
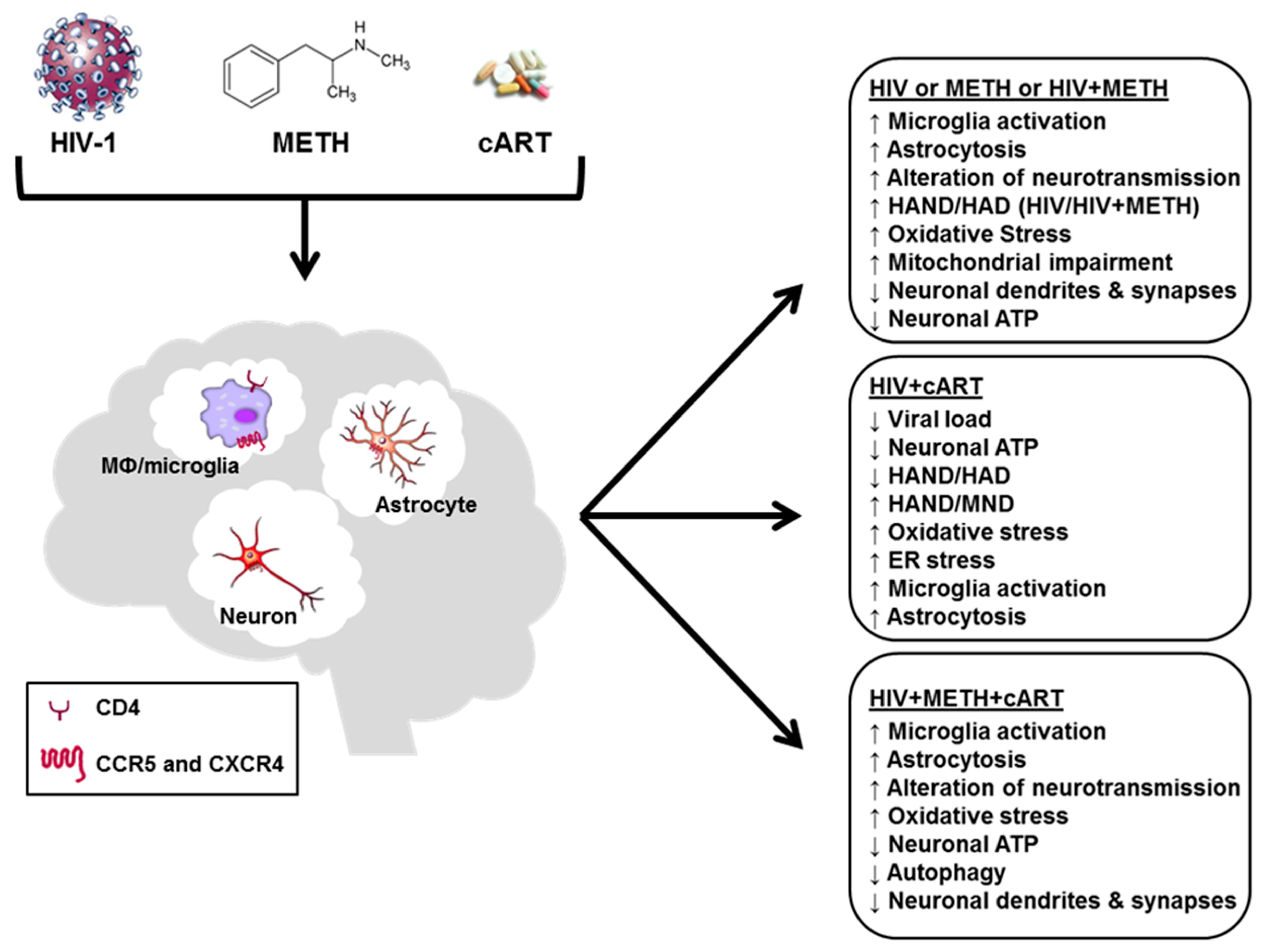
Neuronal Stress and Injury Caused by HIV-1, cART and Drug Abuse: Converging Contributions to HAND

{kind=link}
Abstract
1. Introduction
2. HIV Infection, Neurotoxicity and HAND
3. Neurotoxicity of Antiretroviral Drugs
4. Neurotoxicity of METH
5. Neuronal Injury by HIV + cART + METH
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fauci, A.S. The aids epidemic—Considerations for the 21st century. N. Engl. J. Med. 1999, 341, 1046–1050. [Google Scholar] [CrossRef] [PubMed]
- Piot, P.; Bartos, M.; Ghys, P.D.; Walker, N.; Schwartlander, B. The global impact of HIV/AIDS. Nature 2001, 410, 968–973. [Google Scholar] [CrossRef] [PubMed]
- Fact Sheet November 2016. Available online: http://www.unaids.org/en/resources/fact-sheet (accessed on 9 January 2017).
- The Joint United Nations Programme on HIV/AIDS (UNAIDS). Unaids Report on the Global Aids Epidemic 2013. Available online: http://www.unaids.org/sites/default/files/media_asset/UNAIDS_Global_Report_2013_en_1.pdf (accessed on 9 October 2013).
- The Joint United Nations Programme on HIV/AIDS (UNAIDS). Global Aids Response Progress Reporting 2015. Available online: http://www.unaids.org/sites/default/files/media_asset/JC2702_GARPR2015guidelines_en.pdf (accessed on5 May 2015).
- Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/hiv/ (accessed on 11 January 2017).
- Antinori, A.; Arendt, G.; Becker, J.T.; Brew, B.J.; Byrd, D.A.; Cherner, M.; Clifford, D.B.; Cinque, P.; Epstein, L.G.; Goodkin, K.; et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology 2007, 69, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M. Hiv‘s double strike at the brain: Neuronal toxicity and compromised neurogenesis. Front. Biosci. 2008, 13, 2484–2494. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McArthur, J.C.; Steiner, J.; Sacktor, N.; Nath, A. Human immunodeficiency virus-associated neurocognitive disorders: Mind the gap. Ann. Neurol. 2010, 67, 699–714. [Google Scholar] [PubMed]
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R., Jr.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: Charter study. Neurology 2010, 75, 2087–2096. [Google Scholar] [CrossRef]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder—Pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Drugs of Abuse. Available online: https://www.drugabuse.gov/drugs-abuse (accessed on 10 January 2017).
- Peterson, P.K.; Gekker, G.; Schut, R.; Hu, S.; Balfour, H.H., Jr.; Chao, C.C. Enhancement of HIV-1 replication by opiates and cocaine: The cytokine connection. Adv. Exp. Med. Biol. 1993, 335, 181–188. [Google Scholar] [PubMed]
- Carey, C.L.; Woods, S.P.; Rippeth, J.D.; Gonzalez, R.; Heaton, R.K.; Grant, I. Additive deleterious effects of methamphetamine dependence and immunosuppression on neuropsychological functioning in HIV infection. AIDS Behav. 2006, 10, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Byrd, D.A.; Fellows, R.P.; Morgello, S.; Franklin, D.; Heaton, R.K.; Deutsch, R.; Atkinson, J.H.; Clifford, D.B.; Collier, A.C.; Marra, C.M.; et al. Neurocognitive impact of substance use in HIV infection. J. Acquir. Immune Defic. Syndr. 2011, 58, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Buch, S.; Yao, H.; Guo, M.; Mori, T.; Mathias-Costa, B.; Singh, V.; Seth, P.; Wang, J.; Su, T.P. Cocaine and HIV-1 interplay in CNS: Cellular and molecular mechanisms. Curr. HIV Res. 2012, 10, 425–428. [Google Scholar] [CrossRef]
- Silverstein, P.S.; Shah, A.; Weemhoff, J.; Kumar, S.; Singh, D.P.; Kumar, A. HIV-1 gp120 and drugs of abuse: Interactions in the central nervous system. Curr. HIV Res. 2012, 10, 369–383. [Google Scholar] [CrossRef]
- Chang, S.L.; Connaghan, K.P.; Wei, Y.; Li, M.D. Neurohiv and use of addictive substances. Int. Rev. Neurobiol. 2014, 118, 403–440. [Google Scholar] [PubMed]
- Pilakka-Kanthikeel, S.; Nair, M.P. Interaction of drugs of abuse and microrna with HIV: A brief review. Front. Microbiol. 2015, 6, 967. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, F.; Vlahov, D.; Donahoe, R.M.; Friedland, G. The role of substance abuse in HIV disease progression: Reconciling differences from laboratory and epidemiologic investigations. Clin. Infect. Dis. 2005, 41, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.J.; Morris, S.R.; Kent, C.K.; Stansell, J.; Klausner, J.D. Methamphetamine use and sexual activity among HIV-infected patients in care—San Francisco, 2004. Aids Patient Care STDs 2006, 20, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Urbina, A.; Jones, K. Crystal methamphetamine, its analogues, and HIV infection: Medical and psychiatric aspects of a new epidemic. Clin. Infect. Dis. 2004, 38, 890–894. [Google Scholar] [CrossRef] [PubMed]
- Hinkin, C.H.; Barclay, T.R.; Castellon, S.A.; Levine, A.J.; Durvasula, R.S.; Marion, S.D.; Myers, H.F.; Longshore, D. Drug use and medication adherence among HIV-1 infected individuals. AIDS Behav. 2007, 11, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J.; Childers, M.E.; Cherner, M.; Lazzaretto, D.; Letendre, S.; Grant, I. Increased human immunodeficiency virus loads in active methamphetamine users are explained by reduced effectiveness of antiretroviral therapy. J. Infect. Dis. 2003, 188, 1820–1826. [Google Scholar] [CrossRef] [PubMed]
- Langford, T.D.; Letendre, S.L.; Larrea, G.J.; Masliah, E. Changing patterns in the neuropathogenesis of HIV during the haart era. Brain Pathol. 2003, 13, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Rippeth, J.D.; Heaton, R.K.; Carey, C.L.; Marcotte, T.D.; Moore, D.J.; Gonzalez, R.; Wolfson, T.; Grant, I. Methamphetamine dependence increases risk of neuropsychological impairment in HIV infected persons. J. Int. Neuropsychol. Soc. 2004, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chana, G.; Everall, I.P.; Crews, L.; Langford, D.; Adame, A.; Grant, I.; Cherner, M.; Lazzaretto, D.; Heaton, R.; Ellis, R.; et al. Cognitive deficits and degeneration of interneurons in HIV+ methamphetamine users. Neurology 2006, 67, 1486–1489. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.L.; Krasnova, I.N. Interactions of HIV and methamphetamine: Cellular and molecular mechanisms of toxicity potentiation. Neurotox. Res. 2007, 12, 181–204. [Google Scholar] [CrossRef] [PubMed]
- Eisfeld, C.; Reichelt, D.; Evers, S.; Husstedt, I. Csf penetration by antiretroviral drugs. CNS Drugs 2013, 27, 31–55. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.K.; Franklin, D.R., Jr.; Deutsch, R.; Letendre, S.; Ellis, R.J.; Casaletto, K.; Marquine, M.J.; Woods, S.P.; Vaida, F.; Atkinson, J.H.; et al. Neurocognitive change in the era of HIV combination antiretroviral therapy: The longitudinal charter study. Clin. Infect. Dis. 2015, 60, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.K.; Franklin, D.R.; Ellis, R.J.; McCutchan, J.A.; Letendre, S.L.; Leblanc, S.; Corkran, S.H.; Duarte, N.A.; Clifford, D.B.; Woods, S.P.; et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: Differences in rates, nature, and predictors. J. Neurovirol. 2011, 17, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.R.; Ellis, R.J.; Chen, H.; Yeh, T.M.; Lee, A.J.; Schifitto, G.; Wu, K.; Bosch, R.J.; McArthur, J.C.; Simpson, D.M.; et al. Peripheral neuropathy in HIV: Prevalence and risk factors. AIDS 2011, 25, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.R.; Lee, A.J.; Ellis, R.J.; Chen, H.; Wu, K.; Bosch, R.J.; Clifford, D.B. HIV peripheral neuropathy progression: Protection with glucose-lowering drugs? J. Neurovirol. 2012, 18, 428–433. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Robertson, K.R.; Su, Z.; Margolis, D.M.; Krambrink, A.; Havlir, D.V.; Evans, S.; Skiest, D.J. Neurocognitive effects of treatment interruption in stable HIV-positive patients in an observational cohort. Neurology 2010, 74, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Underwood, J.; Robertson, K.R.; Winston, A. Could antiretroviral neurotoxicity play a role in the pathogenesis of cognitive impairment in treated HIV disease? AIDS 2014, 29, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Carr, A. Toxicity of antiretroviral therapy and implications for drug development. Nat. Rev. Drug Discov. 2003, 2, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Keswani, S.C.; Chander, B.; Hasan, C.; Griffin, J.W.; McArthur, J.C.; Hoke, A. Fk506 is neuroprotective in a model of antiretroviral toxic neuropathy. Ann. Neurol. 2003, 53, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Lewin, S.R.; Hoy, J.; Crowe, S.M.; McDonald, C.F. The role of bronchoscopy in the diagnosis and treatment of pulmonary disease in HIV-infected patients. Aust. N. Z. J. Med. 1995, 25, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.; Liner, J.; Meeker, R.B. Antiretroviral neurotoxicity. J. Neurovirol. 2012, 18, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Akay, C.; Cooper, M.; Odeleye, A.; Jensen, B.K.; White, M.G.; Vassoler, F.; Gannon, P.J.; Mankowski, J.; Dorsey, J.L.; Buch, A.M.; et al. Antiretroviral drugs induce oxidative stress and neuronal damage in the central nervous system. J. Neurovirol. 2014, 20, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Navia, B.A.; Jordan, B.D.; Price, R.W. The aids dementia complex: I. Clinical features. Ann. Neurol. 1986, 19, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Price, R.W.; Brew, B.; Sidtis, J.; Rosenblum, M.; Scheck, A.C.; Cleary, P. The brain in aids: Central nervous system HIV-1 infection and aids dementia complex. Science 1988, 239, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Garden, G.A.; Lipton, S.A. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 2001, 410, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Sacktor, N.; Lyles, R.H.; Skolasky, R.; Kleeberger, C.; Selnes, O.A.; Miller, E.N.; Becker, J.T.; Cohen, B.; McArthur, J.C. HIV-associated neurologic disease incidence changes: Multicenter aids cohort study, 1990–1998. Neurology 2001, 56, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J.; Deutsch, R.; Heaton, R.K.; Marcotte, T.D.; McCutchan, J.A.; Nelson, J.A.; Abramson, I.; Thal, L.J.; Atkinson, J.H.; Wallace, M.R.; et al. Neurocognitive impairment is an independent risk factor for death in hiv infection. San Diego HIV neurobehavioral research center group. Arch. Neurol. 1997, 54, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, P.H.; Smith, D.G.; Satchell, C.; Cooper, D.A.; Brew, B. Evidence for independent development of resistance to HIV-1 reverse transcriptase inhibitors in the cerebrospinal fluid. AIDS 2000, 14, 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Cysique, L.A.; Maruff, P.; Brew, B.J. Variable benefit in neuropsychological function in HIV-infected haart-treated patients. Neurology 2006, 66, 1447–1450. [Google Scholar] [CrossRef] [PubMed]
- Giancola, M.L.; Lorenzini, P.; Balestra, P.; Larussa, D.; Baldini, F.; Corpolongo, A.; Narciso, P.; Bellagamba, R.; Tozzi, V.; Antinori, A. Neuroactive antiretroviral drugs do not influence neurocognitive performance in less advanced hiv-infected patients responding to highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2006, 41, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Sacktor, N. Influence of highly active antiretroviral therapy on persistence of HIV in the central nervous system. Curr. Opin. Neurol. 2006, 19, 358–361. [Google Scholar] [CrossRef] [PubMed]
- McArthur, J.C.; Haughey, N.; Gartner, S.; Conant, K.; Pardo, C.; Nath, A.; Sacktor, N. Human immunodeficiency virus-associated dementia: An evolving disease. J. Neurovirol. 2003, 9, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Brew, B.J.; Crowe, S.M.; Landay, A.; Cysique, L.A.; Guillemin, G. Neurodegeneration and ageing in the haart era. J. Neuroimmune Pharmacol. 2009, 4, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Welch, K.; Morse, A. The clinical profile of end-stage aids in the era of highly active antiretroviral therapy. AIDS Patient Care STDs 2002, 16, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Zheng, J.; Okamoto, S.; Gendelman, H.E.; Lipton, S.A. HIV-1 infection and aids: Consequences for the central nervous system. Cell Death Differ. 2005, 12, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Kramer-Hammerle, S.; Rothenaigner, I.; Wolff, H.; Bell, J.E.; Brack-Werner, R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005, 111, 194–213. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A. Treating aids dementia [letter; comment]. Science 1997, 276, 1629–1630. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Power, C. Regulation of neural cell survival by HIV-1 infection. Neurobiol. Dis. 2006, 21, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Petito, C.K.; Cho, E.S.; Lemann, W.; Navia, B.A.; Price, R.W. Neuropathology of acquired immunodeficiency syndrome (AIDS): An autopsy review. J. Neuropathol. Exp. Neurol. 1986, 45, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Heaton, R.K.; Marcotte, T.D.; Ellis, R.J.; Wiley, C.A.; Mallory, M.; Achim, C.L.; McCutchan, J.A.; Nelson, J.A.; Atkinson, J.H.; et al. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. Hnrc group. The HIV neurobehavioral research center. Ann. Neurol. 1997, 42, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Glass, J.D.; Fedor, H.; Wesselingh, S.L.; McArthur, J.C. Immunocytochemical quantitation of human immunodeficiency virus in the brain: Correlations with dementia. Ann. Neurol. 1995, 38, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Achim, C.L.; Wang, R.; Miners, D.K.; Wiley, C.A. Brain viral burden in HIV infection. J. Neuropathol. Exp. Neurol. 1994, 53, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.A.; Masliah, E.; Achim, C.L. Measurement of cns HIV burden and its association with neurologic damage. Adv. Neuroimmunol. 1994, 4, 319–325. [Google Scholar] [CrossRef]
- Wesselingh, S.L.; Takahashi, K.; Glass, J.D.; McArthur, J.C.; Griffin, J.W.; Griffin, D.E. Cellular localization of tumor necrosis factor mrna in neurological tissue from HIV-infected patients by combined reverse transcriptase/polymerase chain reaction in situ hybridization and immunohistochemistry. J. Neuroimmunol. 1997, 74, 1–8. [Google Scholar] [CrossRef]
- Heyes, M.P.; Brew, B.J.; Martin, A.; Price, R.W.; Salazar, A.M.; Sidtis, J.J.; Yergey, J.A.; Mouradian, M.M.; Sadler, A.E.; Keilp, J.; et al. Quinolinic acid in cerebrospinal fluid and serum in HIV-1 infection: Relationship to clinical and neurological status. Ann. Neurol. 1991, 29, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Shiramizu, B.; Gartner, S.; Williams, A.; Shikuma, C.; Ratto-Kim, S.; Watters, M.; Aguon, J.; Valcour, V. Circulating proviral HIV DNA and HIV-associated dementia. AIDS 2005, 19, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Shiramizu, B.; Ratto-Kim, S.; Sithinamsuwan, P.; Nidhinandana, S.; Thitivichianlert, S.; Watt, G.; Desouza, M.; Chuenchitra, T.; Sukwit, S.; Chitpatima, S.; et al. HIV DNA and dementia in treatment-naive HIV-1-infected individuals in bangkok, thailand. Int. J. Med. Sci. 2006, 4, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Ketzler, S.; Weis, S.; Haug, H.; Budka, H. Loss of neurons in the frontal cortex in aids brains. Acta Neuropathol. 1990, 80, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Everall, I.P.; Luthert, P.J.; Lantos, P.L. Neuronal loss in the frontal cortex in HIV infection. Lancet 1991, 337, 1119–1121. [Google Scholar] [CrossRef]
- Reyes, M.G.; Faraldi, F.; Senseng, C.S.; Flowers, C.; Fariello, R. Nigral degeneration in acquired immune deficiency syndrome (AIDS). Acta Neuropathol. 1991, 82, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Graus, F.; Ribalta, T.; Abos, J.; Alom, J.; Cruz-Sanchez, F.; Mallolas, J.; Miro, J.M.; Cardesa, A.; Tolosa, E. Subacute cerebellar syndrome as the first manifestation of aids dementia complex. Acta Neurol. Scand. 1990, 81, 118–120. [Google Scholar] [CrossRef] [PubMed]
- Everall, I.; Luthert, P.; Lantos, P. A review of neuronal damage in human immunodeficiency virus infection: Its assessment, possible mechanism and relationship to dementia. J. Neuropathol. Exp. Neurol. 1993, 52, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Adle-Biassette, H.; Chrétien, F.; Wingertsmann, L.; Héry, C.; Ereau, T.; Scaravilli, F.; Tardieu, M.; Gray, F. Neuronal apoptosis does not correlate with dementia in HIV infection but is related to microglial activation and axonal damage. Neuropathol. Appl. Neurobiol. 1999, 25, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Petito, C.K.; Roberts, B. Evidence of apoptotic cell death in HIV encephalitis. Am. J. Pathol. 1995, 146, 1121–1130. [Google Scholar] [PubMed]
- Rostasy, K.; Monti, L.; Yiannoutsos, C.; Wu, J.; Bell, J.; Hedreen, J.; Navia, B.A. Nfkappab activation, TNF-alpha expression, and apoptosis in the aids-dementia-complex. J. Neurovirol. 2000, 6, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Anthony, I.C.; Ramage, S.N.; Carnie, F.W.; Simmonds, P.; Bell, J.E. Influence of haart on HIV-related cns disease and neuroinflammation. J. Neuropathol. Exp. Neurol. 2005, 64, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Everall, I.P.; Hansen, L.A.; Masliah, E. The shifting patterns of HIV encephalitis neuropathology. Neurotox. Res. 2005, 8, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Green, D.A.; Masliah, E.; Vinters, H.V.; Beizai, P.; Moore, D.J.; Achim, C.L. Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. AIDS 2005, 19, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.D.; Rota, T.R.; Schooley, R.T.; Kaplan, J.C.; Allan, J.D.; Groopman, J.E.; Resnick, L.; Felsenstein, D.; Andrews, C.A.; Hirsch, M.S. Isolation of HTLV-III from cerebrospinal fluid and neural tissues of patients with neurologic syndromes related to the acquired immunodeficiency syndrome. N. Engl. J. Med. 1985, 313, 1493–1497. [Google Scholar] [CrossRef] [PubMed]
- Gartner, S. HIV infection and dementia. Science 2000, 287, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Scarano, F.; Martin-Garcia, J. The neuropathogenesis of AIDS. Nat. Rev. Immunol. 2005, 5, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Koenig, S.; Gendelman, H.E.; Orenstein, J.M.; Dal Canto, M.C.; Pezeshkpour, G.H.; Yungbluth, M.; Janotta, F.; Aksamit, A.; Martin, M.A.; Fauci, A.S. Detection of aids virus in macrophages in brain tissue from aids patients with encephalopathy. Science 1986, 233, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Dragic, T.; Litwin, V.; Allaway, G.P.; Martin, S.R.; Huang, Y.; Nagashima, K.A.; Cayanan, C.; Maddon, P.J.; Koup, R.A.; Moore, J.P.; et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 1996, 381, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Oberlin, E.; Amara, A.; Bachelerie, F.; Bessia, C.; Virelizier, J.L.; Arenzana-Seisdedos, F.; Schwartz, O.; Heard, J.M.; Clark-Lewis, I.; Legler, D.F.; et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature 1996, 382, 833–835. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Chen, Y.; Farzan, M.; Choe, H.; Ohagen, A.; Gartner, S.; Busciglio, J.; Yang, X.; Hofmann, W.; Newman, W.; et al. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature 1997, 385, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Asensio, V.C.; Campbell, I.L. Chemokines in the CNS: Plurifunctional mediators in diverse states. Trends Neurosci. 1999, 22, 504–512. [Google Scholar] [CrossRef]
- Miller, R.J.; Meucci, O. Aids and the brain: Is there a chemokine connection? Trends Neurosci. 1999, 22, 471–479. [Google Scholar] [CrossRef]
- Hesselgesser, J.; Taub, D.; Baskar, P.; Greenberg, M.; Hoxie, J.; Kolson, D.L.; Horuk, R. Neuronal apoptosis induced by HIV-1 gp120 and the chemokine SDF-1 alpha is mediated by the chemokine receptor CXCR4. Curr. Biol. 1998, 8, 595–598. [Google Scholar] [CrossRef]
- Meucci, O.; Fatatis, A.; Simen, A.A.; Bushell, T.J.; Gray, P.W.; Miller, R.J. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc. Natl. Acad. Sci. USA 1998, 95, 14500–14505. [Google Scholar] [CrossRef] [PubMed]
- Meucci, O.; Fatatis, A.; Simen, A.A.; Miller, R.J. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc. Natl. Acad. Sci. USA 2000, 97, 8075–8080. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Lipton, S.A. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc. Natl. Acad. Sci. USA 1999, 96, 8212–8216. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Ma, Q.; Medders, K.E.; Desai, M.K.; Lipton, S.A. HIV-1 coreceptors CCR5 and CXCR4 both mediate neuronal cell death but CCR5 paradoxically can also contribute to protection. Cell Death Differ. 2007, 14, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Thylin, M.R.; Ghorpade, A.; Xiong, H.; Persidsky, Y.; Cotter, R.; Niemann, D.; Che, M.; Zeng, Y.C.; Gelbard, H.A.; et al. Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. J. Neuroimmunol. 1999, 98, 185–200. [Google Scholar] [CrossRef]
- Maung, R.; Hoefer, M.M.; Sanchez, A.B.; Sejbuk, N.E.; Medders, K.E.; Desai, M.K.; Catalan, I.C.; Dowling, C.C.; de Rozieres, C.M.; Garden, G.A.; et al. CCR5 knockout prevents neuronal injury and behavioral impairment induced in a transgenic mouse model by a CXCR4-using HIV-1 glycoprotein 120. J. Immunol. 2014, 193, 1895–1910. [Google Scholar] [CrossRef] [PubMed]
- Brenneman, D.E.; Westbrook, G.L.; Fitzgerald, S.P.; Ennist, D.L.; Elkins, K.L.; Ruff, M.R.; Pert, C.B. Neuronal cell killing by the envelope protein of HIV and its prevention by vasoactive intestinal peptide. Nature 1988, 335, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Iskander, S.; Walsh, K.A.; Hammond, R.R. Human CNS cultures exposed to HIV-1 gp120 reproduce dendritic injuries of HIV-1-associated dementia. J. Neuroinflamm. 2004, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Walsh, K.A.; Megyesi, J.F.; Wilson, J.X.; Crukley, J.; Laubach, V.E.; Hammond, R.R. Antioxidant protection from HIV-1 gp120-induced neuroglial toxicity. J. Neuroinflamm. 2004, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Ohagen, A.; Ghosh, S.; He, J.; Huang, K.; Chen, Y.; Yuan, M.; Osathanondh, R.; Gartner, S.; Shi, B.; Shaw, G.; et al. Apoptosis induced by infection of primary brain cultures with diverse human immunodeficiency virus type 1 isolates: Evidence for a role of the envelope. J. Virol. 1999, 73, 897–906. [Google Scholar] [PubMed]
- Chen, W.; Sulcove, J.; Frank, I.; Jaffer, S.; Ozdener, H.; Kolson, D.L. Development of a human neuronal cell model for human immunodeficiency virus (HIV)-infected macrophage-induced neurotoxicity: Apoptosis induced by HIV type 1 primary isolates and evidence for involvement of the Bcl-2/Bcl-xl-sensitive intrinsic apoptosis pathway. J. Virol. 2002, 76, 9407–9419. [Google Scholar] [PubMed]
- Garden, G.A.; Guo, W.; Jayadev, S.; Tun, C.; Balcaitis, S.; Choi, J.; Montine, T.J.; Moller, T.; Morrison, R.S. HIV associated neurodegeneration requires p53 in neurons and microglia. FASEB J. 2004, 18, 1141–1143. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Haughey, N.J.; Nath, A. Cell death in HIV dementia. Cell Death Differ. 2005, 12, 893–904. [Google Scholar] [CrossRef] [PubMed]
- New, D.R.; Ma, M.; Epstein, L.G.; Nath, A.; Gelbard, H.A. Human immunodeficiency virus type 1 tat protein induces death by apoptosis in primary human neuron cultures. J. Neurovirol. 1997, 3, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Adamson, D.C.; Wildemann, B.; Sasaki, M.; Glass, J.D.; McArthur, J.C.; Christov, V.I.; Dawson, T.M.; Dawson, V.L. Immunologic no synthase: Elevation in severe aids dementia and induction by HIV-1 gp41. Science 1996, 274, 1917–1921. [Google Scholar] [CrossRef] [PubMed]
- Piller, S.C.; Jans, P.; Gage, P.W.; Jans, D.A. Extracellular HIV-1 virus protein r causes a large inward current and cell death in cultured hippocampal neurons: Implications for aids pathology. Proc. Natl. Acad. Sci. USA 1998, 95, 4595–4600. [Google Scholar] [CrossRef] [PubMed]
- Koedel, U.; Kohleisen, B.; Sporer, B.; Lahrtz, F.; Ovod, V.; Fontana, A.; Erfle, V.; Pfister, H.W. HIV type 1 nef protein is a viral factor for leukocyte recruitment into the central nervous system. J. Immunol. 1999, 163, 1237–1245. [Google Scholar] [PubMed]
- Ellis, R.; Langford, D.; Masliah, E. HIV and antiretroviral therapy in the brain: Neuronal injury and repair. Nat. Rev. Neurosci. 2007, 8, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Krathwohl, M.D.; Kaiser, J.L. HIV-1 promotes quiescence in human neural progenitor cells. J. Infect. Dis. 2004, 190, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.; Kang, Y.J.; Brechtel, C.W.; Siviglia, E.; Russo, R.; Clemente, A.; Harrop, A.; McKercher, S.; Kaul, M.; Lipton, S.A. HIV/gp120 decreases adult neural progenitor cell proliferation via checkpoint kinase-mediated cell-cycle withdrawal and G1 arrest. Cell Stem Cell 2007, 1, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Giulian, D.; Vaca, K.; Noonan, C.A. Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science 1990, 250, 1593–1596. [Google Scholar] [CrossRef] [PubMed]
- Genis, P.; Jett, M.; Bernton, E.W.; Boyle, T.; Gelbard, H.A.; Dzenko, K.; Keane, R.W.; Resnick, L.; Mizrachi, Y.; Volsky, D.J.; et al. Cytokines and arachidonic metabolites produced during human immunodeficiency virus (HIV)-infected macrophage-astroglia interactions: Implications for the neuropathogenesis of HIV disease. J. Exp. Med. 1992, 176, 1703–1718. [Google Scholar] [CrossRef] [PubMed]
- Medders, K.E.; Sejbuk, N.E.; Maung, R.; Desai, M.K.; Kaul, M. Activation of p38 mapk is required in monocytic and neuronal cells for HIV glycoprotein 120-induced neurotoxicity. J. Immunol. 2010, 185, 4883–4895. [Google Scholar] [CrossRef] [PubMed]
- Sui, Z.; Fan, S.; Sniderhan, L.; Reisinger, E.; Litzburg, A.; Schifitto, G.; Gelbard, H.A.; Dewhurst, S.; Maggirwar, S.B. Inhibition of mixed lineage kinase 3 prevents HIV-1 tat-mediated neurotoxicity and monocyte activation. J. Immunol. 2006, 177, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Giulian, D.; Wendt, E.; Vaca, K.; Noonan, C.A. The envelope glycoprotein of human immunodeficiency virus type 1 stimulates release of neurotoxins from monocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 2769–2773. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, E.B.; Kaiser, P.K.; Offermann, J.T.; Lipton, S.A. HIV-1 coat protein neurotoxicity prevented by calcium channel antagonists. Science 1990, 248, 364–367. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, L.A.; Agrawal, A.; Jordan-Sciutto, K.L.; Dichter, M.A.; Lynch, D.R.; Kolson, D.L. Human immunodeficiency virus (HIV)-induced neurotoxicity: Roles for the nmda receptor subtypes. J. Neurosci. 2006, 26, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W.; Labruyere, J.; Wang, G.; Wozniak, D.F.; Price, M.T.; Sesma, M.A. Nmda antagonist neurotoxicity: Mechanism and prevention. Science 1991, 254, 1515–1518. [Google Scholar] [CrossRef] [PubMed]
- Doble, A. The role of excitotoxicity in neurodegenerative disease: Implications for therapy. Pharmacol. Ther. 1999, 81, 163–221. [Google Scholar] [CrossRef]
- Olney, J.W. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 1969, 164, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Tenneti, L.; D’Emilia, D.M.; Troy, C.M.; Lipton, S.A. Role of caspases in N-methyl-d-aspartate-induced apoptosis in cerebrocortical neurons. J. Neurochem. 1998, 71, 946–959. [Google Scholar] [CrossRef]
- Garden, G.A.; Budd, S.L.; Tsai, E.; Hanson, L.; Kaul, M.; D’Emilia, D.M.; Friedlander, R.M.; Yuan, J.; Masliah, E.; Lipton, S.A. Caspase cascades in human immunodeficiency virus-associated neurodegeneration. J. Neurosci. 2002, 22, 4015–4024. [Google Scholar] [PubMed]
- Jordan-Sciutto, K.L.; Wang, G.; Murphey-Corb, M.; Wiley, C.A. Cell cycle proteins exhibit altered expression patterns in lentiviral-associated encephalitis. J. Neurosci. 2002, 22, 2185–2195. [Google Scholar] [PubMed]
- Jana, A.; Pahan, K. Human immunodeficiency virus type 1 gp120 induces apoptosis in human primary neurons through redox-regulated activation of neutral sphingomyelinase. J. Neurosci. 2004, 24, 9531–9540. [Google Scholar] [CrossRef] [PubMed]
- Haughey, N.J.; Mattson, M.P. Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins tat and gp120. J. Acquir. Immune Defic. Syndr. 2002, 31 (Suppl. 2), S55–S61. [Google Scholar] [CrossRef] [PubMed]
- Haughey, N.J.; Cutler, R.G.; Tamara, A.; McArthur, J.C.; Vargas, D.L.; Pardo, C.A.; Turchan, J.; Nath, A.; Mattson, M.P. Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann. Neurol. 2004, 55, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Lindl, K.A.; Akay, C.; Wang, Y.; White, M.G.; Jordan-Sciutto, K.L. Expression of the endoplasmic reticulum stress response marker, BiP, in the central nervous system of HIV-positive individuals. Neuropathol. Appl. Neurobiol. 2007, 33, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Federally Approved HIV/AIDS Medical Practice Guidelines. Available online: https://aidsinfo.nih.gov/guidelines (accessed on 2 December 2016).
- Shah, A.; Gangwani, M.R.; Chaudhari, N.S.; Glazyrin, A.; Bhat, H.K.; Kumar, A. Neurotoxicity in the post-haart era: Caution for the antiretroviral therapeutics. Neurotox. Res. 2016, 30, 677–697. [Google Scholar] [CrossRef] [PubMed]
- Badowski, M.E.; Perez, S.E.; Biagi, M.; Littler, J.A. New antiretroviral treatment for HIV. Infect. Dis. Ther. 2016, 5, 329–352. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, N.; Fabbiani, M.; Di, G.S.; Fanti, I.; Baldonero, E.; Bracciale, L.; Tamburrini, E.; Cauda, R.; De, L.A.; Silveri, M.C. Efavirenz associated with cognitive disorders in otherwise asymptomatic HIV-infected patients. Neurology 2011, 76, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Canizares, S.; Cherner, M.; Ellis, R.J. HIV and aging: Effects on the central nervous system. Semin. Neurol. 2014, 34, 27–34. [Google Scholar] [CrossRef] [PubMed]
- DeVaughn, S.; Muller-Oehring, E.M.; Markey, B.; Bronte-Stewart, H.M.; Schulte, T. Aging with HIV-1 infection: Motor functions, cognition, and attention—A comparison with parkinson’s disease. Neuropsychol. Rev. 2015, 25, 424–438. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Vaida, F.; Wong, J.; Sanders, C.A.; Kao, Y.T.; Croteau, D.; Clifford, D.B.; Collier, A.C.; Gelman, B.B.; Marra, C.M.; et al. Long-term efavirenz use is associated with worse neurocognitive functioning in HIV-infected patients. J. Neurovirol. 2016, 22, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.; Dalakas, M.C. Mitochondrial toxicity of antiviral drugs. Nat. Med. 1995, 1, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Van Dyke, R.B.; Wang, L.; Williams, P.L.; Pediatric AIDS Clinical Trials Group 219C Team. Toxicities associated with dual nucleoside reverse-transcriptase inhibitor regimens in hiv-infected children. J. Infect. Dis. 2008, 198, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Apostolova, N.; Blas-Garcia, A.; Esplugues, J.V. Mitochondrial toxicity in haart: An overview of in vitro evidence. Curr. Pharm. Des. 2011, 17, 2130–2144. [Google Scholar] [CrossRef] [PubMed]
- Arnaudo, E.; Dalakas, M.; Shanske, S.; Moraes, C.T.; DiMauro, S.; Schon, E.A. Depletion of muscle mitochondrial DNA in aids patients with zidovudine-induced myopathy. Lancet 1991, 337, 508–510. [Google Scholar] [CrossRef]
- Cherry, C.L.; Gahan, M.E.; McArthur, J.C.; Lewin, S.R.; Hoy, J.F.; Wesselingh, S.L. Exposure to dideoxynucleosides is reflected in lowered mitochondrial DNA in subcutaneous fat. J. Acquir. Immune Defic. Syndr. 2002, 30, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Cote, H.C.; Brumme, Z.L.; Craib, K.J.; Alexander, C.S.; Wynhoven, B.; Ting, L.; Wong, H.; Harris, M.; Harrigan, P.R.; O’Shaughnessy, M.V.; et al. Changes in mitochondrial DNA as a marker of nucleoside toxicity in HIV-infected patients. N. Engl. J. Med. 2002, 346, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Funes, H.A.; Apostolova, N.; Alegre, F.; Blas-Garcia, A.; Alvarez, A.; Marti-Cabrera, M.; Esplugues, J.V. Neuronal bioenergetics and acute mitochondrial dysfunction: A clue to understanding the central nervous system side effects of efavirenz. J. Infect. Dis. 2014, 210, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Eriksson, S.; Wang, L. Down-regulation of mitochondrial thymidine kinase 2 and deoxyguanosine kinase by didanosine: Implication for mitochondrial toxicities of anti-HIV nucleoside analogs. Biochem. Biophys. Res. Commun. 2014, 450, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Song, F.; Gao, Z.; Ding, W.; Qiao, L.; Yang, S.; Chen, X.; Jin, R.; Chen, D. Long-term exposure of mice to nucleoside analogues disrupts mitochondrial DNA maintenance in cortical neurons. PLoS ONE 2014, 9, e85637. [Google Scholar] [CrossRef] [PubMed]
- Dragovic, G.; Jevtovic, D. Nucleoside reverse transcriptase inhibitor usage and the incidence of peripheral neuropathy in HIV/AIDS patients. Antivir. Chem. Chemother. 2003, 14, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C. Peripheral neuropathy and antiretroviral drugs. J. Peripher. Nerv. Syst. 2001, 6, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Venhoff, N.; Lebrecht, D.; Deveaud, C.; Beauvoit, B.; Bonnet, J.; Muller, K.; Kirschner, J.; Venhoff, A.C.; Walker, U.A. Oral uridine supplementation antagonizes the peripheral neuropathy and encephalopathy induced by antiretroviral nucleoside analogues. AIDS 2010, 24, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Ewings, E.L.; Gerschenson, M.; St Claire, M.C.; Nagashima, K.; Skopets, B.; Harbaugh, S.W.; Harbaugh, J.W.; Poirier, M.C. Genotoxic and functional consequences of transplacental zidovudine exposure in fetal monkey brain mitochondria. J. Acquir. Immune Defic. Syndr. 2000, 24, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Haik, S.; Gauthier, L.R.; Granotier, C.; Peyrin, J.M.; Lages, C.S.; Dormont, D.; Boussin, F.D. Fibroblast growth factor 2 up regulates telomerase activity in neural precursor cells. Oncogene 2000, 19, 2957–2966. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sanchez, A.B.; Varano, G.P.; de Rozieres, C.M.; Maung, R.; Catalan, I.C.; Dowling, C.C.; Sejbuk, N.E.; Hoefer, M.M.; Kaul, M. Antiretrovirals, methamphetamine, and HIV-1 envelope protein gp120 compromise neuronal energy homeostasis in association with various degrees of synaptic and neuritic damage. Antimicrob. Agents Chemother. 2016, 60, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Floris-Moore, M.A.; Mollan, K.; Wilkin, A.M.; Johnson, M.A.; Kashuba, A.D.; Wohl, D.A.; Patterson, K.B.; Francis, O.; Kronk, C.; Eron, J.J. Antiretroviral activity and safety of once-daily etravirine in treatment-naive HIV-infected adults: 48-week results. Antivir. Ther. 2016, 21, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.A.; Jin, J.; Ferrell, D.; Sadic, E.; Obregon, D.; Smith, A.J.; Tan, J.; Giunta, B. Efavirenz promotes beta-secretase expression and increased abeta1–40,42 via oxidative stress and reduced microglial phagocytosis: Implications for hiv associated neurocognitive disorders (hand). PLoS ONE 2014, 9, e95500. [Google Scholar] [CrossRef] [PubMed]
- Decloedt, E.H.; Maartens, G. Neuronal toxicity of efavirenz: A systematic review. Expert Opin. Drug Saf. 2013, 12, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Purnell, P.R.; Fox, H.S. Efavirenz induces neuronal autophagy and mitochondrial alterations. J. Pharmacol. Exp. Ther. 2014, 351, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Blas-Garcia, A.; Polo, M.; Alegre, F.; Funes, H.A.; Martinez, E.; Apostolova, N.; Esplugues, J.V. Lack of mitochondrial toxicity of darunavir, raltegravir and rilpivirine in neurons and hepatocytes: A comparison with efavirenz. J. Antimicrob. Chemother. 2014, 69, 2995–3000. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, L.; Toborek, M. Dysregulation of endoplasmic reticulum stress and autophagic responses by the antiretroviral drug efavirenz. Mol. Pharmacol. 2015, 88, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Gatanaga, H.; Hayashida, T.; Tsuchiya, K.; Yoshino, M.; Kuwahara, T.; Tsukada, H.; Fujimoto, K.; Sato, I.; Ueda, M.; Horiba, M.; et al. Successful efavirenz dose reduction in HIV type 1-infected individuals with cytochrome p450 2b6 *6 and *26. Clin. Infect. Dis. 2007, 45, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. Department of Health and Human Services. Available online: http://aidsinfo.nih.gov/contentfiles/lvguidelines/AdultandAdolescentGL.pdf (accessed on 11 January 2017).
- Bonfanti, P.; Valsecchi, L.; Parazzini, F.; Carradori, S.; Pusterla, L.; Fortuna, P.; Timillero, L.; Alessi, F.; Ghiselli, G.; Gabbuti, A.; et al. Incidence of adverse reactions in HIV patients treated with protease inhibitors: A cohort study. Coordinamento italiano studio allergia e infezione da HIV (CISAI) group. J. Acquir. Immune Defic. Syndr. 2000, 23, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M. Pharmacogenetics of cytochrome p450 and its applications in drug therapy: The past, present and future. Trends Pharmacol. Sci. 2004, 25, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Baeza, A.; Carvajal, F.; Bayon, C.; Perez-Valero, I.; Estebanez, M.; Bernardino, J.I.; Monge, S.; Lagarde, M.; Hernando, A.; Arnalich, F.; et al. Pattern of neurocognitive function in patients receiving boosted protease inhibitor monotherapy: A detailed neuropsychological study. J. Neurovirol. 2014, 20, 362–370. [Google Scholar] [CrossRef] [PubMed]
- James, C.W.; McNelis, K.C.; Matalia, M.D.; Cohen, D.M.; Szabo, S. Central nervous system toxicity and amprenavir oral solution. Ann. Pharmacother. 2002, 36, 174. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, J.A.; Jones, G.; Worthington, C.; Krentz, H.B.; Keppler, O.T.; Hoke, A.; Gill, M.J.; Power, C. Sensory neuropathy in human immunodeficiency virus/acquired immunodeficiency syndrome patients: Protease inhibitor-mediated neurotoxicity. Ann. Neurol. 2006, 59, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Vivithanaporn, P.; Asahchop, E.L.; Acharjee, S.; Baker, G.B.; Power, C. HIV protease inhibitors disrupt astrocytic glutamate transporter function and neurobehavioral performance. AIDS 2016, 30, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Gannon, P.J.; Akay-Espinoza, C.; Yee, A.C.; Briand, L.A.; Erickson, M.A.; Gelman, B.B.; Gao, Y.; Haughey, N.J.; Zink, M.C.; Clements, J.E.; et al. HIV protease inhibitors alter amyloid precursor protein processing via beta-site amyloid precursor protein cleaving enzyme-1 translational up-regulation. Am. J. Pathol. 2017, 187, 91–109. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.; Elion, R.; Ruane, P.; Shamblaw, D.; DeJesus, E.; Rashbaum, B.; Chuck, S.L.; Yale, K.; Liu, H.C.; Warren, D.R.; et al. Randomized, phase 2 evaluation of two single-tablet regimens elvitegravir/cobicistat/ emtricitabine/tenofovir disoproxil fumarate versus efavirenz/emtricitabine/tenofovir disoproxil fumarate for the initial treatment of HIV infection. AIDS 2011, 25, F7–F12. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.; Larsen, G.; Montaner, J.S. Exacerbation of depression associated with starting raltegravir: A report of four cases. AIDS 2008, 22, 1890–1892. [Google Scholar] [CrossRef] [PubMed]
- Teppler, H.; Brown, D.D.; Leavitt, R.Y.; Sklar, P.; Wan, H.; Xu, X.; Lievano, F.; Lehman, H.P.; Mast, T.C.; Nguyen, B.Y. Long-term safety from the raltegravir clinical development program. Curr. HIV Res. 2011, 9, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Fung, H.B.; Guo, Y. Enfuvirtide: A fusion inhibitor for the treatment of hiv infection. Clin. Ther. 2004, 26, 352–378. [Google Scholar] [CrossRef]
- Lalezari, J.P.; Henry, K.; O’Hearn, M.; Montaner, J.S.; Piliero, P.J.; Trottier, B.; Walmsley, S.; Cohen, C.; Kuritzkes, D.R.; Eron, J.J., Jr.; et al. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in north and South America. N. Engl. J. Med. 2003, 348, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- Cherry, C.L.; Duncan, A.J.; Mackie, K.F.; Wesselingh, S.L.; Brew, B.J. A report on the effect of commencing enfuvirtide on peripheral neuropathy. AIDS Res. Hum. Retrovir. 2008, 24, 1027–1030. [Google Scholar] [CrossRef] [PubMed]
- Lazzarin, A.; Clotet, B.; Cooper, D.; Reynes, J.; Arasteh, K.; Nelson, M.; Katlama, C.; Stellbrink, H.J.; Delfraissy, J.F.; Lange, J.; et al. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in europe and australia. N. Engl. J. Med. 2003, 348, 2186–2195. [Google Scholar] [CrossRef] [PubMed]
- Garvey, L.; Nelson, M.; Latch, N.; Erlwein, O.W.; Allsop, J.M.; Mitchell, A.; Kaye, S.; Watson, V.; Back, D.; Taylor-Robinson, S.D.; et al. CNS effects of a CCR5 inhibitor in HIV-infected subjects: A pharmacokinetic and cerebral metabolite study. J. Antimicrob. Chemother. 2012, 67, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Boesecke, C.; Pett, S.L. Clinical studies with chemokine receptor-5 (CCR5)-inhibitors. Curr. Opin. HIV AIDS 2012, 7, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.M.; Beck, S.E.; Pate, K.A.; Queen, S.E.; Dorsey, J.L.; Adams, R.J.; Avery, L.B.; Hubbard, W.; Tarwater, P.M.; Mankowski, J.L. Neuroprotective maraviroc monotherapy in simian immunodeficiency virus-infected macaques: Reduced replicating and latent SIV in the brain. AIDS 2013, 27, F21–F28. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.J.; Grom, A.A.; Thompson, S.D.; Lieuwen, D.; Passo, M.H.; Glass, D.N. Contrasting cytokine profiles in the synovium of different forms of juvenile rheumatoid arthritis and juvenile spondyloarthropathy: Prominence of interleukin 4 in restricted disease. J. Rheumatol. 1998, 25, 1388–1398. [Google Scholar] [PubMed]
- Barr, A.M.; Panenka, W.J.; MacEwan, G.W.; Thornton, A.E.; Lang, D.J.; Honer, W.G.; Lecomte, T. The need for speed: An update on methamphetamine addiction. J. Psychiatry Neurosci. 2006, 31, 301–313. [Google Scholar] [PubMed]
- Kaye, S.; McKetin, R.; Duflou, J.; Darke, S. Methamphetamine and cardiovascular pathology: A review of the evidence. Addiction 2007, 102, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Sekine, Y.; Ouchi, Y.; Sugihara, G.; Takei, N.; Yoshikawa, E.; Nakamura, K.; Iwata, Y.; Tsuchiya, K.J.; Suda, S.; Suzuki, K.; et al. Methamphetamine causes microglial activation in the brains of human abusers. J. Neurosci. 2008, 28, 5756–5761. [Google Scholar] [CrossRef] [PubMed]
- Jaehne, E.J.; Salem, A.; Irvine, R.J. Pharmacological and behavioral determinants of cocaine, methamphetamine, 3,4-methylenedioxymethamphetamine, and para-methoxyamphetamine-induced hyperthermia. Psychopharmacology 2007, 194, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Albertson, T.E.; Derlet, R.W.; Van Hoozen, B.E. Methamphetamine and the expanding complications of amphetamines. West. J. Med. 1999, 170, 214–219. [Google Scholar] [PubMed]
- Murray, J.B. Psychophysiological aspects of amphetamine-methamphetamine abuse. J. Psychol. 1998, 132, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.C.; Woods, S.P.; Matt, G.E.; Meyer, R.A.; Heaton, R.K.; Atkinson, J.H.; Grant, I. Neurocognitive effects of methamphetamine: A critical review and meta-analysis. Neuropsychol. Rev. 2007, 17, 275–297. [Google Scholar] [CrossRef] [PubMed]
- Theodore, S.; Cass, W.A.; Nath, A.; Maragos, W.F. Progress in understanding basal ganglia dysfunction as a common target for methamphetamine abuse and HIV-1 neurodegeneration. Curr. HIV Res. 2007, 5, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.; House, M.A. Cardiovascular effects of methamphetamine. J. Cardiovasc. Nurs. 1992, 6, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.A., Jr.; Arsura, E.L.; Strategos, S. Methamphetamine-related stroke: Four cases. J. Emerg. Med. 1999, 17, 469–471. [Google Scholar] [CrossRef]
- McCann, U.D.; Wong, D.F.; Yokoi, F.; Villemagne, V.; Dannals, R.F.; Ricaurte, G.A. Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: Evidence from positron emission tomography studies with [11c]win-35,428. J. Neurosci. 1998, 18, 8417–8422. [Google Scholar] [PubMed]
- Wilson, J.M.; Kalasinsky, K.S.; Levey, A.I.; Bergeron, C.; Reiber, G.; Anthony, R.M.; Schmunk, G.A.; Shannak, K.; Haycock, J.W.; Kish, S.J. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat. Med. 1996, 2, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Chang, L.; Wang, G.J.; Fowler, J.S.; Ding, Y.S.; Sedler, M.; Logan, J.; Franceschi, D.; Gatley, J.; Hitzemann, R.; et al. Low level of brain dopamine d2 receptors in methamphetamine abusers: Association with metabolism in the orbitofrontal cortex. Am. J. Psychiatry 2001, 158, 2015–2021. [Google Scholar] [CrossRef] [PubMed]
- Sekine, Y.; Ouchi, Y.; Takei, N.; Yoshikawa, E.; Nakamura, K.; Futatsubashi, M.; Okada, H.; Minabe, Y.; Suzuki, K.; Iwata, Y.; et al. Brain serotonin transporter density and aggression in abstinent methamphetamine abusers. Arch. Gen. Psychiatry 2006, 63, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Chang, L.; Wang, G.J.; Fowler, J.S.; Franceschi, D.; Sedler, M.J.; Gatley, S.J.; Hitzemann, R.; Ding, Y.S.; Wong, C.; et al. Higher cortical and lower subcortical metabolism in detoxified methamphetamine abusers. Am. J. Psychiatry 2001, 158, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Cass, W.A. Decreases in evoked overflow of dopamine in rat striatum after neurotoxic doses of methamphetamine. J. Pharmacol. Exp. Ther. 1997, 280, 105–113. [Google Scholar] [PubMed]
- Thompson, P.M.; Hayashi, K.M.; Simon, S.L.; Geaga, J.A.; Hong, M.S.; Sui, Y.; Lee, J.Y.; Toga, A.W.; Ling, W.; London, E.D. Structural abnormalities in the brains of human subjects who use methamphetamine. J. Neurosci. 2004, 24, 6028–6036. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.M.; Kuhn, D.M. Attenuated microglial activation mediates tolerance to the neurotoxic effects of methamphetamine. J. Neurochem. 2005, 92, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Ricaurte, G.A.; Guillery, R.W.; Seiden, L.S.; Schuster, C.R.; Moore, R.Y. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res. 1982, 235, 93–103. [Google Scholar] [CrossRef]
- Fukui, K.; Nakajima, T.; Kariyama, H.; Kashiba, A.; Kato, N.; Tohyama, I.; Kimura, H. Selective reduction of serotonin immunoreactivity in some forebrain regions of rats induced by acute methamphetamine treatment; quantitative morphometric analysis by serotonin immunocytochemistry. Brain Res. 1989, 482, 198–203. [Google Scholar] [CrossRef]
- Hotchkiss, A.J.; Gibb, J.W. Long-term effects of multiple doses of methamphetamine on tryptophan hydroxylase and tyrosine hydroxylase activity in rat brain. J. Pharmacol. Exp. Ther. 1980, 214, 257–262. [Google Scholar] [PubMed]
- Wagner, G.C.; Ricaurte, G.A.; Seiden, L.S.; Schuster, C.R.; Miller, R.J.; Westley, J. Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res. 1980, 181, 151–160. [Google Scholar] [CrossRef]
- Nakayama, M.; Koyama, T.; Yamashita, I. Long-lasting decrease in dopamine uptake sites following repeated administration of methamphetamine in the rat striatum. Brain Res. 1993, 601, 209–212. [Google Scholar] [CrossRef]
- Hirata, H.; Ladenheim, B.; Rothman, R.B.; Epstein, C.; Cadet, J.L. Methamphetamine-induced serotonin neurotoxicity is mediated by superoxide radicals. Brain Res. 1995, 677, 345–347. [Google Scholar] [CrossRef]
- Deng, X.; Cadet, J.L. Methamphetamine administration causes overexpression of nnos in the mouse striatum. Brain Res. 1999, 851, 254–257. [Google Scholar] [CrossRef]
- Frey, K.; Kilbourn, M.; Robinson, T. Reduced striatal vesicular monoamine transporters after neurotoxic but not after behaviorally-sensitizing doses of methamphetamine. Eur. J. Pharmacol. 1997, 334, 273–279. [Google Scholar] [CrossRef]
- Eisch, A.J.; Marshall, J.F. Methamphetamine neurotoxicity: Dissociation of striatal dopamine terminal damage from parietal cortical cell body injury. Synapse 1998, 30, 433–445. [Google Scholar] [CrossRef]
- O’Dell, S.J.; Marshall, J.F. Repeated administration of methamphetamine damages cells in the somatosensory cortex: Overlap with cytochrome oxidase-rich barrels. Synapse 2000, 37, 32–37. [Google Scholar] [PubMed]
- Schmued, L.C.; Bowyer, J.F. Methamphetamine exposure can produce neuronal degeneration in mouse hippocampal remnants. Brain Res. 1997, 759, 135–140. [Google Scholar] [CrossRef]
- Deng, X.; Ladenheim, B.; Jayanthi, S.; Cadet, J.L. Methamphetamine administration causes death of dopaminergic neurons in the mouse olfactory bulb. Biol. Psychiatry 2007, 61, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Jayanthi, S.; Deng, X.; Ladenheim, B.; McCoy, M.T.; Cluster, A.; Cai, N.S.; Cadet, J.L. Calcineurin/NFAT-induced up-regulation of the Fas ligand/Fas death pathway is involved in methamphetamine-induced neuronal apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Thiriet, N.; Deng, X.; Solinas, M.; Ladenheim, B.; Curtis, W.; Goldberg, S.R.; Palmiter, R.D.; Cadet, J.L. Neuropeptide y protects against methamphetamine-induced neuronal apoptosis in the mouse striatum. J. Neurosci. 2005, 25, 5273–5279. [Google Scholar] [CrossRef] [PubMed]
- Seiden, L.S.; Sabol, K.E.; Ricaurte, G.A. Amphetamine: Effects on catecholamine systems and behavior. Annu. Rev. Pharmacol. Toxicol. 1993, 33, 639–677. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.D.; Eiden, L.E. Functional identification and molecular cloning of a human brain vesicle monoamine transporter. J. Neurochem. 1993, 61, 2314–2317. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Sonders, M.S.; Poulsen, N.W.; Galli, A. Mechanisms of neurotransmitter release by amphetamines: A review. Prog. Neurobiol. 2005, 75, 406–433. [Google Scholar] [CrossRef] [PubMed]
- Nash, J.F.; Yamamoto, B.K. Methamphetamine neurotoxicity and striatal glutamate release: Comparison to 3,4-methylenedioxymethamphetamine. Brain Res. 1992, 581, 237–243. [Google Scholar] [CrossRef]
- Yu, Q.; Zhang, D.; Walston, M.; Zhang, J.; Liu, Y.; Watson, R.R. Chronic methamphetamine exposure alters immune function in normal and retrovirus-infected mice. Int. Immunopharmacol. 2002, 2, 951–962. [Google Scholar] [CrossRef]
- In, S.W.; Son, E.W.; Rhee, D.K.; Pyo, S. Methamphetamine administration produces immunomodulation in mice. J. Toxicol. Environ. Health A 2005, 68, 2133–2145. [Google Scholar] [CrossRef] [PubMed]
- Talloczy, Z.; Martinez, J.; Joset, D.; Ray, Y.; Gacser, A.; Toussi, S.; Mizushima, N.; Nosanchuk, J.; Goldstein, H.; Loike, J.; et al. Methamphetamine inhibits antigen processing, presentation, and phagocytosis. PLoS Pathog. 2008, 4, e28. [Google Scholar] [CrossRef]
- Krasnova, I.N.; Cadet, J.L. Methamphetamine toxicity and messengers of death. Brain Res. Rev. 2009, 60, 379–407. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Kumar, S.; Simon, S.D.; Singh, D.P.; Kumar, A. HIV gp120- and methamphetamine-mediated oxidative stress induces astrocyte apoptosis via cytochrome p450 2e1. Cell Death Dis. 2013, 4, e850. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, B.M.; Guadagnini, D.; Tsukumo, D.M.; Schenka, A.A.; Latuf-Filho, P.; Vassallo, J.; Dias, J.C.; Kubota, L.T.; Carvalheira, J.B.; Saad, M.J. Modulation of gut microbiota by antibiotics improves insulin signalling in high-Fat fed mice. Diabetologia 2012, 55, 2823–2834. [Google Scholar] [CrossRef] [PubMed]
- Vearrier, D.; Greenberg, M.I.; Miller, S.N.; Okaneku, J.T.; Haggerty, D.A. Methamphetamine: History, pathophysiology, adverse health effects, current trends, and hazards associated with the clandestine manufacture of methamphetamine. Dis. Mon. 2012, 58, 38–89. [Google Scholar] [CrossRef] [PubMed]
- Kanthasamy, A.; Anantharam, V.; Ali, S.F.; Kanthasamy, A.G. Methamphetamine induces autophagy and apoptosis in a mesencephalic dopaminergic neuronal culture model: Role of cathepsin-D in methamphetamine-induced apoptotic cell death. Ann. N. Y. Acad. Sci. 2006, 1074, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Larsen, K.E.; Fon, E.A.; Hastings, T.G.; Edwards, R.H.; Sulzer, D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J. Neurosci. 2002, 22, 8951–8960. [Google Scholar] [PubMed]
- Pasquali, L.; Lazzeri, G.; Isidoro, C.; Ruggieri, S.; Paparelli, A.; Fornai, F. Role of autophagy during methamphetamine neurotoxicity. Ann. N. Y. Acad. Sci. 2008, 1139, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zhu, L.; Shen, Q.; Bai, X.; Di, X. Recent advances in methamphetamine neurotoxicity mechanisms and its molecular pathophysiology. Behav. Neurol. 2015, 2015, 103969. [Google Scholar] [CrossRef] [PubMed]
- Soontornniyomkij, V.; Kesby, J.P.; Morgan, E.E.; Bischoff-Grethe, A.; Minassian, A.; Brown, G.G.; Grant, I.; Translational Methamphetamine AIDS Research Center (TMARC) Group. Effects of hiv and methamphetamine on brain and behavior: Evidence from human studies and animal models. J. Neuroimmune Pharmacol. 2016, 11, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Marquez, C.; Mitchell, S.J.; Hare, C.B.; John, M.; Klausner, J.D. Methamphetamine use, sexual activity, patient-provider communication, and medication adherence among HIV-infected patients in care, San Francisco 2004–2006. AIDS Care 2009, 21, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Langford, D.; Adame, A.; Grigorian, A.; Grant, I.; McCutchan, J.A.; Ellis, R.J.; Marcotte, T.D.; Masliah, E. Patterns of selective neuronal damage in methamphetamine-user AIDS patients. J. Acquir. Immune Defic. Syndr. 2003, 34, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.J.; Maung, R.; Sejbuk, N.E.; Ake, C.; Kaul, M. Alteration of methamphetamine-induced stereotypic behaviour in transgenic mice expressing HIV-1 envelope protein gp120. J. Neurosci. Methods 2010, 186, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, G.; Buescher, J.L.; Fox, H.S. Methamphetamine and inflammatory cytokines increase neuronal na+/k+-atpase isoform 3: Relevance for HIV associated neurocognitive disorders. PLoS ONE 2012, 7, e37604. [Google Scholar] [CrossRef] [PubMed]
- Bortell, N.; Morsey, B.; Basova, L.; Fox, H.S.; Marcondes, M.C. Phenotypic changes in the brain of SIV-infected macaques exposed to methamphetamine parallel macrophage activation patterns induced by the common gamma-chain cytokine system. Front. Microbiol. 2015, 6, 900. [Google Scholar] [CrossRef] [PubMed]
- Hoefer, M.M.; Sanchez, A.B.; Maung, R.; de Rozieres, C.M.; Catalan, I.C.; Dowling, C.C.; Thaney, V.E.; Pina-Crespo, J.; Zhang, D.; Roberts, A.J.; et al. Combination of methamphetamine and HIV-1 gp120 causes distinct long-term alterations of behavior, gene expression, and injury in the central nervous system. Exp. Neurol. 2015, 263, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Almeida, J.; Bolanos, J.P.; Moncada, S. Different responses of astrocytes and neurons to nitric oxide: The role of glycolytically generated atp in astrocyte protection. Proc. Natl. Acad. Sci. USA 2001, 98, 15294–15299. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Moncada, S.; Bolanos, J.P. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat. Cell Biol. 2004, 6, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.M.; Chen, P.C.; Hsieh, H.C.; Calkins, M.J. Mitochondrial defects arise from nucleoside/nucleotide reverse transcriptase inhibitors in neurons: Potential contribution to HIV-associated neurocognitive disorders. Biochim. Biophys. Acta 2017, 1863, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Wong-Riley, M.T. Cytochrome oxidase: An endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989, 12, 94–101. [Google Scholar] [CrossRef]
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L. Axonal transport: Cargo-specific mechanisms of motility and regulation. Neuron 2014, 84, 292–309. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Zakaria, H.M.; Simone, A.; Sheng, Z.H. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr. Biol. 2012, 22, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Tavernarakis, N. Mitophagy in neurodegeneration and aging. Front. Genet. 2012, 3, 297. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Amp-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [PubMed]
- Oakhill, J.S.; Steel, R.; Chen, Z.P.; Scott, J.W.; Ling, N.; Tam, S.; Kemp, B.E. Ampk is a direct adenylate charge-regulated protein kinase. Science 2011, 332, 1433–1435. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. Mtor signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Fields, J.; Dumaop, W.; Rockenstein, E.; Mante, M.; Spencer, B.; Grant, I.; Ellis, R.; Letendre, S.; Patrick, C.; Adame, A.; et al. Age-dependent molecular alterations in the autophagy pathway in hive patients and in a gp120 tg mouse model: Reversal with beclin-1 gene transfer. J. Neurovirol. 2013, 19, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Balgi, A.D.; Fonseca, B.D.; Donohue, E.; Tsang, T.C.; Lajoie, P.; Proud, C.G.; Nabi, I.R.; Roberge, M. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mtorc1 signaling. PLoS ONE 2009, 4, e7124. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, X.; Tang, S.J. Interactions of opioids and hiv infection in the pathogenesis of chronic pain. Front. Microbiol. 2016, 7, 103. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, N.K.; Williams, R.; Peng, F.; Tsai, Y.J.; Dhillon, S.; Nicolay, B.; Gadgil, M.; Kumar, A.; Buch, S.J. Cocaine-mediated enhancement of virus replication in macrophages: Implications for human immunodeficiency virus-associated dementia. J. Neurovirol. 2007, 13, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Wang, X.; Chen, H.; Song, L.; Ye, L.; Wang, S.H.; Wang, Y.J.; Zhou, L.; Ho, W.Z. Methamphetamine enhances hiv infection of macrophages. Am. J. Pathol. 2008, 172, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Ye, L.; Li, J.; Song, L.; Fulambarkar, N.; Ho, W. Morphine suppresses IFN signaling pathway and enhances AIDS virus infection. PLoS ONE 2012, 7, e31167. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Rivera, L.; Perez-Laspiur, J.; Colon, K.; Melendez, L.M. Inhibition of interferon response by cystatin b: Implication in hiv replication of macrophage reservoirs. J. Neurovirol. 2011, 18, 20–29. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Purohit, V.; Rapaka, R.S.; Rutter, J.; Shurtleff, D. Do opioids activate latent HIV-1 by down-regulating anti-HIV micrornas? J. Neuroimmune Pharmacol. 2012, 7, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Sampey, G.C.; Meyering, S.S.; Asad, Z.M.; Saifuddin, M.; Hakami, R.M.; Kashanchi, F. Exosomes and their role in CNS viral infections. J. Neurovirol. 2014, 20, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Yao, H.; Chaudhuri, A.D.; Duan, M.; Yelamanchili, S.V.; Wen, H.; Cheney, P.D.; Fox, H.S.; Buch, S. Exosome-mediated shuttling of microrna-29 regulates HIV tat and morphine-mediated neuronal dysfunction. Cell Death Dis. 2012, 3, e381. [Google Scholar] [CrossRef] [PubMed]
- Rahimian, P.; He, J.J. Exosome-associated release, uptake, and neurotoxicity of HIV-1 tat protein. J. Neurovirol. 2016, 22, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Raymond, A.D.; Diaz, P.; Chevelon, S.; Agudelo, M.; Yndart-Arias, A.; Ding, H.; Kaushik, A.; Jayant, R.D.; Nikkhah-Moshaie, R.; Roy, U.; et al. Microglia-derived hiv NEF+ exosome impairment of the blood-brain barrier is treatable by nanomedicine-based delivery of nef peptides. J. Neurovirol. 2016, 22, 129–139. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez, A.B.; Kaul, M. Neuronal Stress and Injury Caused by HIV-1, cART and Drug Abuse: Converging Contributions to HAND. Brain Sci. 2017, 7, 25. https://doi.org/10.3390/brainsci7030025
Sanchez AB, Kaul M. Neuronal Stress and Injury Caused by HIV-1, cART and Drug Abuse: Converging Contributions to HAND. Brain Sciences. 2017; 7(3):25. https://doi.org/10.3390/brainsci7030025
Chicago/Turabian StyleSanchez, Ana B., and Marcus Kaul. 2017. "Neuronal Stress and Injury Caused by HIV-1, cART and Drug Abuse: Converging Contributions to HAND" Brain Sciences 7, no. 3: 25. https://doi.org/10.3390/brainsci7030025
APA StyleSanchez, A. B., & Kaul, M. (2017). Neuronal Stress and Injury Caused by HIV-1, cART and Drug Abuse: Converging Contributions to HAND. Brain Sciences, 7(3), 25. https://doi.org/10.3390/brainsci7030025