.I.; Schmucker, S.; Drouot, N.; Clayton-Smith, J.; Pagnamenta, A.T.; Metcalfe, K.A.; Isidor, B.; Louvier, U.W.; et al. Mutations in the hect domain of nedd4l lead to akt-mtor pathway deregulation and cause periventricular nodular heterotopia. Nat. Genet. 2016, 48, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Poirier, K.; Lebrun, N.; Broix, L.; Tian, G.; Saillour, Y.; Boscheron, C.; Parrini, E.; Valence, S.; Pierre, B.S.; Oger, M.; et al. Mutations IN TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 2013, 45, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Friocourt, G.; Marcorelles, P.; Saugier-Veber, P.; Quille, M.L.; Marret, S.; Laquerriere, A. Role of cytoskeletal abnormalities in the neuropathology and pathophysiology of type i lissencephaly. Acta Neuropathol. 2011, 121, 149–170. [Google Scholar] [CrossRef] [PubMed]
- Breuss, M.; Fritz, T.; Gstrein, T.; Chan, K.; Ushakova, L.; Yu, N.; Vonberg, F.W.; Werner, B.; Elling, U.; Keays, D.A. Mutations in the murine homologue of tubb5 cause microcephaly by perturbing cell cycle progression and inducing p53-associated apoptosis. Development 2016, 143, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using crispr/cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. Rna-guided human genome engineering via CAS9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of crispr-cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Kalebic, N.; Taverna, E.; Tavano, S.; Wong, F.K.; Suchold, D.; Winkler, S.; Huttner, W.B.; Sarov, M. Crispr/cas9-induced disruption of gene expression in mouse embryonic brain and single neural stem cells in vivo. EMBO Rep. 2016, 17, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Azzarelli, R.; Pacary, E.; Garg, R.; Garcez, P.; van den Berg, D.; Riou, P.; Ridley, A.J.; Friedel, R.H.; Parsons, M.; Guillemot, F. An antagonistic interaction between plexinb2 and rnd3 controls rhoa activity and cortical neuron migration. Nat. Commun. 2014, 5, 3405. [Google Scholar] [CrossRef] [PubMed]
- Azzarelli, R.; Guillemot, F.; Pacary, E. Function and regulation of rnd proteins in cortical projection neuron migration. Front. Neurosci. 2015, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Azzarelli, R.; Kerloch, T.; Pacary, E. Regulation of cerebral cortex development by rho gtpases: Insights from in vivo studies. Front. Cell. Neurosci. 2014, 8, 445. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Kurokawa, K.; Kiyokawa, E.; Matsuda, M. Analysis of the spatiotemporal activation of rho gtpases using raichu probes. Methods Enzymol. 2006, 406, 315–332. [Google Scholar] [PubMed]
- Rash, B.G.; Ackman, J.B.; Rakic, P. Bidirectional radial Ca(2+) activity regulates neurogenesis and migration during early cortical column formation. Sci. Adv. 2016, 2, e1501733. [Google Scholar] [CrossRef] [PubMed]
- Gee, J.M.; Gibbons, M.B.; Taheri, M.; Palumbos, S.; Morris, S.C.; Smeal, R.M.; Flynn, K.F.; Economo, M.N.; Cizek, C.G.; Capecchi, M.R.; et al. Imaging activity in astrocytes and neurons with genetically encoded calcium indicators following in utero electroporation. Front. Mol. Neurosci. 2015, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Dal Maschio, M.; Ghezzi, D.; Bony, G.; Alabastri, A.; Deidda, G.; Brondi, M.; Sato, S.S.; Zaccaria, R.P.; Di Fabrizio, E.; Ratto, G.M.; et al. High-performance and site-directed in utero electroporation by a triple-electrode probe. Nat. Commun. 2012, 3, 960. [Google Scholar] [CrossRef] [PubMed]
- Szczurkowska, J.; Cwetsch, A.W.; dal Maschio, M.; Ghezzi, D.; Ratto, G.M.; Cancedda, L. Targeted in vivo genetic manipulation of the mouse or rat brain by in utero electroporation with a triple-electrode probe. Nat. Protoc. 2016, 11, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Osumi, N.; Inoue, T. Gene transfer into cultured mammalian embryos by electroporation. Methods 2001, 24, 35–42. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azzarelli, R.; Oleari, R.; Lettieri, A.; Andre', V.; Cariboni, A. In Vitro, Ex Vivo and In Vivo Techniques to Study Neuronal Migration in the Developing Cerebral Cortex. Brain Sci. 2017, 7, 48. https://doi.org/10.3390/brainsci7050048
Azzarelli R, Oleari R, Lettieri A, Andre' V, Cariboni A. In Vitro, Ex Vivo and In Vivo Techniques to Study Neuronal Migration in the Developing Cerebral Cortex. Brain Sciences. 2017; 7(5):48. https://doi.org/10.3390/brainsci7050048
Chicago/Turabian StyleAzzarelli, Roberta, Roberto Oleari, Antonella Lettieri, Valentina Andre', and Anna Cariboni. 2017. "In Vitro, Ex Vivo and In Vivo Techniques to Study Neuronal Migration in the Developing Cerebral Cortex" Brain Sciences 7, no. 5: 48. https://doi.org/10.3390/brainsci7050048
APA StyleAzzarelli, R., Oleari, R., Lettieri, A., Andre', V., & Cariboni, A. (2017). In Vitro, Ex Vivo and In Vivo Techniques to Study Neuronal Migration in the Developing Cerebral Cortex. Brain Sciences, 7(5), 48. https://doi.org/10.3390/brainsci7050048