
A Neurophysiological Perspective on a Preventive Treatment against Schizophrenia Using Transcranial Electric Stimulation of the Corticothalamic Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Is There an Anatomical Target for Advanced Schizophrenia?
3. Overview of Therapeutic Neurophysiological Procedures: DBS versus TES
4. Three Candidates for Preventive TES
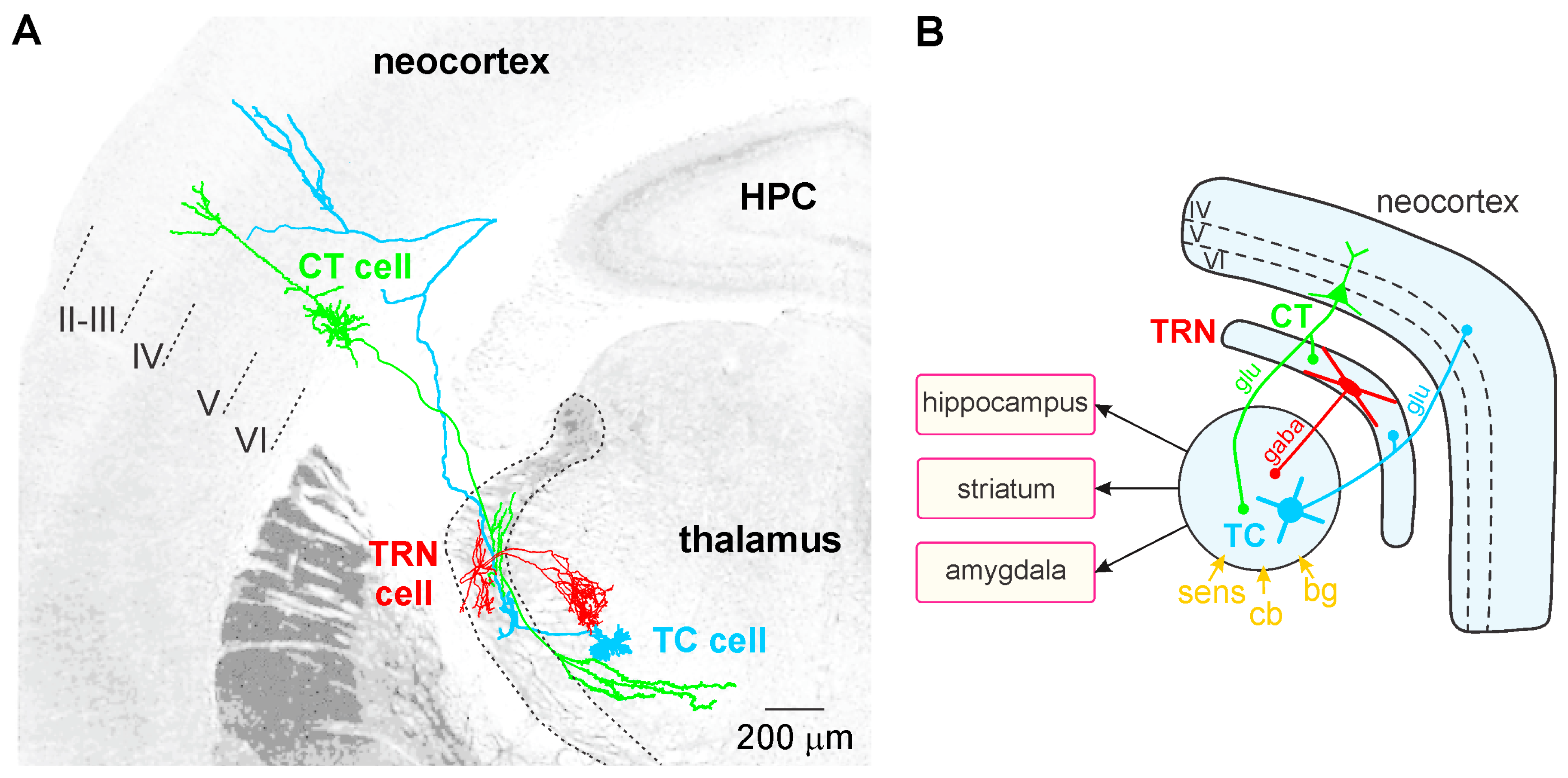
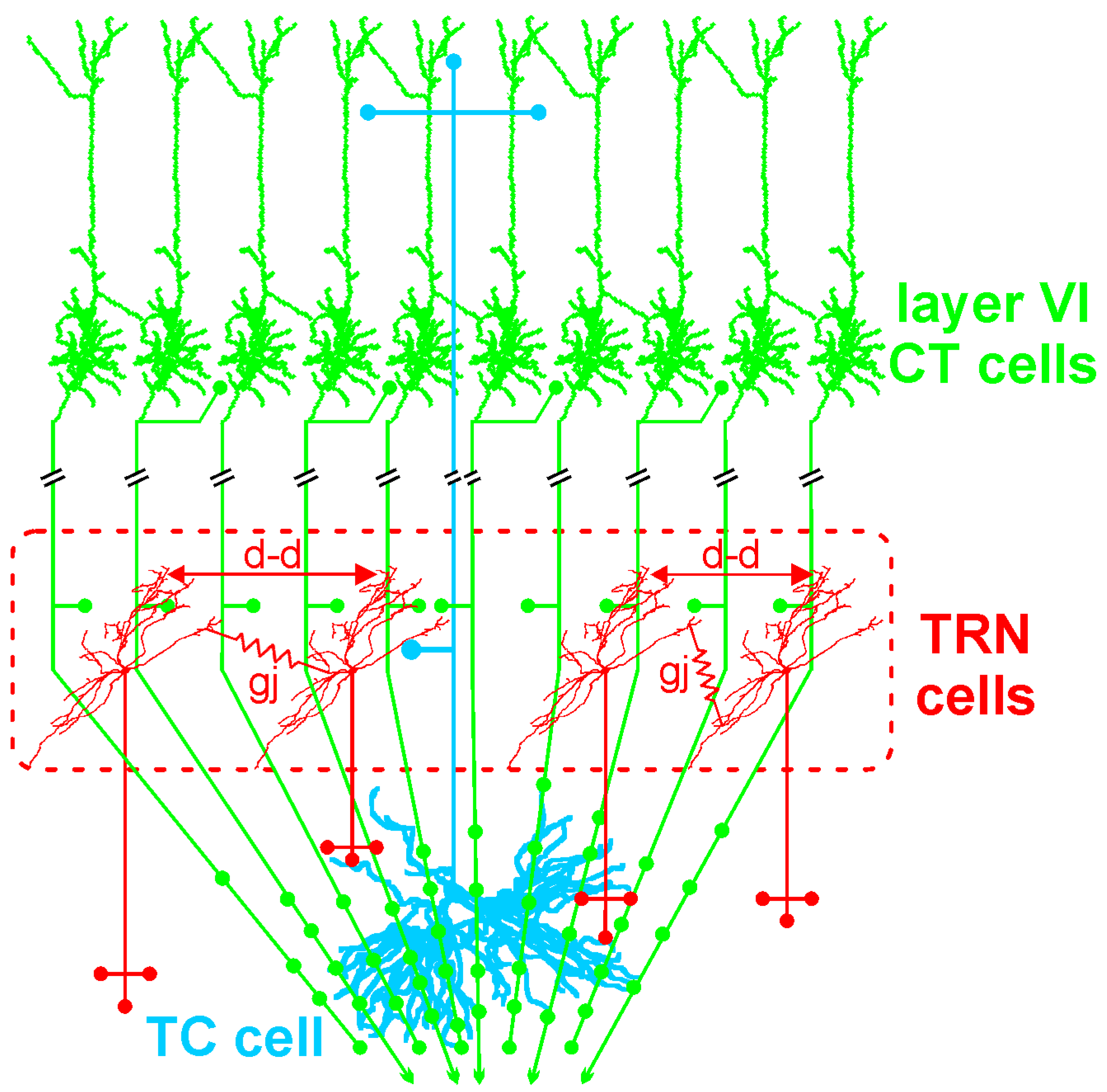
4.1. The Corticothalamic Pathway and the Thalamus
4.2. Glutamatergic Transmission
5. Gamma Frequency (30–80 Hz) Oscillations, a Potential Pathophysiological and Therapeutic Bioelectrical Marker
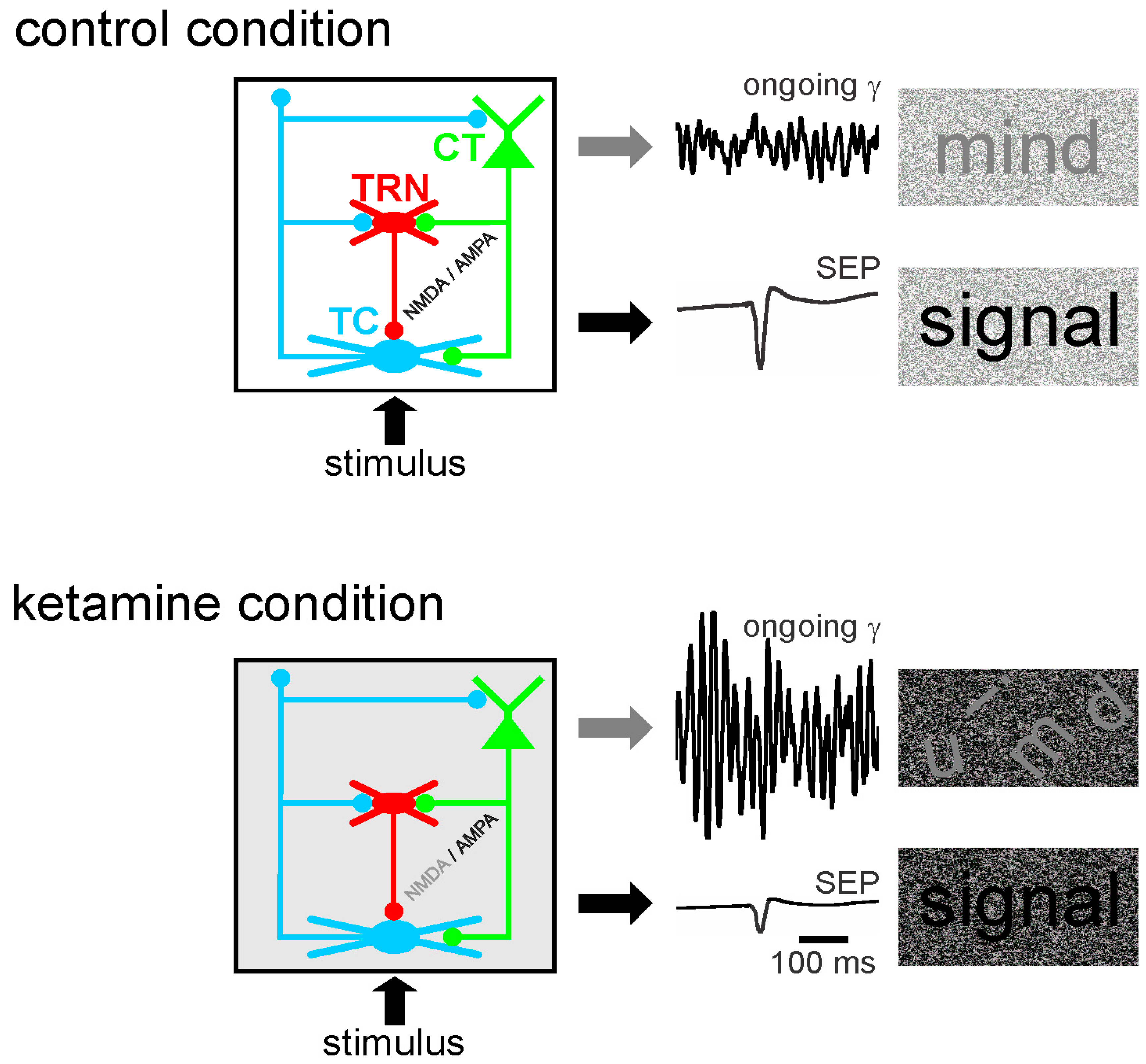
6. Potential Mechanisms of TES
7. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Insel, T.R. Assessing the economic costs of serious mental illness. Am. J. Psychiatry 2008, 165, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Wittchen, H.U.; Jacobi, F.; Rehm, J.; Gustavsson, A.; Svensson, M.; Jonsson, B.; Olesen, J.; Allgulander, C.; Alonso, J.; Faravelli, C.; et al. The size and burden of mental disorders and other disorders of the brain in Europe 2010. Eur. Neuropsychopharmacol. 2011, 21, 655–679. [Google Scholar] [CrossRef] [PubMed]
- Ferrarelli, F.; Tononi, G. Reduced sleep spindle activity point to a TRN-MD thalamus-PFC circuit dysfunction in schizophrenia. Schizophr. Res. 2017, 180, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, C.S.; Demiralp, T. Human eeg gamma oscillations in neuropsychiatric disorders. Clin. Neurophysiol. 2005, 116, 2719–2733. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.G. Cortical development and thalamic pathology in schizophrenia. Schizophr. Bull. 1997, 23, 483–501. [Google Scholar] [CrossRef] [PubMed]
- Minzenberg, M.J.; Firl, A.J.; Yoon, J.H.; Gomes, G.C.; Reinking, C.; Carter, C.S. Gamma oscillatory power is impaired during cognitive control independent of medication status in first-episode schizophrenia. Neuropsychopharmacology 2010, 35, 2590–2599. [Google Scholar] [CrossRef] [PubMed]
- Pergola, G.; Selvaggi, P.; Trizio, S.; Bertolino, A.; Blasi, G. The role of the thalamus in schizophrenia from a neuroimaging perspective. Neurosci. Biobehav. Rev. 2015, 54, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Pinault, D. Dysfunctional thalamus-related networks in schizophrenia. Schizophr. Bull. 2011, 37, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Uhlhaas, P.J.; Haenschel, C.; Nikolic, D.; Singer, W. The role of oscillations and synchrony in cortical networks and their putative relevance for the pathophysiology of schizophrenia. Schizophr. Bull. 2008, 34, 927–943. [Google Scholar] [CrossRef] [PubMed]
- Woodward, N.D.; Karbasforoushan, H.; Heckers, S. Thalamocortical dysconnectivity in schizophrenia. Am. J. Psychiatry 2012, 169, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, T.P.; Liu, B.; Zhou, Y.; Chou, K.H.; Lo, C.Y.; Hung, C.C.; Chen, W.L.; Jiang, T.; Lin, C.P. Disrupted thalamo-cortical connectivity in schizophrenia: A morphometric correlation analysis. Schizophr. Res. 2014, 153, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Folsom, T.D. The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr. Bull. 2009, 35, 528–548. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.J. The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain 1999, 122 Pt 4, 593–624. [Google Scholar] [CrossRef] [PubMed]
- Insel, T.R. Rethinking schizophrenia. Nature 2010, 468, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Margolis, R.L.; Reading, S.A.; Pletnikov, M.; Coyle, J.T. Neurobiology of schizophrenia. Neuron 2006, 52, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.K.; Litman, R.E.; Pickar, D. Adverse effects of antipsychotic drugs. Drug Saf. 1993, 9, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Muench, J.; Hamer, A.M. Adverse effects of antipsychotic medications. Am. Fam. Physician 2010, 81, 617–622. [Google Scholar] [PubMed]
- Kambeitz, J.; Kambeitz-Ilankovic, L.; Cabral, C.; Dwyer, D.B.; Calhoun, V.D.; van den Heuvel, M.P.; Falkai, P.; Koutsouleris, N.; Malchow, B. Aberrant functional whole-brain network architecture in patients with schizophrenia: A meta-analysis. Schizophr. Bull. 2016, 42 (Suppl. 1), S13–S21. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Dean, B. Mapping the pathophysiology of schizophrenia: Interactions between multiple cellular pathways. Front. Cell. Neurosci. 2013, 7, 238. [Google Scholar] [CrossRef] [PubMed]
- Goodman, W.K.; Insel, T.R. Deep brain stimulation in psychiatry: Concentrating on the road ahead. Biol. Psychiatry 2009, 65, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.; Caspi, A.; Noy, S. The treatment of schizophrenia: From premorbid manifestations to the first episode of psychosis. Dialogues Clin. Neurosci. 2005, 7, 7–16. [Google Scholar] [PubMed]
- Brown, A.S.; Patterson, P.H. Maternal infection and schizophrenia: Implications for prevention. Schizophr. Bull. 2011, 37, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Suvisaari, J.M.; Haukka, J.K.; Tanskanen, A.J.; Lonnqvist, J.K. Decline in the incidence of schizophrenia in finnish cohorts born from 1954 to 1965. Arch. Gen. Psychiatry 1999, 56, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Boksa, P. Effects of prenatal infection on brain development and behavior: A review of findings from animal models. Brain Behav. Immun. 2010, 24, 881–897. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Derkits, E.J. Prenatal infection and schizophrenia: A review of epidemiologic and translational studies. Am. J. Psychiatry 2010, 167, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, H.J.; Mortensen, E.L.; Reinisch, J.M.; Mednick, S.A. Association between prenatal exposure to bacterial infection and risk of schizophrenia. Schizophr. Bull. 2009, 35, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U. Developmental neuroinflammation and schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 42, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Giovanoli, S.; Engler, H.; Engler, A.; Richetto, J.; Feldon, J.; Riva, M.A.; Schedlowski, M.; Meyer, U. Preventive effects of minocycline in a neurodevelopmental two-hit model with relevance to schizophrenia. Transl. Psychiatry 2016, 6, e772. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Donnelly, R.; Elkabes, S.; Zhang, P.; Davini, D.; David, B.T.; Ponzio, N.M. Maternal immune stimulation during pregnancy shapes the immunological phenotype of offspring. Brain Behav. Immun. 2013, 33, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Reisinger, S.; Khan, D.; Kong, E.; Berger, A.; Pollak, A.; Pollak, D.D. The poly(i:C)-induced maternal immune activation model in preclinical neuropsychiatric drug discovery. Pharmacol. Ther. 2015, 149, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, L.; Rehavi, M.; Nachman, R.; Weiner, I. Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: A novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology 2003, 28, 1778–1789. [Google Scholar] [CrossRef] [PubMed]
- Buzsaki, G. Rhythms of the Brain; Oxford University Press: New York, NY, USA, 2006. [Google Scholar]
- Antal, A.; Herrmann, C.S. Transcranial alternating current and random noise stimulation: Possible mechanisms. Neural Plast. 2016, 2016, 3616807. [Google Scholar] [CrossRef] [PubMed]
- Brunelin, J.; Poulet, E.; Bediou, B.; Kallel, L.; Dalery, J.; D’Amato, T.; Saoud, M. Low frequency repetitive transcranial magnetic stimulation improves source monitoring deficit in hallucinating patients with schizophrenia. Schizophr. Res. 2006, 81, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Puri, B.K.; Davey, N.J.; Ellaway, P.H.; Lewis, S.W. An investigation of motor function in schizophrenia using transcranial magnetic stimulation of the motor cortex. Br. J. Psychiatry 1996, 169, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Kallel, L.; Mondino, M.; Brunelin, J. Effects of theta-rhythm transcranial alternating current stimulation (4.5 Hz-tACS) in patients with clozapine-resistant negative symptoms of schizophrenia: A case series. J. Neural Transm. 2016, 123, 1213–1217. [Google Scholar] [CrossRef] [PubMed]
- Mondino, M.; Brunelin, J.; Palm, U.; Brunoni, A.R.; Poulet, E.; Fecteau, S. Transcranial direct current stimulation for the treatment of refractory symptoms of schizophrenia. Current evidence and future directions. Curr. Pharm. Des. 2015, 21, 3373–3383. [Google Scholar] [CrossRef] [PubMed]
- Mondino, M.; Haesebaert, F.; Poulet, E.; Suaud-Chagny, M.F.; Brunelin, J. Fronto-temporal transcranial direct current stimulation (tDCS) reduces source-monitoring deficits and auditory hallucinations in patients with schizophrenia. Schizophr. Res. 2015, 161, 515–516. [Google Scholar] [CrossRef] [PubMed]
- Vercammen, A.; Rushby, J.A.; Loo, C.; Short, B.; Weickert, C.S.; Weickert, T.W. Transcranial direct current stimulation influences probabilistic association learning in schizophrenia. Schizophr. Res. 2011, 131, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.J.; Antal, A.; Bikson, M.; Boggio, P.S.; Brunoni, A.R.; Celnik, P.; Cohen, L.G.; Fregni, F.; Herrmann, C.S.; Kappenman, E.S.; et al. A technical guide to tDCS, and related non-invasive brain stimulation tools. Clin. Neurophysiol. 2016, 127, 1031–1048. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, R.; Renaud, P.; Zorn, L.; Goffin, L.; Bayle, B.; Foucher, J.; Lamy, J.; Armspach, J.P.; de Mathelin, M. A custom robot for transcranial magnetic stimulation: First assessment on healthy subjects. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2013, 2013, 5352–5355. [Google Scholar] [PubMed]
- Lefaucheur, J.P.; Andre-Obadia, N.; Antal, A.; Ayache, S.S.; Baeken, C.; Benninger, D.H.; Cantello, R.M.; Cincotta, M.; de Carvalho, M.; de Ridder, D.; et al. Evidence-based guidelines on the therapeutic use of repetitive transcranial magnetic stimulation (RTMS). Clin. Neurophysiol. 2014, 125, 2150–2206. [Google Scholar] [CrossRef] [PubMed]
- Yung, A.R.; McGorry, P.D. The prodromal phase of first-episode psychosis: Past and current conceptualizations. Schizophr. Bull. 1996, 22, 353–370. [Google Scholar] [CrossRef] [PubMed]
- Kahn, R.S.; Sommer, I.E. The neurobiology and treatment of first-episode schizophrenia. Mol. Psychiatry 2015, 20, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Perkins, D.O.; Gu, H.; Boteva, K.; Lieberman, J.A. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: A critical review and meta-analysis. Am. J. Psychiatry 2005, 162, 1785–1804. [Google Scholar] [CrossRef] [PubMed]
- Kulhara, P.; Banerjee, A.; Dutt, A. Early intervention in schizophrenia. Indian J. Psychiatry 2008, 50, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.K.; Walker, E.F.; Compton, M.T. Early signs, diagnosis and therapeutics of the prodromal phase of schizophrenia and related psychotic disorders. Expert Rev. Neurother. 2010, 10, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, R.J. Neuroleptics and the natural course of schizophrenia. Schizophr. Bull. 1991, 17, 325–351. [Google Scholar] [CrossRef] [PubMed]
- Basar, E. Brain oscillations in neuropsychiatric disease. Dialogues Clin. Neurosci. 2013, 15, 291–300. [Google Scholar] [PubMed]
- Baldeweg, T.; Spence, S.; Hirsch, S.R.; Gruzelier, J. Gamma-band electroencephalographic oscillations in a patient with somatic hallucinations. Lancet 1998, 352, 620–621. [Google Scholar] [CrossRef]
- Becker, C.; Gramann, K.; Muller, H.J.; Elliott, M.A. Electrophysiological correlates of flicker-induced color hallucinations. Conscious. Cogn. 2009, 18, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Mulert, C.; Kirsch, V.; Pascual-Marqui, R.; McCarley, R.W.; Spencer, K.M. Long-range synchrony of gamma oscillations and auditory hallucination symptoms in schizophrenia. Int. J. Psychophysiol. 2011, 79, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Spencer, K.M.; Nestor, P.G.; Perlmutter, R.; Niznikiewicz, M.A.; Klump, M.C.; Frumin, M.; Shenton, M.E.; McCarley, R.W. Neural synchrony indexes disordered perception and cognition in schizophrenia. Proc. Natl. Acad. Sci. USA 2004, 101, 17288–17293. [Google Scholar] [CrossRef] [PubMed]
- Andreou, C.; Nolte, G.; Leicht, G.; Polomac, N.; Hanganu-Opatz, I.L.; Lambert, M.; Engel, A.K.; Mulert, C. Increased resting-state gamma-band connectivity in first-episode schizophrenia. Schizophr. Bull. 2015, 41, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Flynn, G.; Alexander, D.; Harris, A.; Whitford, T.; Wong, W.; Galletly, C.; Silverstein, S.; Gordon, E.; Williams, L.M. Increased absolute magnitude of gamma synchrony in first-episode psychosis. Schizophr. Res. 2008, 105, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Ramyead, A.; Kometer, M.; Studerus, E.; Koranyi, S.; Ittig, S.; Gschwandtner, U.; Fuhr, P.; Riecher-Rossler, A. Aberrant current source-density and lagged phase synchronization of neural oscillations as markers for emerging psychosis. Schizophr. Bull. 2015, 41, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Ramyead, A.; Studerus, E.; Kometer, M.; Uttinger, M.; Gschwandtner, U.; Fuhr, P.; Riecher-Rossler, A. Prediction of psychosis using neural oscillations and machine learning in neuroleptic-naive at-risk patients. World J. Biol. Psychiatry 2016, 17, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Rivolta, D.; Heidegger, T.; Scheller, B.; Sauer, A.; Schaum, M.; Birkner, K.; Singer, W.; Wibral, M.; Uhlhaas, P.J. Ketamine dysregulates the amplitude and connectivity of high-frequency oscillations in cortical-subcortical networks in humans: Evidence from resting-state magnetoencephalography-recordings. Schizophr. Bull. 2015, 41, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.M.; Jones, N.C.; O’Brien, T.J.; Pinault, D. The n-methyl d-aspartate glutamate receptor antagonist ketamine disrupts the functional state of the corticothalamic pathway. Cereb. Cortex 2016. [Google Scholar] [CrossRef] [PubMed]
- Hakami, T.; Jones, N.C.; Tolmacheva, E.A.; Gaudias, J.; Chaumont, J.; Salzberg, M.; O’Brien, T.J.; Pinault, D. Nmda receptor hypofunction leads to generalized and persistent aberrant gamma oscillations independent of hyperlocomotion and the state of consciousness. PLoS ONE 2009, 4, e6755. [Google Scholar] [CrossRef] [PubMed]
- Kulikova, S.P.; Tolmacheva, E.A.; Anderson, P.; Gaudias, J.; Adams, B.E.; Zheng, T.; Pinault, D. Opposite effects of ketamine and deep brain stimulation on rat thalamocortical information processing. Eur. J. Neurosci. 2012, 36, 3407–3419. [Google Scholar] [CrossRef] [PubMed]
- Pinault, D. N-methyl d-aspartate receptor antagonists ketamine and MK-801 induce wake-related aberrant gamma oscillations in the rat neocortex. Biol. Psychiatry 2008, 63, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Treen, D.; Batlle, S.; Molla, L.; Forcadell, E.; Chamorro, J.; Bulbena, A.; Perez, V. Are there glutamate abnormalities in subjects at high risk mental state for psychosis? A review of the evidence. Schizophr. Res. 2016, 171, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Manoach, D.S.; Pan, J.Q.; Purcell, S.M.; Stickgold, R. Reduced sleep spindles in schizophrenia: A treatable endophenotype that links risk genes to impaired cognition? Biol. Psychiatry 2016, 80, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Tesler, N.; Gerstenberg, M.; Franscini, M.; Jenni, O.G.; Walitza, S.; Huber, R. Reduced sleep spindle density in early onset schizophrenia: A preliminary finding. Schizophr. Res. 2015, 166, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Monti, J.M.; Monti, D. Sleep disturbance in schizophrenia. Int. Rev. Psychiatry 2005, 17, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Kamath, J.; Virdi, S.; Winokur, A. Sleep disturbances in schizophrenia. Psychiatr. Clin. N. Am. 2015, 38, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Giesel, F.L.; Mehndiratta, A.; Hempel, A.; Hempel, E.; Kress, K.R.; Essig, M.; Schroder, J. Improvement of auditory hallucinations and reduction of primary auditory area’s activation following tms. Eur. J. Radiol. 2012, 81, 1273–1275. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.E.; Gueorguieva, R.; Hawkins, K.A.; Varanko, M.; Boutros, N.N.; Wu, Y.T.; Carroll, K.; Krystal, J.H. Temporoparietal transcranial magnetic stimulation for auditory hallucinations: Safety, efficacy and moderators in a fifty patient sample. Biol. Psychiatry 2005, 58, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Luber, B.; Kinnunen, L.H.; Rakitin, B.C.; Ellsasser, R.; Stern, Y.; Lisanby, S.H. Facilitation of performance in a working memory task with rtms stimulation of the precuneus: Frequency- and time-dependent effects. Brain Res. 2007, 1128, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Luber, B.; Steffener, J.; Tucker, A.; Habeck, C.; Peterchev, A.V.; Deng, Z.D.; Basner, R.C.; Stern, Y.; Lisanby, S.H. Extended remediation of sleep deprived-induced working memory deficits using FMRI-guided transcranial magnetic stimulation. Sleep 2013, 36, 857–871. [Google Scholar] [CrossRef] [PubMed]
- George, M.S.; Padberg, F.; Schlaepfer, T.E.; O’Reardon, J.P.; Fitzgerald, P.B.; Nahas, Z.H.; Marcolin, M.A. Controversy: Repetitive transcranial magnetic stimulation or transcranial direct current stimulation shows efficacy in treating psychiatric diseases (depression, mania, schizophrenia, obsessive-complusive disorder, panic, posttraumatic stress disorder). Brain Stimul. 2009, 2, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Lisanby, S.H.; Kinnunen, L.H.; Crupain, M.J. Applications of TMS to therapy in psychiatry. J. Clin. Neurophysiol. 2002, 19, 344–360. [Google Scholar] [CrossRef] [PubMed]
- Haynes, W.I.; Mallet, L. High-frequency stimulation of deep brain structures in obsessive-compulsive disorder: The search for a valid circuit. Eur. J. Neurosci. 2010, 32, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Mallet, L.; Schupbach, M.; N’Diaye, K.; Remy, P.; Bardinet, E.; Czernecki, V.; Welter, M.L.; Pelissolo, A.; Ruberg, M.; Agid, Y.; et al. Stimulation of subterritories of the subthalamic nucleus reveals its role in the integration of the emotional and motor aspects of behavior. Proc. Natl. Acad. Sci. USA 2007, 104, 10661–10666. [Google Scholar] [CrossRef] [PubMed]
- George, M.S.; Nahas, Z.; Borckardt, J.J.; Anderson, B.; Foust, M.J.; Burns, C.; Kose, S.; Short, E.B. Brain stimulation for the treatment of psychiatric disorders. Curr. Opin. Psychiatry 2007, 20, 250–254; discussion 247–259. [Google Scholar] [CrossRef] [PubMed]
- Keller, W.R.; Fischer, B.A.; Carpenter, W.T., Jr. Revisiting the diagnosis of schizophrenia: Where have we been and where are we going? CNS Neurosci. Ther. 2011, 17, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Clausen, J. Ethical brain stimulation—Neuroethics of deep brain stimulation in research and clinical practice. Eur. J. Neurosci. 2010, 32, 1152–1162. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Katayama, Y.; Kobayashi, K.; Oshima, H.; Fukaya, C.; Tsubokawa, T. Deep brain stimulation for the treatment of vegetative state. Eur. J. Neurosci. 2010, 32, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Forrest, A.D.; Coto, C.A.; Siegel, S.J. Animal models of psychosis: Current state and future directions. Curr. Behav. Neurosci. Rep. 2014, 1, 100–116. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.A.; Watson, D.J.; Fone, K.C. Animal models of schizophrenia. Br. J. Pharmacol. 2011, 164, 1162–1194. [Google Scholar] [CrossRef] [PubMed]
- Low, N.C.; Hardy, J. What is a schizophrenic mouse? Neuron 2007, 54, 348–349. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.M.; Miyakawa, T. Schizophrenia-relevant behavioral testing in rodent models: A uniquely human disorder? Biol. Psychiatry 2006, 59, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Douglas, K.S.; Guy, L.S.; Hart, S.D. Psychosis as a risk factor for violence to others: A meta-analysis. Psychol. Bull. 2009, 135, 679–706. [Google Scholar] [CrossRef] [PubMed]
- Hodgins, S.; Piatosa, M.J.; Schiffer, B. Violence among people with schizophrenia: Phenotypes and neurobiology. Curr. Top. Behav. Neurosci. 2014, 17, 329–368. [Google Scholar] [PubMed]
- Iozzino, L.; Ferrari, C.; Large, M.; Nielssen, O.; de Girolamo, G. Prevalence and risk factors of violence by psychiatric acute inpatients: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0128536. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, E.R.; Pearson, D.M.; Srivastava, L.K. Animal models of schizophrenia: A critical review. J. Psychiatry Neurosci. 2001, 26, 395–410. [Google Scholar] [PubMed]
- Robertson, G.S.; Hori, S.E.; Powell, K.J. Schizophrenia: An integrative approach to modelling a complex disorder. J. Psychiatry Neurosci. 2006, 31, 157–167. [Google Scholar] [PubMed]
- Bitanihirwe, B.K.; Peleg-Raibstein, D.; Mouttet, F.; Feldon, J.; Meyer, U. Late prenatal immune activation in mice leads to behavioral and neurochemical abnormalities relevant to the negative symptoms of schizophrenia. Neuropsychopharmacology 2010, 35, 2462–2478. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.; Nyffeler, M.; Schwendener, S.; Knuesel, I.; Yee, B.K.; Feldon, J. Relative prenatal and postnatal maternal contributions to schizophrenia-related neurochemical dysfunction after in utero immune challenge. Neuropsychopharmacology 2008, 33, 441–456. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, A.M.; Jennische, E.; Hansson, H.A.; Holmang, A. Prenatal exposure to interleukin-6 results in inflammatory neurodegeneration in hippocampus with nmda/gaba(a) dysregulation and impaired spatial learning. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R1345–R1356. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, E.A.; Wycis, H.T.; Marks, M.; Lee, A.J. Stereotaxic apparatus for operations on the human brain. Science 1947, 106, 349–350. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.R.; Taylor, J.J.; Lamb, K.; Hanlon, C.A.; Short, E.B.; George, M.S. Role of functional imaging in the development and refinement of invasive neuromodulation for psychiatric disorders. World J. Radiol. 2014, 6, 756–778. [Google Scholar] [CrossRef] [PubMed]
- Benabid, A.L.; Pollak, P.; Gervason, C.; Hoffmann, D.; Gao, D.M.; Hommel, M.; Perret, J.E.; de, R.J. Long-term suppression of tremor by chronic stimulation of the ventral intermediate thalamic nucleus. Lancet 1991, 337, 403–406. [Google Scholar] [CrossRef]
- Benabid, A.L.; Pollak, P.; Louveau, A.; Henry, S.; de, R.J. Combined (thalamotomy and stimulation) stereotactic surgery of the vim thalamic nucleus for bilateral parkinson disease. Appl. Neurophysiol. 1987, 50, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Lockman, J.; Fisher, R.S. Therapeutic brain stimulation for epilepsy. Neurol. Clin. 2009, 27, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Mehdorn, H.M. Deep brain stimulation for dystonia: Review of the literature. J. Neurosurg. Sci. 2016, 60, 199–210. [Google Scholar] [PubMed]
- Boccard, S.G.; Pereira, E.A.; Aziz, T.Z. Deep brain stimulation for chronic pain. J. Clin. Neurosci. 2015, 22, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.A.; Green, A.L.; Aziz, T.Z. Deep brain stimulation for pain. Handb. Clin. Neurol. 2013, 116, 277–294. [Google Scholar] [PubMed]
- Roy, H.A.; Aziz, T.Z. Deep brain stimulation and multiple sclerosis: Therapeutic applications. Mult. Scler. Relat. Disord. 2014, 3, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Mayberg, H.S.; Lozano, A.M.; Voon, V.; McNeely, H.E.; Seminowicz, D.; Hamani, C.; Schwalb, J.M.; Kennedy, S.H. Deep brain stimulation for treatment-resistant depression. Neuron 2005, 45, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Hamani, C.; Nobrega, J.N. Deep brain stimulation in clinical trials and animal models of depression. Eur. J. Neurosci. 2010, 32, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Schrock, L.E.; Mink, J.W.; Woods, D.W.; Porta, M.; Servello, D.; Visser-Vandewalle, V.; Silburn, P.A.; Foltynie, T.; Walker, H.C.; Shahed-Jimenez, J.; et al. Tourette syndrome deep brain stimulation: A review and updated recommendations. Mov. Disord. 2015, 30, 448–471. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Schiff, N.D. Central thalamic deep brain stimulation for cognitive neuromodulation—A review of proposed mechanisms and investigational studies. Eur. J. Neurosci. 2010, 32, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Benabid, A.L.; Torres, N. New targets for dbs. Parkinsonism Relat. Disord. 2012, 18 (Suppl. 1), S21–S23. [Google Scholar] [CrossRef]
- Nassery, A.; Palmese, C.A.; Sarva, H.; Groves, M.; Miravite, J.; Kopell, B.H. Psychiatric and cognitive effects of deep brain stimulation for parkinson’s disease. Curr. Neurol. Neurosci. Rep. 2016, 16, 87. [Google Scholar] [CrossRef] [PubMed]
- Piasecki, S.D.; Jefferson, J.W. Psychiatric complications of deep brain stimulation for parkinson’s disease. J. Clin. Psychiatry 2004, 65, 845–849. [Google Scholar] [CrossRef] [PubMed]
- Kringelbach, M.L.; Jenkinson, N.; Owen, S.L.; Aziz, T.Z. Translational principles of deep brain stimulation. Nat. Rev. Neurosci. 2007, 8, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Deniau, J.M.; Degos, B.; Bosch, C.; Maurice, N. Deep brain stimulation mechanisms: Beyond the concept of local functional inhibition. Eur. J. Neurosci. 2010, 32, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Conca, A.; Koppi, S.; Konig, P.; Swoboda, E.; Krecke, N. Transcranial magnetic stimulation: A novel antidepressive strategy? Neuropsychobiology 1996, 34, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, D.A.; Zangen, A.; George, M.S. Transcranial magnetic stimulation in the treatment of substance addiction. Ann. N. Y. Acad. Sci. 2014, 1327, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Pascual-Leone, A. Transcranial magnetic stimulation in neurology. Lancet Neurol. 2003, 2, 145–156. [Google Scholar] [CrossRef]
- Bikson, M.; Grossman, P.; Thomas, C.; Zannou, A.L.; Jiang, J.; Adnan, T.; Mourdoukoutas, A.P.; Kronberg, G.; Truong, D.; Boggio, P.; et al. Safety of transcranial direct current stimulation: Evidence based update 2016. Brain Stimul. 2016, 9, 641–661. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, H.; Shivakumar, V.; Agarwal, S.M.; Bose, A.; Venugopal, D.; Rajasekaran, A.; Subbanna, M.; Kalmady, S.V.; Narayanaswamy, J.C.; Debnath, M.; et al. Transcranial direct current stimulation and neuroplasticity genes: Implications for psychiatric disorders. Acta Neuropsychiatr. 2016, 28, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.; Strube, W.; Palm, U.; Wobrock, T. Repetitive noninvasive brain stimulation to modulate cognitive functions in schizophrenia: A systematic review of primary and secondary outcomes. Schizophr. Bull. 2016, 42 (Suppl. 1), S95–S109. [Google Scholar] [CrossRef] [PubMed]
- Mondino, M.; Jardri, R.; Suaud-Chagny, M.F.; Saoud, M.; Poulet, E.; Brunelin, J. Effects of fronto-temporal transcranial direct current stimulation on auditory verbal hallucinations and resting-state functional connectivity of the left temporo-parietal junction in patients with schizophrenia. Schizophr. Bull. 2016, 42, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Brunelin, J.; Mondino, M.; Gassab, L.; Haesebaert, F.; Gaha, L.; Suaud-Chagny, M.F.; Saoud, M.; Mechri, A.; Poulet, E. Examining transcranial direct-current stimulation (tDCS) as a treatment for hallucinations in schizophrenia. Am. J. Psychiatry 2012, 169, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Priori, A.; Berardelli, A.; Rona, S.; Accornero, N.; Manfredi, M. Polarization of the human motor cortex through the scalp. Neuroreport 1998, 9, 2257–2260. [Google Scholar] [CrossRef] [PubMed]
- Stagg, C.J.; Nitsche, M.A. Physiological basis of transcranial direct current stimulation. Neuroscientist 2011, 17, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Antal, A.; Boros, K.; Poreisz, C.; Chaieb, L.; Terney, D.; Paulus, W. Comparatively weak after-effects of transcranial alternating current stimulation (tACS) on cortical excitability in humans. Brain Stimul. 2008, 1, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Abd Hamid, A.I.; Gall, C.; Speck, O.; Antal, A.; Sabel, B.A. Effects of alternating current stimulation on the healthy and diseased brain. Front. Neurosci. 2015, 9, 391. [Google Scholar] [CrossRef] [PubMed]
- Bennabi, D.; Pedron, S.; Haffen, E.; Monnin, J.; Peterschmitt, Y.; Van Waes, V. Transcranial direct current stimulation for memory enhancement: From clinical research to animal models. Front. Syst. Neurosci. 2014, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Santarnecchi, E.; Polizzotto, N.R.; Godone, M.; Giovannelli, F.; Feurra, M.; Matzen, L.; Rossi, A.; Rossi, S. Frequency-dependent enhancement of fluid intelligence induced by transcranial oscillatory potentials. Curr. Biol. 2013, 23, 1449–1453. [Google Scholar] [CrossRef] [PubMed]
- Voss, U.; Holzmann, R.; Hobson, A.; Paulus, W.; Koppehele-Gossel, J.; Klimke, A.; Nitsche, M.A. Induction of self awareness in dreams through frontal low current stimulation of gamma activity. Nat. Neurosci. 2014, 17, 810–812. [Google Scholar] [CrossRef] [PubMed]
- Urbano, F.J.; D’Onofrio, S.M.; Luster, B.R.; Beck, P.B.; Hyde, J.R.; Bisagno, V.; Garcia-Rill, E. Pedunculopontine nucleus gamma band activity-preconscious awareness, waking, and rem sleep. Front. Neurol. 2014, 5, 210. [Google Scholar] [CrossRef] [PubMed]
- Moisa, M.; Polania, R.; Grueschow, M.; Ruff, C.C. Brain network mechanisms underlying motor enhancement by transcranial entrainment of gamma oscillations. J. Neurosci. 2016, 36, 12053–12065. [Google Scholar] [CrossRef] [PubMed]
- Brignani, D.; Ruzzoli, M.; Mauri, P.; Miniussi, C. Is transcranial alternating current stimulation effective in modulating brain oscillations? PLoS ONE 2013, 8, e56589. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, C.S.; Struber, D.; Helfrich, R.F.; Engel, A.K. Eeg oscillations: From correlation to causality. Int. J. Psychophysiol. 2016, 103, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Terney, D.; Chaieb, L.; Moliadze, V.; Antal, A.; Paulus, W. Increasing human brain excitability by transcranial high-frequency random noise stimulation. J. Neurosci. 2008, 28, 14147–14155. [Google Scholar] [CrossRef] [PubMed]
- Fertonani, A.; Pirulli, C.; Miniussi, C. Random noise stimulation improves neuroplasticity in perceptual learning. J. Neurosci. 2011, 31, 15416–15423. [Google Scholar] [CrossRef] [PubMed]
- Brittain, J.S.; Probert-Smith, P.; Aziz, T.Z.; Brown, P. Tremor suppression by rhythmic transcranial current stimulation. Curr. Biol. 2013, 23, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Hess, C.W. Modulation of cortical-subcortical networks in parkinson’s disease by applied field effects. Front. Hum. Neurosci. 2013, 7, 565. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.; Luigjes, J.; Howes, O.D.; Egerton, A.; Hirao, K.; Valli, I.; Kambeitz, J.; Fusar-Poli, P.; Broome, M.; McGuire, P. Transition to psychosis associated with prefrontal and subcortical dysfunction in ultra high-risk individuals. Schizophr. Bull. 2012, 38, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Brans, R.G.; van Haren, N.E.; van Baal, G.C.; Staal, W.G.; Schnack, H.G.; Kahn, R.S.; Hulshoff Pol, H.E. Longitudinal mri study in schizophrenia patients and their healthy siblings. Br. J. Psychiatry 2008, 193, 422–423. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.I.; Shenton, M.E.; Kubicki, M.; Jung, W.H.; Lee, T.Y.; Yun, J.Y.; Kim, S.N.; Kwon, J.S. Altered thalamo-cortical white matter connectivity: Probabilistic tractography study in clinical-high risk for psychosis and first-episode psychosis. Schizophr. Bull. 2016, 42, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; Fusar-Poli, P.; Bloomfield, M.; Selvaraj, S.; McGuire, P. From the prodrome to chronic schizophrenia: The neurobiology underlying psychotic symptoms and cognitive impairments. Curr. Pharm. Des. 2012, 18, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Kahn, R.S.; Keefe, R.S. Schizophrenia is a cognitive illness: Time for a change in focus. JAMA Psychiatry 2013, 70, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.; Eack, S.M.; Montrose, D.M.; Tandon, N.; Miewald, J.M.; Prasad, K.M.; Keshavan, M.S. Multivariate prediction of emerging psychosis in adolescents at high risk for schizophrenia. Schizophr. Res. 2012, 141, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Yung, A.R.; McGorry, P.D.; McFarlane, C.A.; Jackson, H.J.; Patton, G.C.; Rakkar, A. Monitoring and care of young people at incipient risk of psychosis. Schizophr. Bull. 1996, 22, 283–303. [Google Scholar] [CrossRef] [PubMed]
- Cronenwett, W.J.; Csernansky, J. Thalamic pathology in schizophrenia. Curr. Top. Behav. Neurosci. 2010, 4, 509–528. [Google Scholar] [PubMed]
- van Haren, N.E.; Cahn, W.; Hulshoff Pol, H.E.; Kahn, R.S. Schizophrenia as a progressive brain disease. Eur. Psychiatry 2008, 23, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Zipursky, R.B.; Reilly, T.J.; Murray, R.M. The myth of schizophrenia as a progressive brain disease. Schizophr. Bull. 2013, 39, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Harvey, P.D. When does cognitive decline occur in the period prior to the first episode of schizophrenia? Psychiatry 2009, 6, 12–14. [Google Scholar] [PubMed]
- Simon, A.E.; Cattapan-Ludewig, K.; Zmilacher, S.; Arbach, D.; Gruber, K.; Dvorsky, D.N.; Roth, B.; Isler, E.; Zimmer, A.; Umbricht, D. Cognitive functioning in the schizophrenia prodrome. Schizophr. Bull. 2007, 33, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Tandon, N.; Shah, J.; Keshavan, M.S.; Tandon, R. Attenuated psychosis and the schizophrenia prodrome: Current status of risk identification and psychosis prevention. Neuropsychiatry 2012, 2, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Vidailhet, P. First-episode psychosis, cognitive difficulties and remediation. Encephale 2013, 39 (Suppl. 2), S83–S92. [Google Scholar] [CrossRef]
- Jahshan, C.; Heaton, R.K.; Golshan, S.; Cadenhead, K.S. Course of neurocognitive deficits in the prodrome and first episode of schizophrenia. Neuropsychology 2010, 24, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Riley, B.; Kendler, K.S. Molecular genetic studies of schizophrenia. Eur. J. Hum. Genet. 2006, 14, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Susser, E.; Neugebauer, R.; Hoek, H.W.; Brown, A.S.; Lin, S.; Labovitz, D.; Gorman, J.M. Schizophrenia after prenatal famine. Further evidence. Arch. Gen. Psychiatry 1996, 53, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Joyce, E. Origins of cognitive dysfunction in schizophrenia: Clues from age at onset. Br. J. Psychiatry 2005, 186, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Susser, E.S. Prenatal nutritional deficiency and risk of adult schizophrenia. Schizophr. Bull. 2008, 34, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S. The risk for schizophrenia from childhood and adult infections. Am. J. Psychiatry 2008, 165, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Muller, N.; Schwarz, M.J. Immune system and schizophrenia. Curr. Immunol. Rev. 2010, 6, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; McDonald, C.; Cannon, M.; Arseneault, L.; Boydell, J.; Murray, R.M. Pathways to schizophrenia: The impact of environmental factors. Int. J. Neuropsychopharmacol. 2004, 7 (Suppl. 1), S7–S13. [Google Scholar] [CrossRef] [PubMed]
- Peters, E.; Day, S.; McKenna, J.; Orbach, G. Delusional ideation in religious and psychotic populations. Br. J. Clin. Psychol. 1999, 38 Pt 1, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Guillery, R.W.; Sherman, S.M. Thalamic relay functions and their role in corticocortical communication: Generalizations from the visual system. Neuron 2002, 33, 163–175. [Google Scholar] [CrossRef]
- Watis, L.; Chen, S.H.; Chua, H.C.; Chong, S.A.; Sim, K. Glutamatergic abnormalities of the thalamus in schizophrenia: A systematic review. J. Neural Transm. 2008, 115, 493–511. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.G. The Thalamus, 2nd ed.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2007. [Google Scholar]
- Sherman, S.M. Thalamus plays a central role in ongoing cortical functioning. Nat. Neurosci. 2016, 19, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.M.; Guillery, R.W. Exploring the Thalamus; Academic Press: San Diego, CA, USA, 2001; pp. xvii, 1312. [Google Scholar]
- Castaigne, P.; Lhermitte, F.; Buge, A.; Escourolle, R.; Hauw, J.J.; Lyon-Caen, O. Paramedian thalamic and midbrain infarct: Clinical and neuropathological study. Ann. Neurol. 1981, 10, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Ingvar, D.H.; Sourander, P. Destruction of the reticular core of the brain stem. A patho-anatomical follow-up of a case of coma of three years’ duration. Arch. Neurol. 1970, 23, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hassler, R.; Ore, G.D.; Dieckmann, G.; Bricolo, A.; Dolce, G. Behavioural and eeg arousal induced by stimulation of unspecific projection systems in a patient with post-traumatic apallic syndrome. Electroencephalogr. Clin. Neurophysiol. 1969, 27, 306–310. [Google Scholar] [CrossRef]
- Hardenacke, K.; Shubina, E.; Buhrle, C.P.; Zapf, A.; Lenartz, D.; Klosterkotter, J.; Visser-Vandewalle, V.; Kuhn, J. Deep brain stimulation as a tool for improving cognitive functioning in alzheimer’s dementia: A systematic review. Front. Psychiatry 2013, 4, 159. [Google Scholar] [CrossRef] [PubMed]
- Pinault, D. The thalamic reticular nucleus: Structure, function and concept. Brain Res. Brain Res. Rev. 2004, 46, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Deschenes, M.; Veinante, P.; Zhang, Z.W. The organization of corticothalamic projections: Reciprocity versus parity. Brain Res. Brain Res. Rev. 1998, 28, 286–308. [Google Scholar] [CrossRef]
- Guillery, R.W.; Feig, S.L.; Lozsadi, D.A. Paying attention to the thalamic reticular nucleus. Trends Neurosci. 1998, 21, 28–32. [Google Scholar] [CrossRef]
- Deschenes, M.; Madariaga-Domich, A.; Steriade, M. Dendrodendritic synapses in the cat reticularis thalami nucleus: A structural basis for thalamic spindle synchronization. Brain Res. 1985, 334, 165–168. [Google Scholar] [CrossRef]
- Ide, L.S. The fine structure of the perigeniculate nucleus in the cat. J. Comp. Neurol. 1982, 210, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Montero, V.M.; Singer, W. Ultrastructure and synaptic relations of neural elements containing glutamic acid decarboxylase (GAD) in the perigeniculate nucleus of the cat. A light and electron microscopic immunocytochemical study. Exp. Brain Res. 1984, 56, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Pinault, D.; Smith, Y.; Deschenes, M. Dendrodendritic and axoaxonic synapses in the thalamic reticular nucleus of the adult rat. J. Neurosci. 1997, 17, 3215–3233. [Google Scholar] [PubMed]
- Landisman, C.E.; Long, M.A.; Beierlein, M.; Deans, M.R.; Paul, D.L.; Connors, B.W. Electrical synapses in the thalamic reticular nucleus. J. Neurosci. 2002, 22, 1002–1009. [Google Scholar] [PubMed]
- Deleuze, C.; Huguenard, J.R. Distinct electrical and chemical connectivity maps in the thalamic reticular nucleus: Potential roles in synchronization and sensation. J. Neurosci. 2006, 26, 8633–8645. [Google Scholar] [CrossRef] [PubMed]
- Golshani, P.; Liu, X.B.; Jones, E.G. Differences in quantal amplitude reflect GluR4- subunit number at corticothalamic synapses on two populations of thalamic neurons. Proc. Natl. Acad. Sci. USA 2001, 98, 4172–4177. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Neely, R.; Landisman, C.E. Activation of group i and group II metabotropic glutamate receptors causes LTD and LTP of electrical synapses in the rat thalamic reticular nucleus. J. Neurosci. 2015, 35, 7616–7625. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jones, E.G. Corticothalamic inhibition in the thalamic reticular nucleus. J. Neurophysiol. 2004, 91, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Bucherelli, C.; Tassoni, G.; Bures, J. Differential effect of functional ablation of thalamic reticular nucleus on the acquisition of passive and active avoidance. Int. J. Neurosci. 1993, 73, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Weese, G.D.; Phillips, J.M.; Brown, V.J. Attentional orienting is impaired by unilateral lesions of the thalamic reticular nucleus in the rat. J. Neurosci. 1999, 19, 10135–10139. [Google Scholar] [PubMed]
- Pinault, D.; Deschenes, M. Anatomical evidence for a mechanism of lateral inhibition in the rat thalamus. Eur. J. Neurosci. 1998, 10, 3462–3469. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.M.; Slater, B.J.; Gribkova, E.D.; Llano, D.A. Open-loop organization of thalamic reticular nucleus and dorsal thalamus: A computational model. J. Neurophysiol. 2015, 114, 2353–2367. [Google Scholar] [CrossRef] [PubMed]
- Pratt, J.A.; Morris, B.J. The thalamic reticular nucleus: A functional hub for thalamocortical network dysfunction in schizophrenia and a target for drug discovery. J. PsychoPharmacol. 2015, 29, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.M.; Guillery, R.W. Distinct functions for direct and transthalamic corticocortical connections. J. Neurophysiol. 2011, 106, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Veinante, P.; Lavallee, P.; Deschenes, M. Corticothalamic projections from layer 5 of the vibrissal barrel cortex in the rat. J. Comp. Neurol. 2000, 424, 197–204. [Google Scholar] [CrossRef]
- Feinberg, I.; Guazzelli, M. Schizophrenia—A disorder of the corollary discharge systems that integrate the motor systems of thought with the sensory systems of consciousness. Br. J. Psychiatry 1999, 174, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.M.; Mathalon, D.H. Corollary discharge dysfunction in schizophrenia: Can it explain auditory hallucinations? Int. J. Psychophysiol. 2005, 58, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Lisman, J. Excitation, inhibition, local oscillations, or large-scale loops: What causes the symptoms of schizophrenia? Curr. Opin. Neurobiol. 2012, 22, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Behuret, S.; Deleuze, C.; Bal, T. Corticothalamic synaptic noise as a mechanism for selective attention in thalamic neurons. Front. Neural Circuits 2015, 9, 80. [Google Scholar] [CrossRef] [PubMed]
- Briggs, F. Organizing principles of cortical layer 6. Front. Neural Circuits 2010, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Sillito, A.M.; Jones, H.E. Corticothalamic interactions in the transfer of visual information. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2002, 357, 1739–1752. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.M.; Koch, C. The control of retinogeniculate transmission in the mammalian lateral geniculate nucleus. Exp. Brain Res. 1986, 63, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Sirota, M.G.; Swadlow, H.A.; Beloozerova, I.N. Three channels of corticothalamic communication during locomotion. J. Neurosci. 2005, 25, 5915–5925. [Google Scholar] [CrossRef] [PubMed]
- Tsumoto, T.; Suda, K. Three groups of cortico-geniculate neurons and their distribution in binocular and monocular segments of cat striate cortex. J. Comp. Neurol. 1980, 193, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.W.; Deschenes, M. Intracortical axonal projections of lamina VI cells of the primary somatosensory cortex in the rat: A single-cell labeling study. J. Neurosci. 1997, 17, 6365–6379. [Google Scholar] [PubMed]
- Da Costa, N.M.; Martin, K.A. Selective targeting of the dendrites of corticothalamic cells by thalamic afferents in area 17 of the cat. J. Neurosci. 2009, 29, 13919–13928. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Matney, C.J.; Blankenship, A.; Hestrin, S.; Brown, S.P. Layer 6 corticothalamic neurons activate a cortical output layer, layer 5a. J. Neurosci. 2014, 34, 9656–9664. [Google Scholar] [CrossRef] [PubMed]
- Staiger, J.F.; Zilles, K.; Freund, T.F. Recurrent axon collaterals of corticothalamic projection neurons in rat primary somatosensory cortex contribute to excitatory and inhibitory feedback-loops. Anat. Embryol. 1996, 194, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Briggs, F.; Callaway, E.M. Layer-specific input to distinct cell types in layer 6 of monkey primary visual cortex. J. Neurosci. 2001, 21, 3600–3608. [Google Scholar] [PubMed]
- Staiger, J.F.; Kotter, R.; Zilles, K.; Luhmann, H.J. Laminar characteristics of functional connectivity in rat barrel cortex revealed by stimulation with caged-glutamate. Neurosci. Res. 2000, 37, 49–58. [Google Scholar] [CrossRef]
- Ledergerber, D.; Larkum, M.E. Properties of layer 6 pyramidal neuron apical dendrites. J. Neurosci. 2010, 30, 13031–13044. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Hatada, S.; Kondo, S.; Karube, F.; Kawaguchi, Y. Neocortical inhibitory terminals innervate dendritic spines targeted by thalamocortical afferents. J. Neurosci. 2007, 27, 1139–1150. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Sherman, S.M. Glutamatergic inhibition in sensory neocortex. Cereb. Cortex 2009, 19, 2281–2289. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Lam, Y.W.; Sherman, S.M. Intracortical convergence of layer 6 neurons. Neuroreport 2012, 23, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Morrow, T.J.; Casey, K.L. Corticofugal influences of s1 cortex on ventrobasal thalamic neurons in the awake rat. J. Neurosci. 1986, 6, 3611–3617. [Google Scholar] [PubMed]
- Miyata, M.; Imoto, K. Different composition of glutamate receptors in corticothalamic and lemniscal synaptic responses and their roles in the firing responses of ventrobasal thalamic neurons in juvenile mice. J. Physiol. 2006, 575, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Crandall, S.R.; Cruikshank, S.J.; Connors, B.W. A corticothalamic switch: Controlling the thalamus with dynamic synapses. Neuron 2015, 86, 768–782. [Google Scholar] [CrossRef] [PubMed]
- Salt, T.E. Mediation of thalamic sensory input by both nmda receptors and non-nmda receptors. Nature 1986, 322, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Salt, T.E.; Wilson, D.G.; Prasad, S.K. Antagonism of n-methylaspartate and synaptic responses of neurones in the rat ventrobasal thalamus by ketamine and mk-801. Br. J. Pharmacol. 1988, 94, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Pinault, D.; Deschenes, M. Control of 40-hz firing of reticular thalamic cells by neurotransmitters. Neuroscience 1992, 51, 259–268. [Google Scholar] [CrossRef]
- Hillenbrand, U.; van Hemmen, J.L. Does corticothalamic feedback control cortical velocity tuning? Neural Comput. 2001, 13, 327–355. [Google Scholar] [CrossRef] [PubMed]
- Yousif, N.; Denham, M. The role of cortical feedback in the generation of the temporal receptive field responses of lateral geniculate nucleus neurons: A computational modelling study. Biol. Cybern 2007, 97, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Destexhe, A. Modelling corticothalamic feedback and the gating of the thalamus by the cerebral cortex. J. Physiol. Paris 2000, 94, 391–410. [Google Scholar] [CrossRef]
- Liu, X.B. Subcellular distribution of ampa and nmda receptor subunit immunoreactivity in ventral posterior and reticular nuclei of rat and cat thalamus. J. Comp. Neurol. 1997, 388, 587–602. [Google Scholar] [CrossRef]
- Crick, F. Function of the thalamic reticular complex: The searchlight hypothesis. Proc. Natl. Acad. Sci. USA 1984, 81, 4586–4590. [Google Scholar] [CrossRef] [PubMed]
- Sillito, A.M.; Jones, H.E.; Gerstein, G.L.; West, D.C. Feature-linked synchronization of thalamic relay cell firing induced by feedback from the visual cortex. Nature 1994, 369, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Temereanca, S.; Simons, D.J. Functional topography of corticothalamic feedback enhances thalamic spatial response tuning in the somatosensory whisker/barrel system. Neuron 2004, 41, 639–651. [Google Scholar] [CrossRef]
- Murphy, P.C.; Sillito, A.M. Corticofugal feedback influences the generation of length tuning in the visual pathway. Nature 1987, 329, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Ghazanfar, A.A.; Krupa, D.J.; Nicolelis, M.A. Role of cortical feedback in the receptive field structure and nonlinear response properties of somatosensory thalamic neurons. Exp. Brain Res. 2001, 141, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Alitto, H.J.; Usrey, W.M. Corticothalamic feedback and sensory processing. Curr. Opin. Neurobiol. 2003, 13, 440–445. [Google Scholar] [CrossRef]
- O’Connor, D.H.; Fukui, M.M.; Pinsk, M.A.; Kastner, S. Attention modulates responses in the human lateral geniculate nucleus. Nat. Neurosci. 2002, 5, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- McAlonan, K.; Cavanaugh, J.; Wurtz, R.H. Guarding the gateway to cortex with attention in visual thalamus. Nature 2008, 456, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Steriade, M.; Deschenes, M. The thalamus as a neuronal oscillator. Brain Res. 1984, 320, 1–63. [Google Scholar] [CrossRef]
- Steriade, M.; Llinas, R.R. The functional states of the thalamus and the associated neuronal interplay. Physiol. Rev. 1988, 68, 649–742. [Google Scholar] [PubMed]
- Timofeev, I.; Chauvette, S. Thalamocortical oscillations: Local control of eeg slow waves. Curr. Top. Med. Chem. 2011, 11, 2457–2471. [Google Scholar] [CrossRef] [PubMed]
- Steriade, M. Grouping of brain rhythms in corticothalamic systems. Neuroscience 2006, 137, 1087–1106. [Google Scholar] [CrossRef] [PubMed]
- Steriade, M.; McCormick, D.A.; Sejnowski, T.J. Thalamocortical oscillations in the sleeping and aroused brain. Science 1993, 262, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Crunelli, V.; Cope, D.W.; Hughes, S.W. Thalamic T-type Ca2+ channels and NREM sleep. Cell. Calcium 2006, 40, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Kim, U.; Sanchez-Vives, M.V.; McCormick, D.A. Functional dynamics of gabaergic inhibition in the thalamus. Science 1997, 278, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.M. A wake-up call from the thalamus. Nat. Neurosci. 2001, 4, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Bal, T.; McCormick, D.A. Mechanisms of oscillatory activity in guinea-pig nucleus reticularis thalami in vitro: A mammalian pacemaker. J. Physiol. 1993, 468, 669–691. [Google Scholar] [CrossRef] [PubMed]
- Fuentealba, P.; Steriade, M. The reticular nucleus revisited: Intrinsic and network properties of a thalamic pacemaker. Prog. Neurobiol. 2005, 75, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Luthi, A. Sleep spindles: Where they come from, what they do. Neuroscientist 2014, 20, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Pinault, D.; Deschenes, M. Voltage-dependent 40-Hz oscillations in rat reticular thalamic neurons in vivo. Neuroscience 1992, 51, 245–258. [Google Scholar] [CrossRef]
- McCormick, D.A.; von Krosigk, M. Corticothalamic activation modulates thalamic firing through glutamate “metabotropic” receptors. Proc. Natl. Acad. Sci. USA 1992, 89, 2774–2778. [Google Scholar] [CrossRef] [PubMed]
- Pinault, D. Cellular interactions in the rat somatosensory thalamocortical system during normal and epileptic 5–9 hz oscillations. J. Physiol. 2003, 552, 881–905. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, N.C.; Paradiso, S.; O’Leary, D.S. “Cognitive dysmetria” as an integrative theory of schizophrenia: A dysfunction in cortical-subcortical-cerebellar circuitry? Schizophr. Bull. 1998, 24, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Dorph-Petersen, K.A.; Lewis, D.A. Postmortem structural studies of the thalamus in schizophrenia. Schizophr. Res. 2017, 180, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Sim, K.; Cullen, T.; Ongur, D.; Heckers, S. Testing models of thalamic dysfunction in schizophrenia using neuroimaging. J. Neural Transm. 2006, 113, 907–928. [Google Scholar] [CrossRef] [PubMed]
- Van Haren, N.E.; Bakker, S.C.; Kahn, R.S. Genes and structural brain imaging in schizophrenia. Curr. Opin. Psychiatry 2008, 21, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Adriano, F.; Spoletini, I.; Caltagirone, C.; Spalletta, G. Updated meta-analyses reveal thalamus volume reduction in patients with first-episode and chronic schizophrenia. Schizophr. Res. 2010, 123, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Harrisberger, F.; Buechler, R.; Smieskova, R.; Lenz, C.; Walter, A.; Egloff, L.; Bendfeldt, K.; Simon, A.E.; Wotruba, D.; Theodoridou, A.; et al. Alterations in the hippocampus and thalamus in individuals at high risk for psychosis. NPJ Schizophr. 2016, 2, 16033. [Google Scholar] [CrossRef] [PubMed]
- Anticevic, A.; Cole, M.W.; Repovs, G.; Murray, J.D.; Brumbaugh, M.S.; Winkler, A.M.; Savic, A.; Krystal, J.H.; Pearlson, G.D.; Glahn, D.C. Characterizing thalamo-cortical disturbances in schizophrenia and bipolar illness. Cereb. Cortex 2014, 24, 3116–3130. [Google Scholar] [CrossRef] [PubMed]
- Janssen, J.; Aleman-Gomez, Y.; Reig, S.; Schnack, H.G.; Parellada, M.; Graell, M.; Moreno, C.; Moreno, D.; Mateos-Perez, J.M.; Udias, J.M.; et al. Regional specificity of thalamic volume deficits in male adolescents with early-onset psychosis. Br. J. Psychiatry 2012, 200, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Woodward, N.D.; Heckers, S. Mapping thalamocortical functional connectivity in chronic and early stages of psychotic disorders. Biol. Psychiatry 2016, 79, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.; Chaddock, C.A.; Egerton, A.; Howes, O.D.; Barker, G.; Bonoldi, I.; Fusar-Poli, P.; Murray, R.; McGuire, P. Functional outcome in people at high risk for psychosis predicted by thalamic glutamate levels and prefronto-striatal activation. Schizophr. Bull. 2015, 41, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Fusar-Poli, P.; Stone, J.M.; Broome, M.R.; Valli, I.; Mechelli, A.; McLean, M.A.; Lythgoe, D.J.; O’Gorman, R.L.; Barker, G.J.; McGuire, P.K. Thalamic glutamate levels as a predictor of cortical response during executive functioning in subjects at high risk for psychosis. Arch. Gen. Psychiatry 2011, 68, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Manoach, D.S.; Demanuele, C.; Wamsley, E.J.; Vangel, M.; Montrose, D.M.; Miewald, J.; Kupfer, D.; Buysse, D.; Stickgold, R.; Keshavan, M.S. Sleep spindle deficits in antipsychotic-naive early course schizophrenia and in non-psychotic first-degree relatives. Front. Hum. Neurosci. 2014, 8, 762. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, R.B.; Ulrich, D.; Huguenard, J.R. Gaba(b) and nmda receptors contribute to spindle-like oscillations in rat thalamus in vitro. J. Neurophysiol. 2001, 86, 1365–1375. [Google Scholar] [PubMed]
- Moghaddam, B. Bringing order to the glutamate chaos in schizophrenia. Neuron 2003, 40, 881–884. [Google Scholar] [CrossRef]
- Stephan, K.E.; Baldeweg, T.; Friston, K.J. Synaptic plasticity and dysconnection in schizophrenia. Biol. Psychiatry 2006, 59, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Behar, T.N.; Scott, C.A.; Greene, C.L.; Wen, X.; Smith, S.V.; Maric, D.; Liu, Q.Y.; Colton, C.A.; Barker, J.L. Glutamate acting at nmda receptors stimulates embryonic cortical neuronal migration. J. Neurosci. 1999, 19, 4449–4461. [Google Scholar] [PubMed]
- Floresco, S.B.; Blaha, C.D.; Yang, C.R.; Phillips, A.G. Dopamine d1 and nmda receptors mediate potentiation of basolateral amygdala-evoked firing of nucleus accumbens neurons. J. Neurosci. 2001, 21, 6370–6376. [Google Scholar] [PubMed]
- Lisman, J.E.; Coyle, J.T.; Green, R.W.; Javitt, D.C.; Benes, F.M.; Heckers, S.; Grace, A.A. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008, 31, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, T.L.; Sachdeva, S.; Stahl, S.M. Genetic data supporting the nmda glutamate receptor hypothesis for schizophrenia. Curr. Pharm. Des. 2012, 18, 1580–1592. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kornhuber, H.H.; Schmid-Burgk, W.; Holzmuller, B. Low cerebrospinal fluid glutamate in schizophrenic patients and a new hypothesis on schizophrenia. Neurosci. Lett. 1980, 20, 379–382. [Google Scholar] [CrossRef]
- Kim, J.S.; Kornhuber, H.H.; Brand, U.; Menge, H.G. Effects of chronic amphetamine treatment on the glutamate concentration in cerebrospinal fluid and brain: Implications for a theory of schizophrenia. Neurosci. Lett. 1981, 24, 93–96. [Google Scholar] [CrossRef]
- Moghaddam, B.; Javitt, D. From revolution to evolution: The glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 2012, 37, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Clinton, S.M.; Meador-Woodruff, J.H. Thalamic dysfunction in schizophrenia: Neurochemical, neuropathological, and in vivo imaging abnormalities. Schizophr. Res. 2004, 69, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, R.; Coyle, J.T. Naag, nmda receptor and psychosis. Curr. Med. Chem. 2012, 19, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- Molina, V.; Sanchez, J.; Reig, S.; Sanz, J.; Benito, C.; Santamarta, C.; Pascau, J.; Sarramea, F.; Gispert, J.D.; Misiego, J.M.; et al. N-acetyl-aspartate levels in the dorsolateral prefrontal cortex in the early years of schizophrenia are inversely related to disease duration. Schizophr. Res. 2005, 73, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.J.; Owen, M.J. Genes for schizophrenia? Recent findings and their pathophysiological implications. Lancet 2003, 361, 417–419. [Google Scholar] [CrossRef]
- Kendler, K.S. What psychiatric genetics has taught us about the nature of psychiatric illness and what is left to learn. Mol. Psychiatry 2013, 18, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Richard, E.A.; Khlestova, E.; Nanu, R.; Lisman, J.E. Potential synergistic action of 19 schizophrenia risk genes in the thalamus. Schizophr. Res. 2017, 180, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, N.R.; Gur, R.E.; Braff, D.L. Consortium on the genetics of schizophrenia (cogs) assessment of endophenotypes for schizophrenia: An introduction to this special issue of schizophrenia research. Schizophr. Res. 2015, 163, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Pilowsky, L.S.; Bressan, R.A.; Stone, J.M.; Erlandsson, K.; Mulligan, R.S.; Krystal, J.H.; Ell, P.J. First in vivo evidence of an nmda receptor deficit in medication-free schizophrenic patients. Mol. Psychiatry 2006, 11, 118–119. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.; Bogerts, B.; Sarnyai, Z.; Walter, M.; Gos, T.; Bernstein, H.G.; Myint, A.M. Bridging the gap between the immune and glutamate hypotheses of schizophrenia and major depression: Potential role of glial nmda receptor modulators and impaired blood-brain barrier integrity. World J. Biol. Psychiatry 2012, 13, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Egerton, A.; Stone, J.M.; Chaddock, C.A.; Barker, G.J.; Bonoldi, I.; Howard, R.M.; Merritt, K.; Allen, P.; Howes, O.D.; Murray, R.M.; et al. Relationship between brain glutamate levels and clinical outcome in individuals at ultra high risk of psychosis. Neuropsychopharmacology 2014, 39, 2891–2899. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.M.; Day, F.; Tsagaraki, H.; Valli, I.; McLean, M.A.; Lythgoe, D.J.; O’Gorman, R.L.; Barker, G.J.; McGuire, P.K.; OASIS. Glutamate dysfunction in people with prodromal symptoms of psychosis: Relationship to gray matter volume. Biol. Psychiatry 2009, 66, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Corlett, P.R.; Honey, G.D.; Fletcher, P.C. From prediction error to psychosis: Ketamine as a pharmacological model of delusions. J. Psychopharmacol. 2007, 21, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Lodge, D.; Anis, N.A. Effects of phencyclidine on excitatory amino acid activation of spinal interneurones in the cat. Eur. J. Pharmacol. 1982, 77, 203–204. [Google Scholar] [CrossRef]
- Tricklebank, M.D.; Singh, L.; Oles, R.J.; Preston, C.; Iversen, S.D. The behavioural effects of mk-801: A comparison with antagonists acting non-competitively and competitively at the nmda receptor. Eur. J. Pharmacol. 1989, 167, 127–135. [Google Scholar] [CrossRef]
- Hetem, L.A.; Danion, J.M.; Diemunsch, P.; Brandt, C. Effect of a subanesthetic dose of ketamine on memory and conscious awareness in healthy volunteers. Psychopharmacology 2000, 152, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C.; Zukin, S.R. Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 1991, 148, 1301–1308. [Google Scholar] [PubMed]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B., Jr.; Charney, D.S. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Lahti, A.C.; Koffel, B.; LaPorte, D.; Tamminga, C.A. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology 1995, 13, 9–19. [Google Scholar] [CrossRef]
- Anticevic, A.; Corlett, P.R.; Cole, M.W.; Savic, A.; Gancsos, M.; Tang, Y.; Repovs, G.; Murray, J.D.; Driesen, N.R.; Morgan, P.T.; et al. N-methyl-d-aspartate receptor antagonist effects on prefrontal cortical connectivity better model early than chronic schizophrenia. Biol. Psychiatry 2015, 77, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Driesen, N.R.; McCarthy, G.; Bhagwagar, Z.; Bloch, M.; Calhoun, V.; D’souza, D.C.; Gueorguieva, R.; He, G.; Ramachandran, R.; Suckow, R.F.; et al. Relationship of resting brain hyperconnectivity and schizophrenia-like symptoms produced by the nmda receptor antagonist ketamine in humans. Mol. Psychiatry 2013, 18, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Leung, L.S. The supramammillo-septal-hippocampal pathway mediates sensorimotor gating impairment and hyperlocomotion induced by MK-801 and ketamine in rats. Psychopharmacology 2007, 191, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Ehrlichman, R.S.; Gandal, M.J.; Maxwell, C.R.; Lazarewicz, M.T.; Finkel, L.H.; Contreras, D.; Turetsky, B.I.; Siegel, S.J. N-methyl-d-aspartic acid receptor antagonist-induced frequency oscillations in mice recreate pattern of electrophysiological deficits in schizophrenia. Neuroscience 2009, 158, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Kocsis, B. State-dependent increase of cortical gamma activity during rem sleep after selective blockade of nr2b subunit containing nmda receptors. Sleep 2012, 35, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.; Kim, Y.; Moghaddam, B. Disruption of prefrontal cortex large scale neuronal activity by different classes of psychotomimetic drugs. J. Neurosci. 2012, 32, 3022–3031. [Google Scholar] [CrossRef] [PubMed]
- Daskalakis, Z.J.; Christensen, B.K.; Fitzgerald, P.B.; Chen, R. Dysfunctional neural plasticity in patients with schizophrenia. Arch. Gen. Psychiatry 2008, 65, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.J.; Weinberger, D.R. Schizophrenia genes, gene expression, and neuropathology: On the matter of their convergence. Mol. Psychiatry 2005, 10, 40–68. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.G.; Wang, H.Y.; Cho, D.S.; Talbot, K.; Gur, R.E.; Berrettini, W.H.; Bakshi, K.; Kamins, J.; Borgmann-Winter, K.E.; Siegel, S.J.; et al. Altered neuregulin 1-erbb4 signaling contributes to nmda receptor hypofunction in schizophrenia. Nat. Med. 2006, 12, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Snyder, M.A.; Gao, W.J. Nmda hypofunction as a convergence point for progression and symptoms of schizophrenia. Front. Cell. Neurosci. 2013, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Lane, H.Y.; Liu, Y.C.; Huang, C.L.; Chang, Y.C.; Liau, C.H.; Perng, C.H.; Tsai, G.E. Sarcosine (n-methylglycine) treatment for acute schizophrenia: A randomized, double-blind study. Biol. Psychiatry 2008, 63, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Singh, V. Meta-analysis of the efficacy of adjunctive nmda receptor modulators in chronic schizophrenia. CNS Drugs 2011, 25, 859–885. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T. The gaba-glutamate connection in schizophrenia: Which is the proximate cause? Biochem. Pharmacol. 2004, 68, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Burgos, G.; Cho, R.Y.; Lewis, D.A. Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol. Psychiatry 2015, 77, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Hashimoto, T.; Volk, D.W. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 2005, 6, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Curley, A.A.; Glausier, J.R.; Volk, D.W. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012, 35, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Uhlhaas, P.J.; Roux, F.; Rodriguez, E.; Rotarska-Jagiela, A.; Singer, W. Neural synchrony and the development of cortical networks. Trends Cogn. Sci. 2010, 14, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Minlebaev, M.; Colonnese, M.; Tsintsadze, T.; Sirota, A.; Khazipov, R. Early gamma oscillations synchronize developing thalamus and cortex. Science 2011, 334, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Brucke, C.; Bock, A.; Huebl, J.; Krauss, J.K.; Schonecker, T.; Schneider, G.H.; Brown, P.; Kuhn, A.A. Thalamic gamma oscillations correlate with reaction time in a go/nogo task in patients with essential tremor. Neuroimage 2013, 75, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Joliot, M.; Ribary, U.; Llinas, R. Human oscillatory brain activity near 40 Hz coexists with cognitive temporal binding. Proc. Natl. Acad. Sci. USA 1994, 91, 11748–11751. [Google Scholar] [CrossRef] [PubMed]
- Tallon-Baudry, C.; Bertrand, O. Oscillatory gamma activity in humans and its role in object representation. Trends Cogn. Sci. 1999, 3, 151–162. [Google Scholar] [CrossRef]
- Varela, F.; Lachaux, J.P.; Rodriguez, E.; Martinerie, J. The brainweb: Phase synchronization and large-scale integration. Nat. Rev. Neurosci. 2001, 2, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Hu, L.; Hung, Y.S.; Mouraux, A.; Iannetti, G.D. Gamma-band oscillations in the primary somatosensory cortex—A direct and obligatory correlate of subjective pain intensity. J. Neurosci. 2012, 32, 7429–7438. [Google Scholar] [CrossRef] [PubMed]
- Buzsaki, G.; Draguhn, A. Neuronal oscillations in cortical networks. Science 2004, 304, 1926–1929. [Google Scholar] [CrossRef] [PubMed]
- Buzsaki, G.; Chrobak, J.J. Temporal structure in spatially organized neuronal ensembles: A role for interneuronal networks. Curr. Opin. Neurobiol. 1995, 5, 504–510. [Google Scholar] [CrossRef]
- Engel, A.K.; Roelfsema, P.R.; Fries, P.; Brecht, M.; Singer, W. Role of the temporal domain for response selection and perceptual binding. Cereb. Cortex 1997, 7, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Fries, P. Neuronal gamma-band synchronization as a fundamental process in cortical computation. Annu. Rev. Neurosci. 2009, 32, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.M.; Konig, P.; Engel, A.K.; Singer, W. Oscillatory responses in cat visual cortex exhibit inter-columnar synchronization which reflects global stimulus properties. Nature 1989, 338, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Singer, W. Time as coding space? Curr. Opin. Neurobiol. 1999, 9, 189–194. [Google Scholar] [CrossRef]
- Buzsaki, G.; Wang, X.J. Mechanisms of gamma oscillations. Annu. Rev. Neurosci. 2012, 35, 203–225. [Google Scholar] [CrossRef] [PubMed]
- Roux, F.; Wibral, M.; Singer, W.; Aru, J.; Uhlhaas, P.J. The phase of thalamic alpha activity modulates cortical gamma-band activity: Evidence from resting-state meg recordings. J. Neurosci. 2013, 33, 17827–17835. [Google Scholar] [CrossRef] [PubMed]
- Bartos, M.; Vida, I.; Jonas, P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat. Rev. Neurosci. 2007, 8, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Uhlhaas, P.J.; Singer, W. Neural synchrony in brain disorders: Relevance for cognitive dysfunctions and pathophysiology. Neuron 2006, 52, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Stephan, K.E.; Friston, K.J.; Frith, C.D. Dysconnection in schizophrenia: From abnormal synaptic plasticity to failures of self-monitoring. Schizophr. Bull. 2009, 35, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Uhlhaas, P.J.; Roux, F.; Singer, W. Thalamocortical synchronization and cognition: Implications for schizophrenia? Neuron 2013, 77, 997–999. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.Y.; Konecky, R.O.; Carter, C.S. Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia. Proc. Natl. Acad. Sci. USA 2006, 103, 19878–19883. [Google Scholar] [CrossRef] [PubMed]
- Hirano, Y.; Oribe, N.; Kanba, S.; Onitsuka, T.; Nestor, P.G.; Spencer, K.M. Spontaneous gamma activity in schizophrenia. JAMA Psychiatry 2015, 72, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Leicht, G.; Vauth, S.; Polomac, N.; Andreou, C.; Rauh, J.; Mussmann, M.; Karow, A.; Mulert, C. Eeg-informed fmri reveals a disturbed gamma-band-specific network in subjects at high risk for psychosis. Schizophr. Bull. 2016, 42, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Nizamie, S.H.; Goyal, N.; Tikka, S.K. Evaluation of resting state gamma power as a response marker in schizophrenia. Psychiatry Clin. Neurosci. 2015, 69, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Leicht, G.; Andreou, C.; Polomac, N.; Lanig, C.; Schottle, D.; Lambert, M.; Mulert, C. Reduced auditory evoked gamma band response and cognitive processing deficits in first episode schizophrenia. World J. Biol. Psychiatry 2015. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Nagai, T.; Kirihara, K.; Koike, S.; Suga, M.; Araki, T.; Kobayashi, T.; Kasai, K. Differential alterations of auditory gamma oscillatory responses between pre-onset high-risk individuals and first-episode schizophrenia. Cereb. Cortex 2016, 26, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Basar, E. A review of gamma oscillations in healthy subjects and in cognitive impairment. Int. J. Psychophysiol. 2013, 90, 99–117. [Google Scholar] [CrossRef] [PubMed]
- De la Salle, S.; Choueiry, J.; Shah, D.; Bowers, H.; McIntosh, J.; Ilivitsky, V.; Knott, V. Effects of ketamine on resting-state eeg activity and their relationship to perceptual/dissociative symptoms in healthy humans. Front. Pharmacol. 2016, 7, 348. [Google Scholar] [CrossRef] [PubMed]
- Callicott, J.H.; Bertolino, A.; Mattay, V.S.; Langheim, F.J.; Duyn, J.; Coppola, R.; Goldberg, T.E.; Weinberger, D.R. Physiological dysfunction of the dorsolateral prefrontal cortex in schizophrenia revisited. Cereb. Cortex 2000, 10, 1078–1092. [Google Scholar] [CrossRef] [PubMed]
- Gandal, M.J.; Edgar, J.C.; Klook, K.; Siegel, S.J. Gamma synchrony: Towards a translational biomarker for the treatment-resistant symptoms of schizophrenia. Neuropharmacology 2012, 62, 1504–1518. [Google Scholar] [CrossRef] [PubMed]
- Pinault, D. N-methyl-d-aspartate receptor antagonists amplify network baseline gamma frequency (30–80 Hz) oscillations: Noise and signal. AIMS Neurosci. 2014, 1, 169–182. [Google Scholar] [CrossRef]
- Molina, L.A.; Skelin, I.; Gruber, A.J. Acute nmda receptor antagonism disrupts synchronization of action potential firing in rat prefrontal cortex. PLoS ONE 2014, 9, e85842. [Google Scholar] [CrossRef] [PubMed]
- Homayoun, H.; Moghaddam, B. Nmda receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J. Neurosci. 2007, 27, 11496–11500. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Kulikova, S.P.; Shibato, J.; Rakwal, R.; Satoh, H.; Pinault, D.; Masuo, Y. DNA microarray unravels rapid changes in transcriptome of mk-801 treated rat brain. World J. Biol. Chem. 2015, 6, 389–408. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Quintero, S.V.; Datta, A.; Amaya, R.; Elwassif, M.; Bikson, M.; Tarbell, J.M. Dbs-relevant electric fields increase hydraulic conductivity of in vitro endothelial monolayers. J. Neural Eng. 2010, 7, 16005. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Watanabe, T.; Takahashi, A.; Saito, N.; Hirato, M.; Sasaki, T. Transcranial electrical stimulation through screw electrodes for intraoperative monitoring of motor evoked potentials. Technical note. J. Neurosurg. 2004, 100, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Fenoy, A.J.; Goetz, L.; Chabardes, S.; Xia, Y. Deep brain stimulation: Are astrocytes a key driver behind the scene? CNS Neurosci. Ther. 2014, 20, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, J.C.; Seijo, F.J.; Alvarez Vega, M.A.; Fernandez Gonzalez, F.; Lozano Aragoneses, B.; Blazquez, M. Therapeutic extradural cortical stimulation for parkinson’s disease: Report of six cases and review of the literature. Clin. Neurol. Neurosurg. 2009, 111, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.J. Mechanisms for electrical stimulation of excitable tissue. Crit. Rev. Biomed. Eng. 1994, 22, 253–305. [Google Scholar] [PubMed]
- Kanai, R.; Paulus, W.; Walsh, V. Transcranial alternating current stimulation (TACS) modulates cortical excitability as assessed by TMS-induced phosphene thresholds. Clin. Neurophysiol. 2010, 121, 1551–1554. [Google Scholar] [CrossRef] [PubMed]
- Struber, D.; Rach, S.; Neuling, T.; Herrmann, C.S. On the possible role of stimulation duration for after-effects of transcranial alternating current stimulation. Front. Cell. Neurosci. 2015, 9, 311. [Google Scholar] [CrossRef] [PubMed]
- Feurra, M.; Pasqualetti, P.; Bianco, G.; Santarnecchi, E.; Rossi, A.; Rossi, S. State-dependent effects of transcranial oscillatory currents on the motor system: What you think matters. J. Neurosci. 2013, 33, 17483–17489. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Calderin, Y.; Anne Weinrich, C.; Schmidt-Samoa, C.; Poland, E.; Dechent, P.; Bahr, M.; Wilke, M. Transcranial alternating current stimulation affects the bold signal in a frequency and task-dependent manner. Hum. Brain Mapp. 2016, 37, 94–121. [Google Scholar] [CrossRef] [PubMed]
- Tauc, L.; Hughes, G.M. Modes of initiation and propagation of spikes in the branching axons of molluscan central neurons. J. Gen. Physiol. 1963, 46, 533–549. [Google Scholar] [CrossRef] [PubMed]
- De Raedt, R.; Vanderhasselt, M.A.; Baeken, C. Neurostimulation as an intervention for treatment resistant depression: From research on mechanisms towards targeted neurocognitive strategies. Clin. Psychol. Rev. 2015, 41, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Fertonani, A.; Miniussi, C. Transcranial electrical stimulation: What we know and do not know about mechanisms. Neuroscientist 2016. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, F. Experiments and models of cortical oscillations as a target for noninvasive brain stimulation. Prog. Brain Res. 2015, 222, 41–73. [Google Scholar] [PubMed]
- Herrmann, C.S.; Rach, S.; Neuling, T.; Struber, D. Transcranial alternating current stimulation: A review of the underlying mechanisms and modulation of cognitive processes. Front. Hum. Neurosci. 2013, 7, 279. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.T.; Rogasch, N.C.; Fitzgerald, P.B.; Hoy, K.E. Tms-eeg: A window into the neurophysiological effects of transcranial electrical stimulation in non-motor brain regions. Neurosci. Biobehav. Rev. 2016, 64, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.P.; Rahman, A.; Lafon, B.; Kronberg, G.; Ling, D.; Parra, L.C.; Bikson, M. Animal models of transcranial direct current stimulation: Methods and mechanisms. Clin. Neurophysiol. 2016, 127, 3425–3454. [Google Scholar] [CrossRef] [PubMed]
- Knotkova, H.; Nitsche, M.A.; Cruciani, R.A. Putative physiological mechanisms underlying tDCS analgesic effects. Front. Hum. Neurosci. 2013, 7, 628. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.M.; Thomson, R.H.; Rosenfeld, J.V.; Fitzgerald, P.B. Brain neuromodulation techniques: A review. Neuroscientist 2016, 22, 406–421. [Google Scholar] [CrossRef] [PubMed]
- Lisanby, S.H.; Belmaker, R.H. Animal models of the mechanisms of action of repetitive transcranial magnetic stimulation (rTMS): Comparisons with electroconvulsive shock (ECS). Depress. Anxiety 2000, 12, 178–187. [Google Scholar] [CrossRef]
- Medeiros, L.F.; de Souza, I.C.; Vidor, L.P.; de Souza, A.; Deitos, A.; Volz, M.S.; Fregni, F.; Caumo, W.; Torres, I.L. Neurobiological effects of transcranial direct current stimulation: A review. Front. Psychiatry 2012, 3, 110. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, M.A.; Cohen, L.G.; Wassermann, E.M.; Priori, A.; Lang, N.; Antal, A.; Paulus, W.; Hummel, F.; Boggio, P.S.; Fregni, F.; et al. Transcranial direct current stimulation: State of the art 2008. Brain Stimul. 2008, 1, 206–223. [Google Scholar] [CrossRef] [PubMed]
- Reato, D.; Rahman, A.; Bikson, M.; Parra, L.C. Effects of weak transcranial alternating current stimulation on brain activity—A review of known mechanisms from animal studies. Front. Hum. Neurosci. 2013, 7, 687. [Google Scholar] [CrossRef] [PubMed]
- Terao, Y.; Ugawa, Y. Basic mechanisms of tms. J. Clin. Neurophysiol. 2002, 19, 322–343. [Google Scholar] [CrossRef] [PubMed]
- Zaghi, S.; Acar, M.; Hultgren, B.; Boggio, P.S.; Fregni, F. Noninvasive brain stimulation with low-intensity electrical currents: Putative mechanisms of action for direct and alternating current stimulation. Neuroscientist 2010, 16, 285–307. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, K.L.; Bhadra, N. Nerve conduction block utilising high-frequency alternating current. Med. Biol. Eng. Comput. 2004, 42, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, B.; Reis, J.; Martinowich, K.; Schambra, H.M.; Ji, Y.; Cohen, L.G.; Lu, B. Direct current stimulation promotes bdnf-dependent synaptic plasticity: Potential implications for motor learning. Neuron 2010, 66, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Monte-Silva, K.; Kuo, M.F.; Hessenthaler, S.; Fresnoza, S.; Liebetanz, D.; Paulus, W.; Nitsche, M.A. Induction of late LTP-like plasticity in the human motor cortex by repeated non-invasive brain stimulation. Brain Stimul. 2013, 6, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Suppa, A.; Huang, Y.Z.; Funke, K.; Ridding, M.C.; Cheeran, B.; Di Lazzaro, V.; Ziemann, U.; Rothwell, J.C. Ten years of theta burst stimulation in humans: Established knowledge, unknowns and prospects. Brain Stimul. 2016, 9, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.; Sellers, K.K.; Frohlich, F. Transcranial alternating current stimulation modulates large-scale cortical network activity by network resonance. J. Neurosci. 2013, 33, 11262–11275. [Google Scholar] [CrossRef] [PubMed]
- D’Atri, A.; De Simoni, E.; Gorgoni, M.; Ferrara, M.; Ferlazzo, F.; Rossini, P.M.; De Gennaro, L. Electrical stimulation of the frontal cortex enhances slow-frequency eeg activity and sleepiness. Neuroscience 2016. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, F. Endogenous and exogenous electric fields as modifiers of brain activity: Rational design of noninvasive brain stimulation with transcranial alternating current stimulation. Dialogues Clin. Neurosci. 2014, 16, 93–102. [Google Scholar] [PubMed]
- Helfrich, R.F.; Schneider, T.R.; Rach, S.; Trautmann-Lengsfeld, S.A.; Engel, A.K.; Herrmann, C.S. Entrainment of brain oscillations by transcranial alternating current stimulation. Curr. Biol. 2014, 24, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Marshall, L.; Helgadottir, H.; Molle, M.; Born, J. Boosting slow oscillations during sleep potentiates memory. Nature 2006, 444, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Rosanova, M.; Casali, A.; Bellina, V.; Resta, F.; Mariotti, M.; Massimini, M. Natural frequencies of human corticothalamic circuits. J. Neurosci. 2009, 29, 7679–7685. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.L.; Iyengar, A.K.; Foulser, A.A.; Boyle, M.R.; Frohlich, F. Endogenous cortical oscillations constrain neuromodulation by weak electric fields. Brain Stimul. 2014, 7, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Vossen, A.; Gross, J.; Thut, G. Alpha power increase after transcranial alternating current stimulation at alpha frequency (alpha-tACS) reflects plastic changes rather than entrainment. Brain Stimul. 2015, 8, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Ozen, S.; Sirota, A.; Belluscio, M.A.; Anastassiou, C.A.; Stark, E.; Koch, C.; Buzsaki, G. Transcranial electric stimulation entrains cortical neuronal populations in rats. J. Neurosci. 2010, 30, 11476–11485. [Google Scholar] [CrossRef] [PubMed]
- Moliadze, V.; Zhao, Y.; Eysel, U.; Funke, K. Effect of transcranial magnetic stimulation on single-unit activity in the cat primary visual cortex. J. Physiol. 2003, 553, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.C.; Palmer, L.M.; Nyffeler, T.; Muri, R.M.; Larkum, M.E. Transcranial magnetic stimulation (TMS) inhibits cortical dendrites. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Monai, H.; Ohkura, M.; Tanaka, M.; Oe, Y.; Konno, A.; Hirai, H.; Mikoshiba, K.; Itohara, S.; Nakai, J.; Iwai, Y.; et al. Calcium imaging reveals glial involvement in transcranial direct current stimulation-induced plasticity in mouse brain. Nat. Commun. 2016, 7, 11100. [Google Scholar] [CrossRef] [PubMed]
- Reato, D.; Bikson, M.; Parra, L.C. Lasting modulation of in vitro oscillatory activity with weak direct current stimulation. J. Neurophysiol. 2015, 113, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- Tehovnik, E.J. Electrical stimulation of neural tissue to evoke behavioral responses. J. Neurosci. Methods 1996, 65, 1–17. [Google Scholar] [CrossRef]
- Bishop, G.H.; O’Leary, J.L. The effects of polarizing currents on cell potentials and their significance in the interpretation of central nervous system activity. Electroencephalogr. Clin. Neurophysiol. 1950, 2, 401–416. [Google Scholar] [CrossRef]
- Francis, J.T.; Gluckman, B.J.; Schiff, S.J. Sensitivity of neurons to weak electric fields. J. Neurosci. 2003, 23, 7255–7261. [Google Scholar] [PubMed]
- Jefferys, J.G. Nonsynaptic modulation of neuronal activity in the brain: Electric currents and extracellular ions. Physiol. Rev. 1995, 75, 689–723. [Google Scholar] [PubMed]
- McIntyre, C.C.; Grill, W.M. Excitation of central nervous system neurons by nonuniform electric fields. Biophys J. 1999, 76, 878–888. [Google Scholar] [CrossRef]
- Terzuolo, C.A.; Bullock, T.H. Measurement of imposed voltage gradient adequate to modulate neuronal firing. Proc. Natl. Acad. Sci. USA 1956, 42, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Nowak, L.G.; Bullier, J. Axons, but not cell bodies, are activated by electrical stimulation in cortical gray matter. I. Evidence from chronaxie measurements. Exp. Brain Res. 1998, 118, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Nowak, L.G.; Bullier, J. Axons, but not cell bodies, are activated by electrical stimulation in cortical gray matter. Ii. Evidence from selective inactivation of cell bodies and axon initial segments. Exp. Brain Res. 1998, 118, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Rapp, M.; Yarom, Y.; Segev, I. Modeling back propagating action potential in weakly excitable dendrites of neocortical pyramidal cells. Proc. Natl. Acad. Sci. USA 1996, 93, 11985–11990. [Google Scholar] [CrossRef] [PubMed]
- Bahner, F.; Weiss, E.K.; Birke, G.; Maier, N.; Schmitz, D.; Rudolph, U.; Frotscher, M.; Traub, R.D.; Both, M.; Draguhn, A. Cellular correlate of assembly formation in oscillating hippocampal networks in vitro. Proc. Natl. Acad. Sci. USA 2011, 108, E607–E616. [Google Scholar] [CrossRef] [PubMed]
- Dugladze, T.; Schmitz, D.; Whittington, M.A.; Vida, I.; Gloveli, T. Segregation of axonal and somatic activity during fast network oscillations. Science 2012, 336, 1458–1461. [Google Scholar] [CrossRef] [PubMed]
- Pinault, D. Backpropagation of action potentials generated at ectopic axonal loci: Hypothesis that axon terminals integrate local environmental signals. Brain Res. Brain Res. Rev. 1995, 21, 42–92. [Google Scholar] [CrossRef]
- Sasaki, T. The axon as a unique computational unit in neurons. Neurosci. Res. 2013, 75, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, M.E.; Best, T.K.; Mensh, B.D.; Kath, W.L.; Spruston, N. Slow integration leads to persistent action potential firing in distal axons of coupled interneurons. Nat. Neurosci. 2011, 14, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Markin, V.S. Electric interaction of parallel non-myelinated nerve fibers. Iv. Role of anatomic non-uniformities of the nerve trunks. Biofizika 1973, 18, 512–518. [Google Scholar] [PubMed]
- Tranchina, D.; Nicholson, C. A model for the polarization of neurons by extrinsically applied electric fields. Biophys. J. 1986, 50, 1139–1156. [Google Scholar] [CrossRef]
- Bucher, D.; Thirumalai, V.; Marder, E. Axonal dopamine receptors activate peripheral spike initiation in a stomatogastric motor neuron. J. Neurosci. 2003, 23, 6866–6875. [Google Scholar] [PubMed]
- Semyanov, A.; Kullmann, D.M. Kainate receptor-dependent axonal depolarization and action potential initiation in interneurons. Nat. Neurosci. 2001, 4, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Fields, R.D. Oligodendrocytes changing the rules: Action potentials in glia and oligodendrocytes controlling action potentials. Neuroscientist 2008, 14, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Dal Maschio, M.; Beltramo, R.; Haydon, P.G.; Benfenati, F.; Fellin, T. Integrated brain circuits: Neuron-astrocyte interaction in sleep-related rhythmogenesis. ScientificWorldJournal 2010, 10, 1634–1645. [Google Scholar] [CrossRef] [PubMed]
- Molnar, G.; Olah, S.; Komlosi, G.; Fule, M.; Szabadics, J.; Varga, C.; Barzo, P.; Tamas, G. Complex events initiated by individual spikes in the human cerebral cortex. PLoS Biol. 2008, 6, e222. [Google Scholar] [CrossRef] [PubMed]
- Pinault, D.; Pumain, R. Antidromic firing occurs spontaneously on thalamic relay neurons: Triggering of somatic intrinsic burst discharges by ectopic action potentials. Neuroscience 1989, 31, 625–637. [Google Scholar] [CrossRef]
- Radman, T.; Su, Y.; An, J.H.; Parra, L.C.; Bikson, M. Spike timing amplifies the effect of electric fields on neurons: Implications for endogenous field effects. J. Neurosci. 2007, 27, 3030–3036. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.V.; Zukin, R.S. Electrical coupling and neuronal synchronization in the mammalian brain. Neuron 2004, 41, 495–511. [Google Scholar] [CrossRef]
- Schmitz, D.; Schuchmann, S.; Fisahn, A.; Draguhn, A.; Buhl, E.H.; Petrasch-Parwez, E.; Dermietzel, R.; Heinemann, U.; Traub, R.D. Axo-axonal coupling. A novel mechanism for ultrafast neuronal communication. Neuron 2001, 31, 831–840. [Google Scholar] [CrossRef]
- Cuntz, H.; Haag, J.; Forstner, F.; Segev, I.; Borst, A. Robust coding of flow-field parameters by axo-axonal gap junctions between fly visual interneurons. Proc. Natl. Acad. Sci. USA 2007, 104, 10229–10233. [Google Scholar] [CrossRef] [PubMed]
- Draguhn, A.; Traub, R.D.; Schmitz, D.; Jefferys, J.G. Electrical coupling underlies high-frequency oscillations in the hippocampus in vitro. Nature 1998, 394, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.J.; Rinzel, J. Self-organized synchronous oscillations in a network of excitable cells coupled by gap junctions. Network 2000, 11, 299–320. [Google Scholar] [CrossRef] [PubMed]
- Maex, R.; De Schutter, E. Mechanism of spontaneous and self-sustained oscillations in networks connected through axo-axonal gap junctions. Eur. J. Neurosci. 2007, 25, 3347–3358. [Google Scholar] [CrossRef] [PubMed]
- Traub, R.D.; Bibbig, A. A model of high-frequency ripples in the hippocampus based on synaptic coupling plus axon-axon gap junctions between pyramidal neurons. J. Neurosci. 2000, 20, 2086–2093. [Google Scholar] [PubMed]
- Traub, R.D.; Schmitz, D.; Jefferys, J.G.; Draguhn, A. High-frequency population oscillations are predicted to occur in hippocampal pyramidal neuronal networks interconnected by axoaxonal gap junctions. Neuroscience 1999, 92, 407–426. [Google Scholar] [CrossRef]
- Debanne, D. Information processing in the axon. Nat. Rev. Neurosci. 2004, 5, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Waxman, S.G. Integrative properties and design principles of axons. Int. Rev. Neurobiol. 1975, 18, 1–40. [Google Scholar] [PubMed]
- Fukuda, T.; Kosaka, T. Gap junctions linking the dendritic network of gabaergic interneurons in the hippocampus. J. Neurosci. 2000, 20, 1519–1528. [Google Scholar] [PubMed]
- Arvanitaki, A. Effects evoked in an axon by the activity of a contiguous one. J. Neurophysiol. 1942, 5, 89–108. [Google Scholar]
- Katz, B.; Schmitt, O.H. Electric interaction between two adjacent nerve fibres. J. Physiol. 1940, 97, 471–488. [Google Scholar] [CrossRef] [PubMed]
- Anastassiou, C.A.; Perin, R.; Markram, H.; Koch, C. Ephaptic coupling of cortical neurons. Nat. Neurosci. 2011, 14, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.Y.; Nicholson, C. Modulation by applied electric fields of purkinje and stellate cell activity in the isolated turtle cerebellum. J. Physiol. 1986, 371, 89–114. [Google Scholar] [CrossRef] [PubMed]
- Jefferys, J.G. Gap junctions and diseases of the nervous system. Trends Neurosci. 1995, 18, 520–521. [Google Scholar] [CrossRef]
- Womelsdorf, T.; Schoffelen, J.M.; Oostenveld, R.; Singer, W.; Desimone, R.; Engel, A.K.; Fries, P. Modulation of neuronal interactions through neuronal synchronization. Science 2007, 316, 1609–1612. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.Y.; Hounsgaard, J.; Nicholson, C. Effects of electric fields on transmembrane potential and excitability of turtle cerebellar purkinje cells in vitro. J. Physiol. 1988, 402, 751–771. [Google Scholar] [CrossRef] [PubMed]
- Deans, J.K.; Powell, A.D.; Jefferys, J.G. Sensitivity of coherent oscillations in rat hippocampus to ac electric fields. J. Physiol. 2007, 583, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Bishop, P.O. Synaptic transmission; an analysis of the electrical activity of the lateral geniculate nucleus in the cat after optic nerve stimulation. Proc. R. Soc. Lond. B Biol. Sci. 1953, 141, 362–392. [Google Scholar] [CrossRef] [PubMed]
- Carras, P.L.; Coleman, P.A.; Miller, R.F. Site of action potential initiation in amphibian retinal ganglion cells. J. Neurophysiol. 1992, 67, 292–304. [Google Scholar] [PubMed]
- Clark, B.D.; Goldberg, E.M.; Rudy, B. Electrogenic tuning of the axon initial segment. Neuroscientist 2009, 15, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.; Ottoson, D. The site of impulse initiation in a nerve cell of a crustacean stretch receptor. J. Physiol. 1958, 143, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Stuart, G.; Hausser, M. Initiation and spread of sodium action potentials in cerebellar purkinje cells. Neuron 1994, 13, 703–712. [Google Scholar] [CrossRef]
- Tauc, L. Site of origin and propagation in spike in the giant neuron of aplysia. J. Gen. Physiol. 1962, 45, 1077–1097. [Google Scholar] [CrossRef] [PubMed]
- Augustine, G.J.; Charlton, M.P.; Smith, S.J. Calcium entry and transmitter release at voltage-clamped nerve terminals of squid. J. Physiol. 1985, 367, 163–181. [Google Scholar] [CrossRef] [PubMed]
- Stuart, G.J.; Sakmann, B. Active propagation of somatic action potentials into neocortical pyramidal cell dendrites. Nature 1994, 367, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Alle, H.; Geiger, J.R. Combined analog and action potential coding in hippocampal mossy fibers. Science 2006, 311, 1290–1293. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Hasenstaub, A.; Duque, A.; Yu, Y.; McCormick, D.A. Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential. Nature 2006, 441, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Hestrin, S.; Armstrong, W.E. Morphology and physiology of cortical neurons in layer i. J. Neurosci. 1996, 16, 5290–5300. [Google Scholar] [PubMed]
- Ina, A.; Sugiyama, M.; Konno, J.; Yoshida, S.; Ohmomo, H.; Nogami, H.; Shutoh, F.; Hisano, S. Cajal-retzius cells and subplate neurons differentially express vesicular glutamate transporters 1 and 2 during development of mouse cortex. Eur. J. Neurosci. 2007, 26, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, F.; McCormick, D.A. Endogenous electric fields may guide neocortical network activity. Neuron 2010, 67, 129–143. [Google Scholar] [CrossRef] [PubMed]
- deng, B.; Wang, L.; Wang, J.; Wei, X.L.; Yu, H.T. Endogenous fields enhanced stochastic resonance in a randomly coupled neuronal network. Chaos Solitons Fractals 2014, 68, 30–39. [Google Scholar] [CrossRef]
- Helias, M.; Deger, M.; Diesmann, M.; Rotter, S. Equilibrium and response properties of the integrate-and-fire neuron in discrete time. Front. Comput. Neurosci. 2010, 3, 29. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Sherman, S.M. Detectability of excitatory versus inhibitory drive in an integrate-and-fire-or-burst thalamocortical relay neuron model. J. Neurosci. 2002, 22, 10242–10250. [Google Scholar] [PubMed]
- Destexhe, A.; Sejnowski, T.J. The initiation of bursts in thalamic neurons and the cortical control of thalamic sensitivity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2002, 357, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Huguenard, J.R.; Prince, D.A. A novel T-type current underlies prolonged Ca(2+)-dependent burst firing in gabaergic neurons of rat thalamic reticular nucleus. J. Neurosci. 1992, 12, 3804–3817. [Google Scholar] [PubMed]
- Destexhe, A.; Contreras, D.; Steriade, M.; Sejnowski, T.J.; Huguenard, J.R. In vivo, in vitro, and computational analysis of dendritic calcium currents in thalamic reticular neurons. J. Neurosci. 1996, 16, 169–185. [Google Scholar] [PubMed]
- Landisman, C.E.; Connors, B.W. Long-term modulation of electrical synapses in the mammalian thalamus. Science 2005, 310, 1809–1813. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Patrick, S.L.; Richardson, K.A.; Connors, B.W. Two functionally distinct networks of gap junction-coupled inhibitory neurons in the thalamic reticular nucleus. J. Neurosci. 2014, 34, 13170–13182. [Google Scholar] [CrossRef] [PubMed]
- Cudeiro, J.; Sillito, A.M. Looking back: Corticothalamic feedback and early visual processing. Trends Neurosci. 2006, 29, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Poo, M.M.; Dan, Y. Burst spiking of a single cortical neuron modifies global brain state. Science 2009, 324, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Brecht, M.; Schneider, M.; Sakmann, B.; Margrie, T.W. Whisker movements evoked by stimulation of single pyramidal cells in rat motor cortex. Nature 2004, 427, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Haken, H. Synergetic Computers and Cognition, 2nd ed.; Springer: Berlin, Germany, 2004. [Google Scholar]
- Kelso, J.A.S. Dynamic Patterns: The Self-Organization of Brain and Behavior, 1995th ed.; MIT Press: Cambridge, MA, USA, 1995. [Google Scholar]
- Wach, C.; Krause, V.; Moliadze, V.; Paulus, W.; Schnitzler, A.; Pollok, B. The effect of 10 hz transcranial alternating current stimulation (TACS) on corticomuscular coherence. Front. Hum. Neurosci. 2013, 7, 511. [Google Scholar] [CrossRef] [PubMed]
- Berenyi, A.; Belluscio, M.; Mao, D.; Buzsaki, G. Closed-loop control of epilepsy by transcranial electrical stimulation. Science 2012, 337, 735–737. [Google Scholar] [CrossRef] [PubMed]
- Elgamal, S.; McKinnon, M.C.; Ramakrishnan, K.; Joffe, R.T.; MacQueen, G. Successful computer-assisted cognitive remediation therapy in patients with unipolar depression: A proof of principle study. Psychol. Med. 2007, 37, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Mueser, K.T.; Penn, D.L.; Addington, J.; Brunette, M.F.; Gingerich, S.; Glynn, S.M.; Lynde, D.W.; Gottlieb, J.D.; Meyer-Kalos, P.; McGurk, S.R.; et al. The navigate program for first-episode psychosis: Rationale, overview, and description of psychosocial components. Psychiatr. Serv. 2015, 66, 680–690. [Google Scholar] [CrossRef] [PubMed]
- Wykes, T.; Reeder, C.; Landau, S.; Everitt, B.; Knapp, M.; Patel, A.; Romeo, R. Cognitive remediation therapy in schizophrenia: Randomised controlled trial. Br. J. Psychiatry 2007, 190, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Van der Valk, R.; van de Waerdt, S.; Meijer, C.J.; van den Hout, I.; de Haan, L. Feasibility of mindfulness-based therapy in patients recovering from a first psychotic episode: A pilot study. Early Interv. Psychiatry 2013, 7, 64–70. [Google Scholar] [CrossRef] [PubMed]
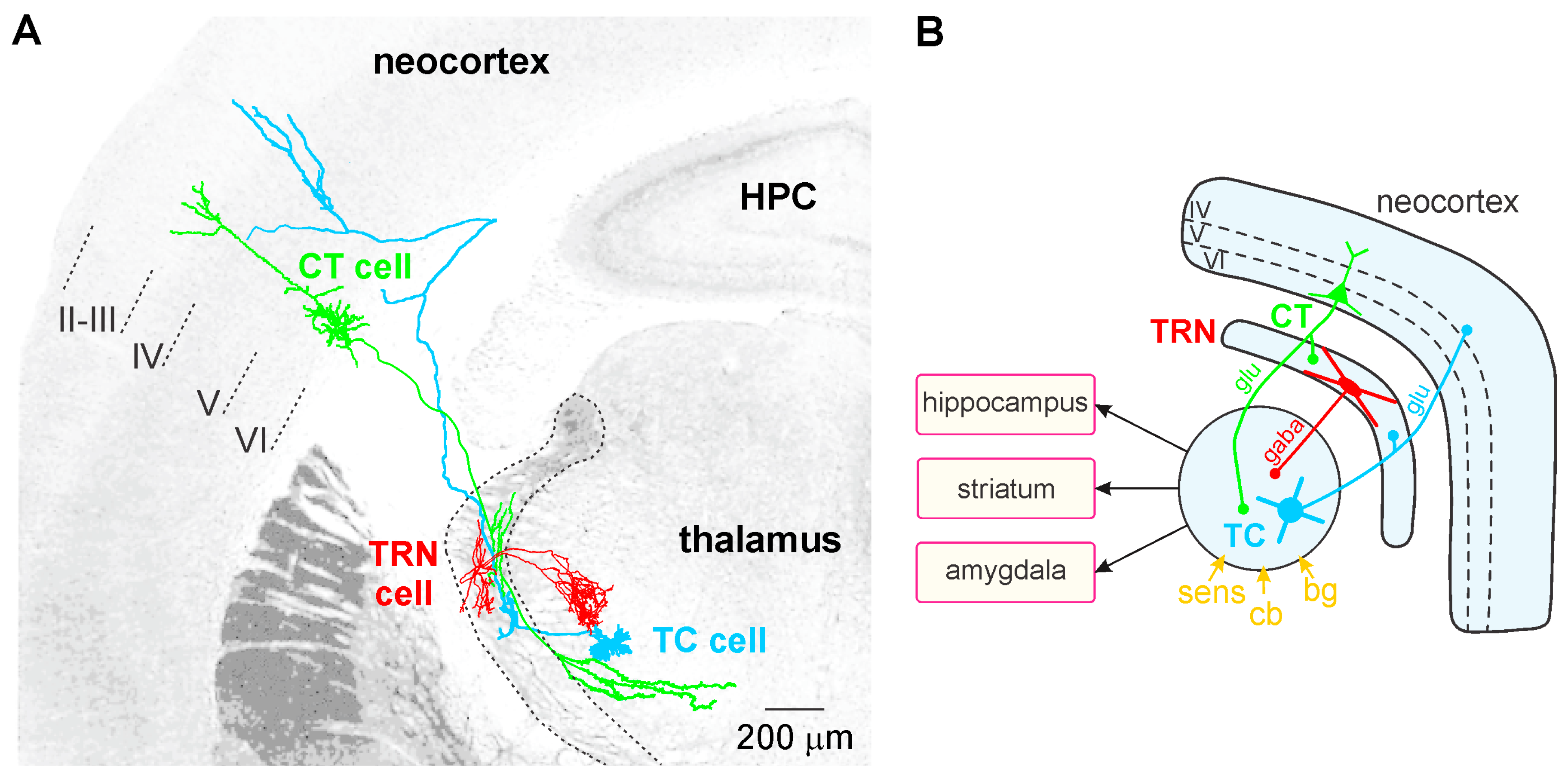
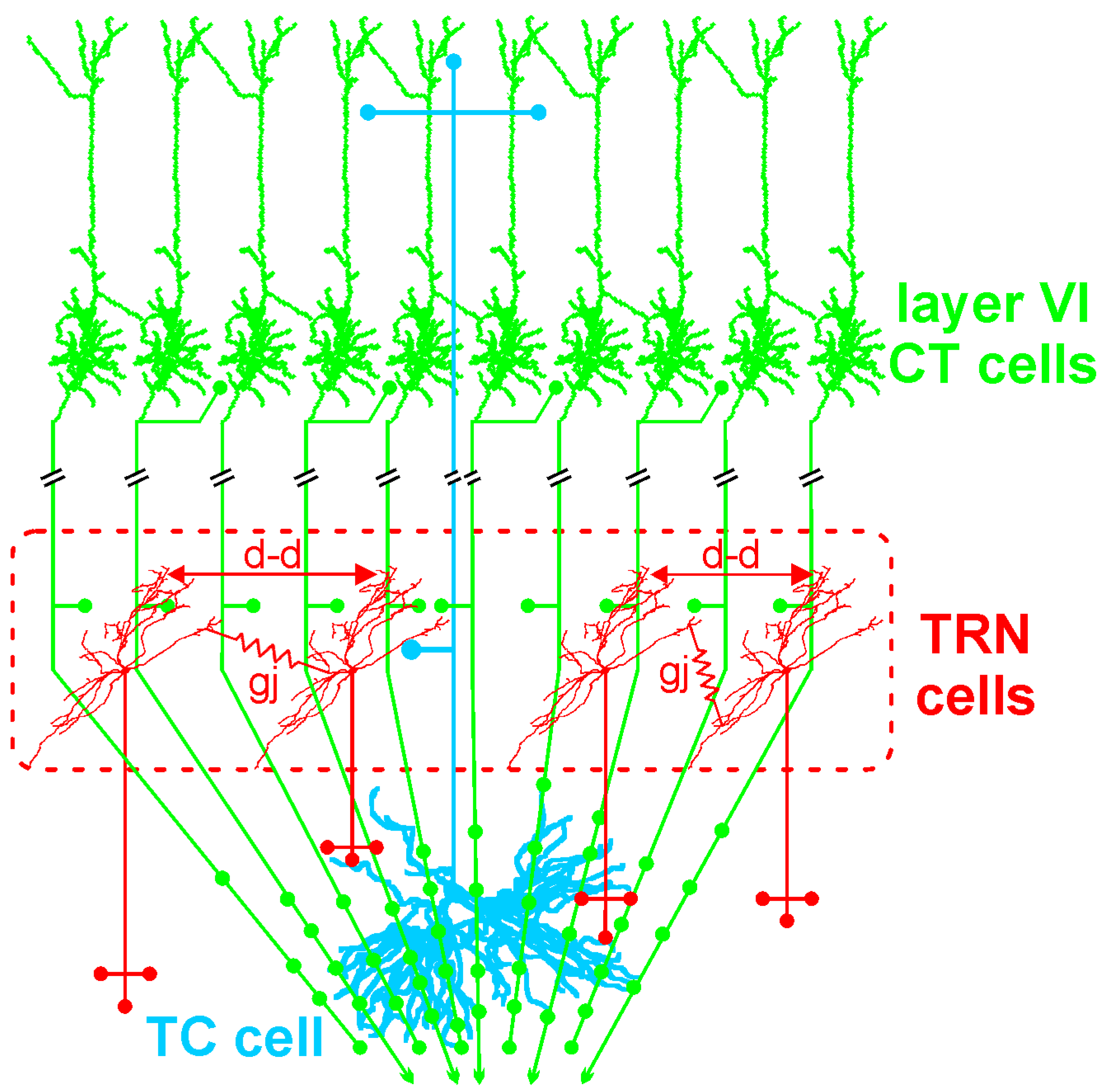
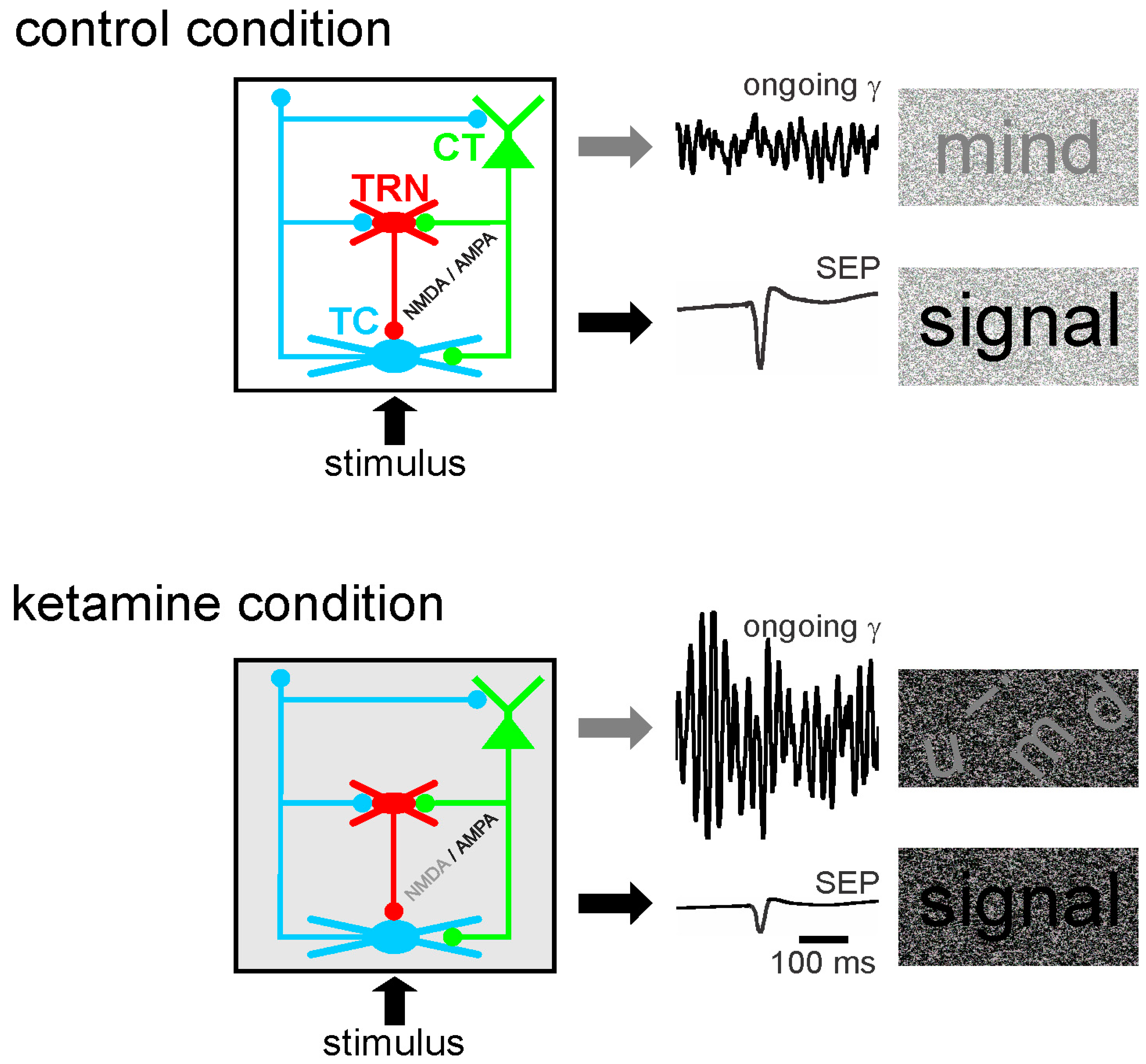
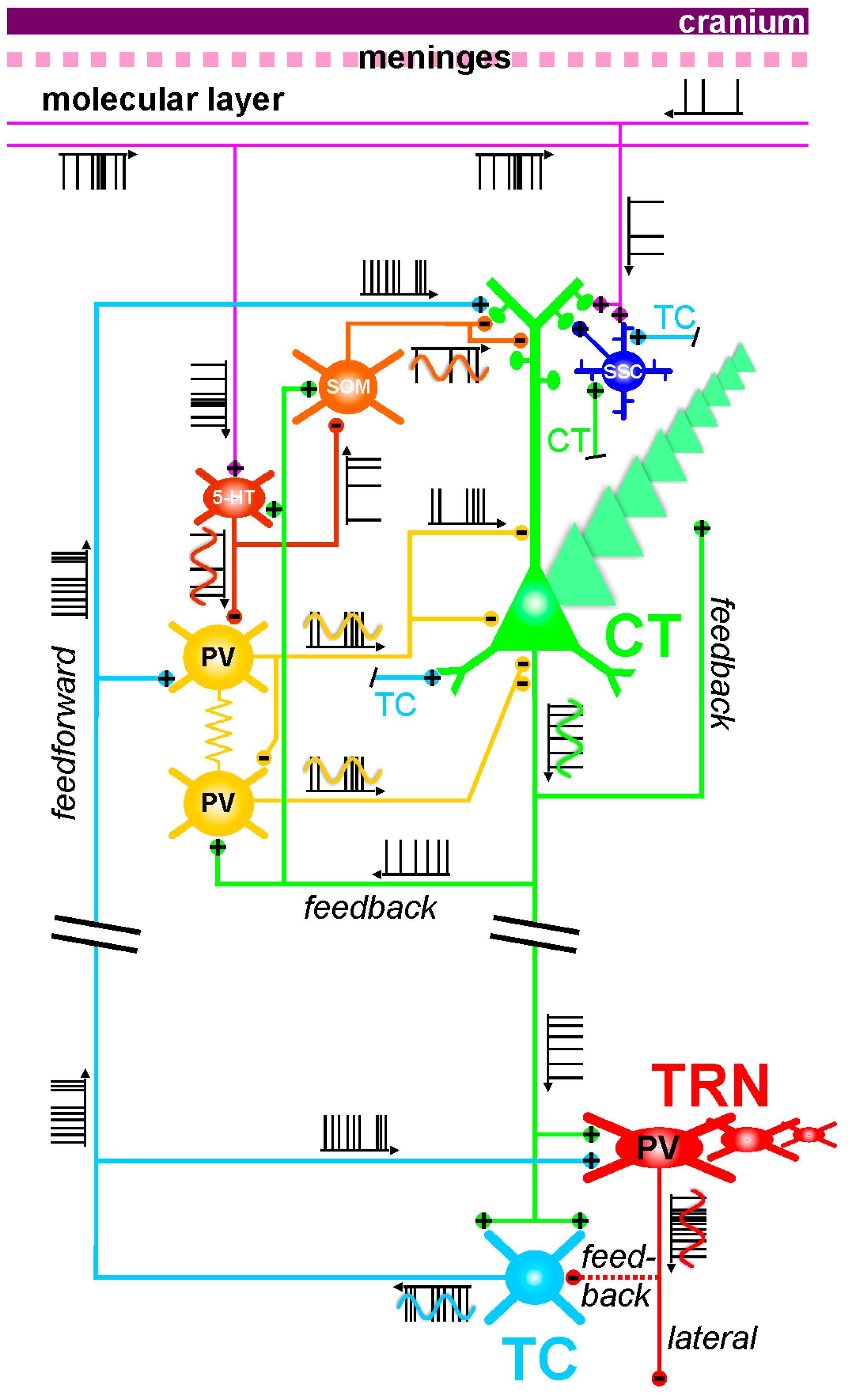
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinault, D. A Neurophysiological Perspective on a Preventive Treatment against Schizophrenia Using Transcranial Electric Stimulation of the Corticothalamic Pathway. Brain Sci. 2017, 7, 34. https://doi.org/10.3390/brainsci7040034
Pinault D. A Neurophysiological Perspective on a Preventive Treatment against Schizophrenia Using Transcranial Electric Stimulation of the Corticothalamic Pathway. Brain Sciences. 2017; 7(4):34. https://doi.org/10.3390/brainsci7040034
Chicago/Turabian StylePinault, Didier. 2017. "A Neurophysiological Perspective on a Preventive Treatment against Schizophrenia Using Transcranial Electric Stimulation of the Corticothalamic Pathway" Brain Sciences 7, no. 4: 34. https://doi.org/10.3390/brainsci7040034
APA StylePinault, D. (2017). A Neurophysiological Perspective on a Preventive Treatment against Schizophrenia Using Transcranial Electric Stimulation of the Corticothalamic Pathway. Brain Sciences, 7(4), 34. https://doi.org/10.3390/brainsci7040034