A Proposed Mechanism for Development of CTE Following Concussive Events: Head Impact, Water Hammer Injury, Neurofilament Release, and Autoimmune Processes


Abstract
1. Introduction
2. Methods
3. Results
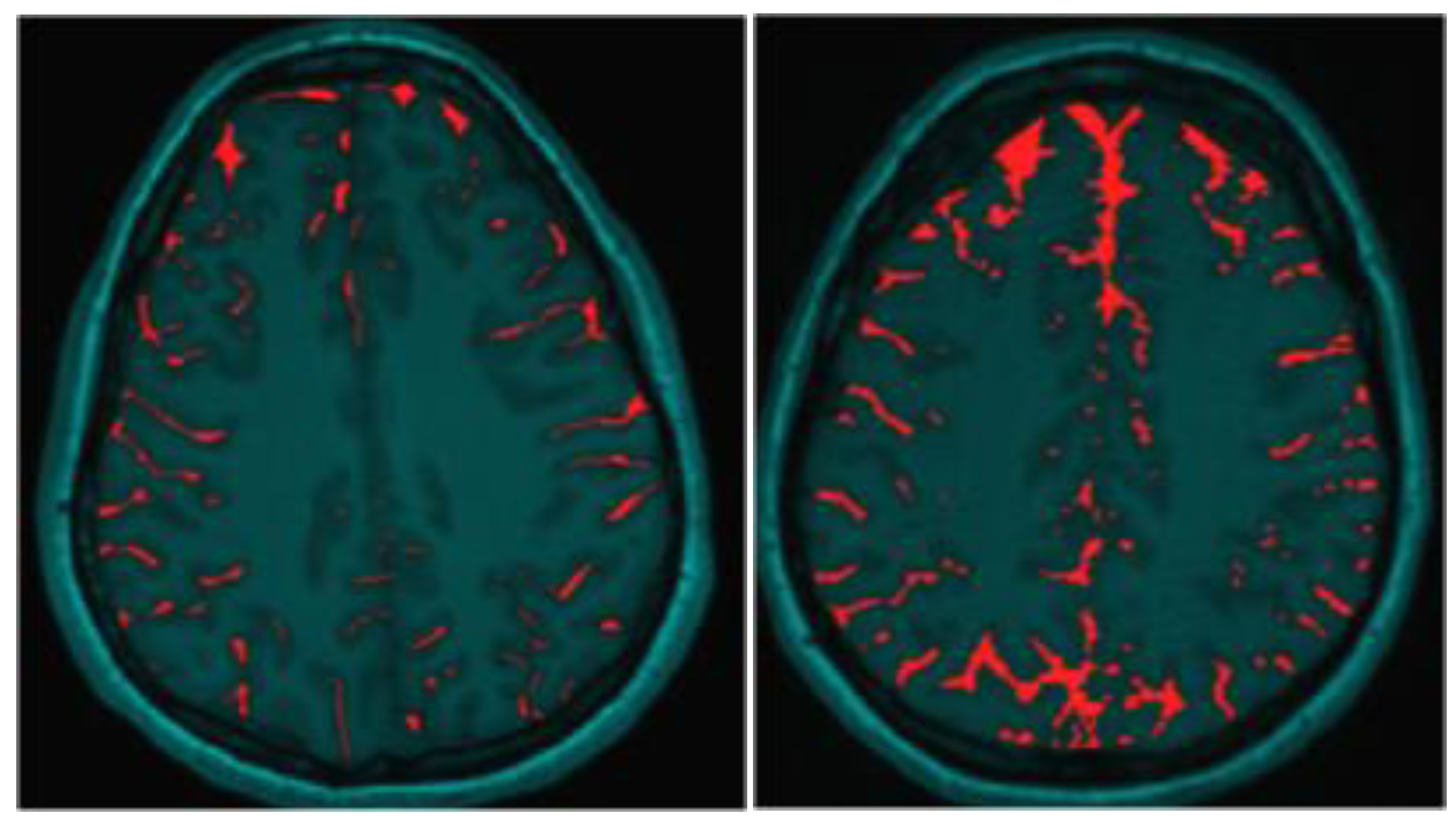
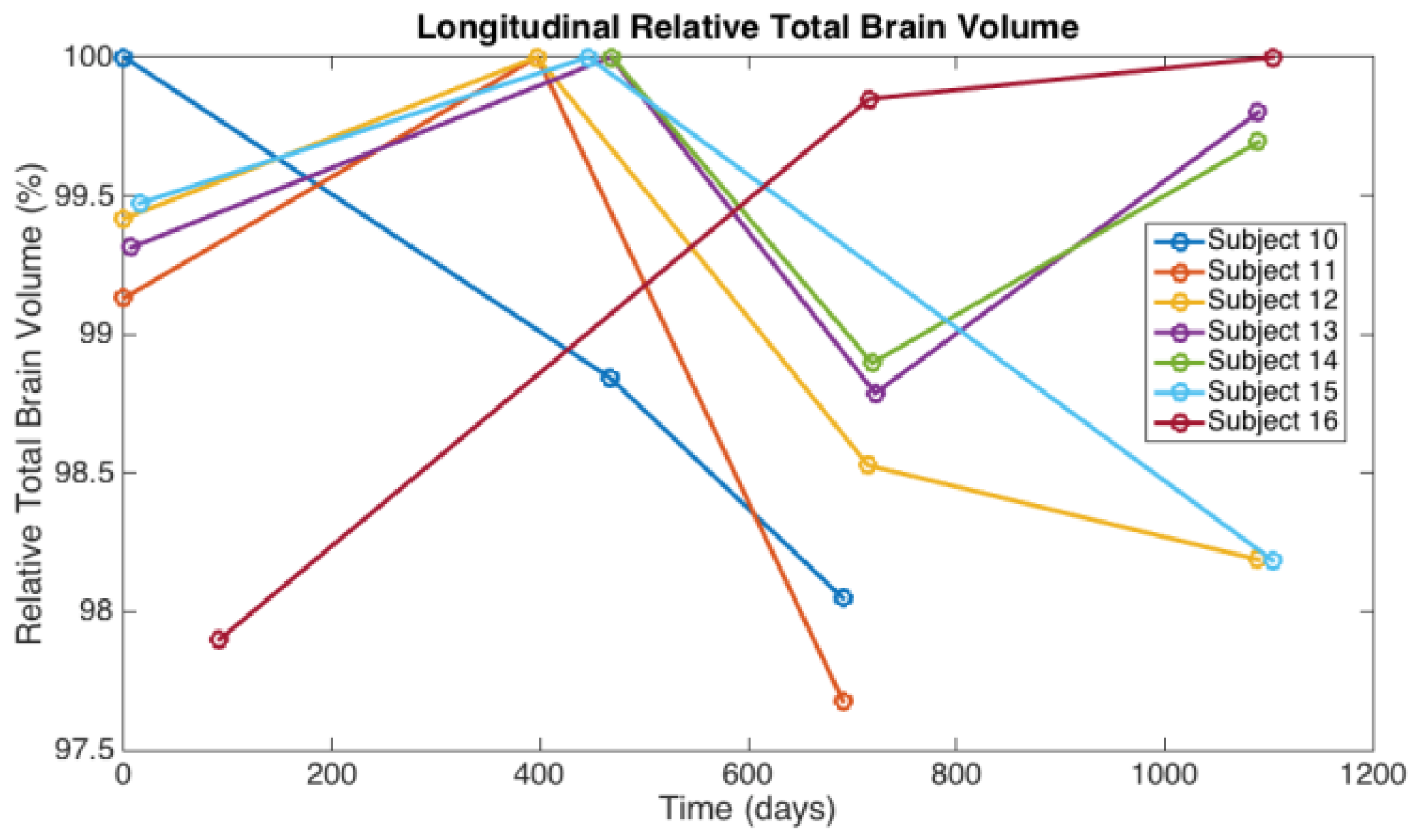
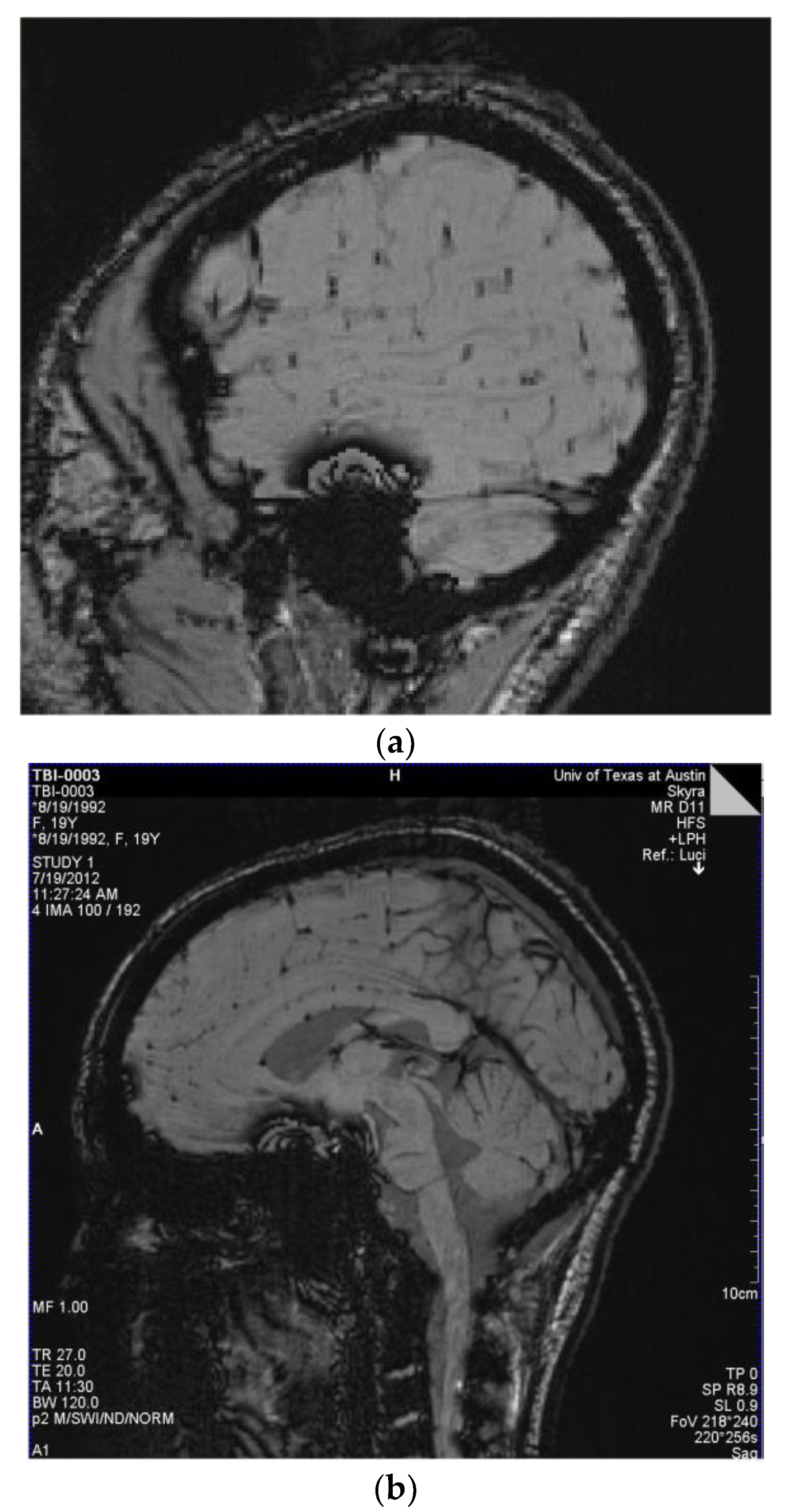
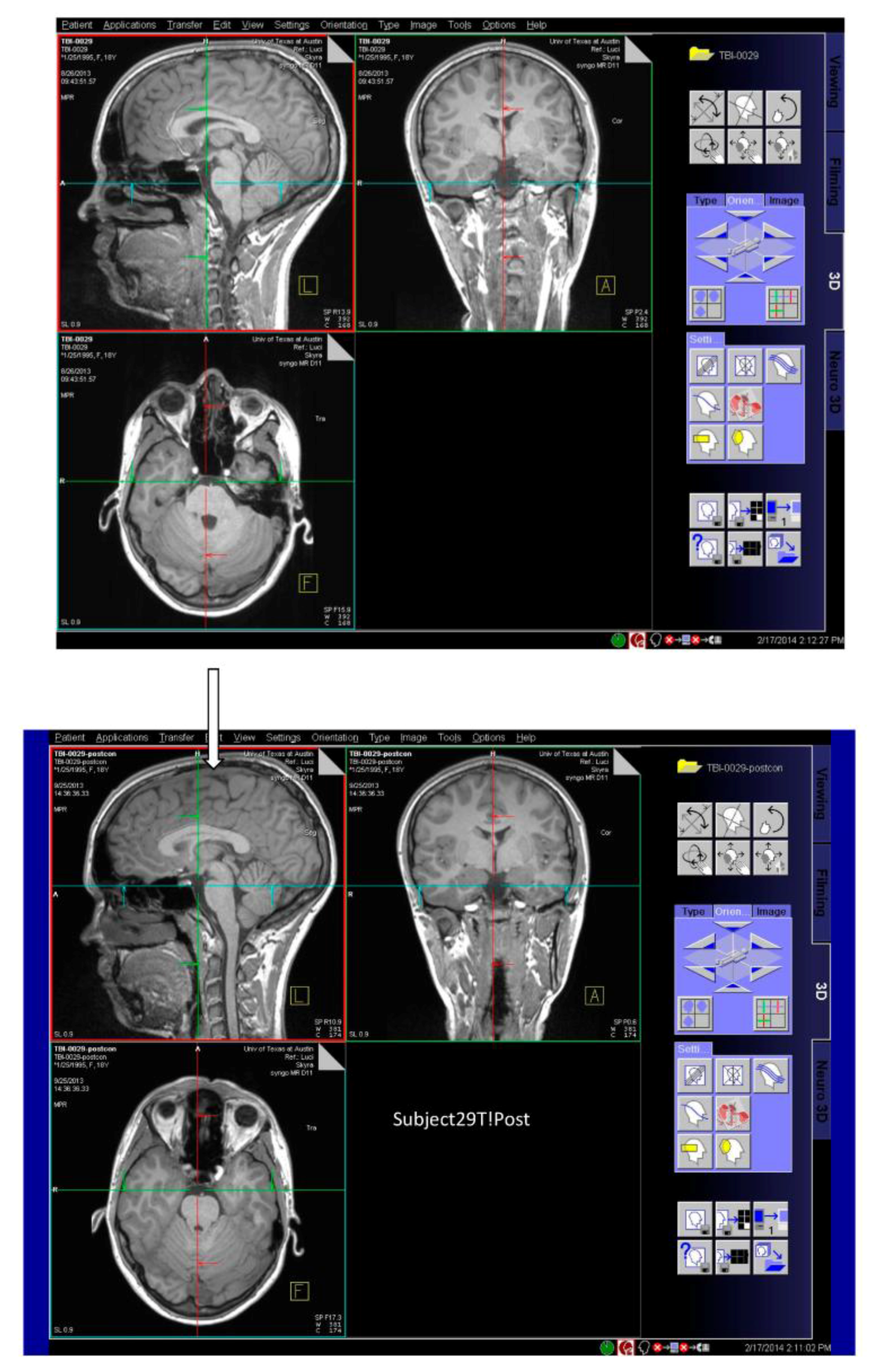
3.1. Observations from MR Imaging
3.2. Observations from IMPACT and C3 Logix Assessments
4. Discussion
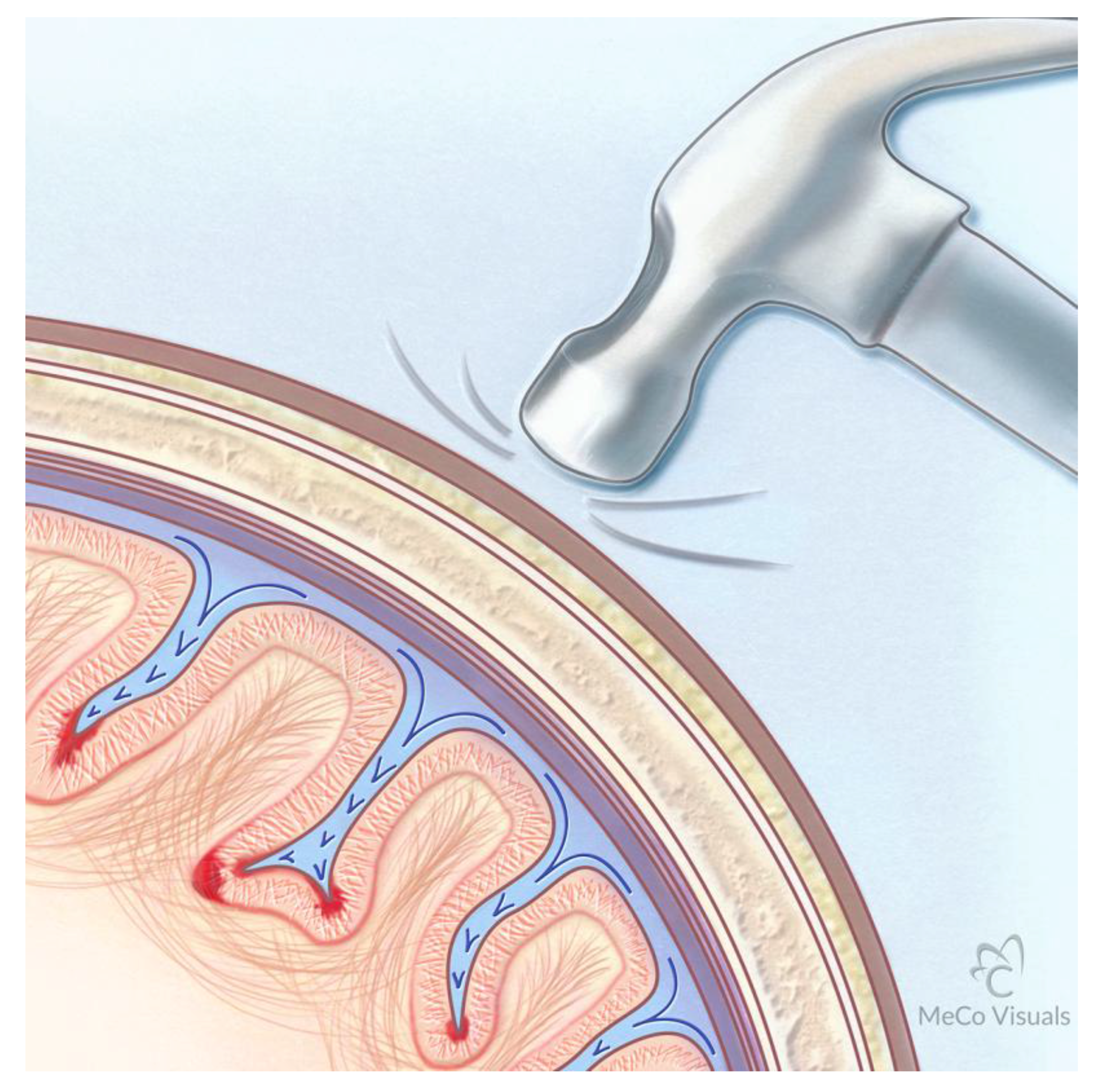
Proposed Mechanism for Development of CTE Following Repetitive Traumatic Events
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Institute of Medicine. Cognitive Rehabilitation Therapy for Traumatic Brain Injury: Evaluating the Evidence; Koehler, R., Wilhelm, E., Shoulson, I., Eds.; Institute of Medicine: Washington, DC, USA, 2011. [Google Scholar]
- Joy, J.E.; Patlak, M. Is Soccer Bad for Children’s Heads?: Summary of the IOM Workshop on Neuropsychological Consequences of Head Impact in Youth Soccer; Institute of Medicine: Washington, DC, USA, 2002. [Google Scholar]
- Centers for Disease Control and Prevention. Nonfatal Traumatic Brain Injuries Related to Sports and Recreation Activites Among Persons Aged ≤ 19 Years—United States, 2001–2009. MMWR Morb. Mortal. Wkly. Rep. 2011, 60, 1137–1142. [Google Scholar]
- Frommer, L.J.; Gurka, K.K.; Cross, K.M.; Ingersoll, C.D.; Comstock, R.D.; Saliba, S.A. Sex differences in concussion symptoms of high school athletes. J. Athl. Train. 2011, 46, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Yard, E.E.; Schroeder, M.J.; Fields, S.K.; Collins, C.L.; Comstock, R.D. The epidemiology of United States high school soccer injuries, 2005–2007. Am. J. Sports Med. 2008, 36, 1930–1937. [Google Scholar] [CrossRef] [PubMed]
- Mansell, J.L.; Tierney, R.T.; Higgins, M.; McDevitt, J.; Toone, N.; Glutting, J. Concussive signs and symptoms following head impacts in collegiate athletes. Brain Inj. 2010, 24, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Ikonomovic, M.D.; Gandy, S. Traumatic brain injury-football, warfare, and long-term effects. N. Engl. J. Med. 2010, 363, 1293–1296. [Google Scholar] [CrossRef] [PubMed]
- Babbs, C.F. Biomechanics of heading a soccer ball: Implications for player safety. Sci.World J. 2001, 1, 281–322. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.E.; Fisher, A.M.; Tagge, C.A.; Zhang, X.L.; Velisek, L.; Sullivan, J.A.; Upreti, C.; Kracht, J.M.; Ericsson, M.; Wojnarowicz, M.W.; et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 2012, 4, 134ra60. [Google Scholar] [CrossRef]
- Rubenstein, R.; Chang, B.; Yue, J.K.; Chiu, A.; Winkler, E.A.; Puccio, A.M.; Diaz-Arrastia, R.; Yuh, E.L.; Mukherjee, P.; Valadka, A.B.; et al. Comparing Plasma Phospho Tau, Total Tau, and Phospho Tau-Total Tau Ratio as Acute and Chronic Traumatic Brain Injury Biomarkers. JAMA Neurol. 2017, 74, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Mez, J.; Daneshvar, D.H.; Kiernan, P.T.; Abdolmohammadi, B.; Alvarez, V.E.; Huber, B.R.; Alosco, M.L.; Solomon, T.M.; Nowinski, C.J.; McHale, L.; et al. Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football. JAMA 2017, 318, 360–370. [Google Scholar] [PubMed]
- Shively, S.; Scher, A.I.; Perl, D.P.; Diaz-Arrastia, R. Dementia resulting from traumatic brain injury: What is the pathology? Arch. Neurol. 2012, 69, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, S.K.; Hasegawa, R.B.; Rabinowitz, A.R.; Whyte, J.; Roan, C.L.; Tabatabaei, A.; Baiocchi, M.; Karlawish, J.H.; Master, C.L.; Small, D.S. Association of Playing High School Football With Cognition and Mental Health Later in Life. JAMA Neurol. 2017, 74, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Shahim, P.; Gren, M.; Liman, V.; Andreasson, U.; Norgren, N.; Tegner, Y.; Mattsson, N.; Andreasen, N.; Ost, M.; Zetterberg, H.; et al. Serum neurofilament light protein predicts clinical outcome in traumatic brain injury. Sci. Rep. 2016, 6, 36791. [Google Scholar] [CrossRef] [PubMed]
- Shahim, P.; Tegner, Y.; Gustafsson, B.; Gren, M.; Arlig, J.; Olsson, M.; Lehto, N.; Engstrom, A.; Hoglund, K.; Portelius, E.; et al. Neurochemical Aftermath of Repetitive Mild Traumatic Brain Injury. JAMA Neurol. 2016, 73, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Bernick, C. Signs of Long-Term Brain Injury in Blood of Boxers, Mixed Martial Arts Fighters. In Proceedings of the American Academy of Neurology Sports Concussion Conference, Jacksonville, FL, USA, 14–16 July 2017. [Google Scholar]
- Neselius, S.; Zetterberg, H.; Blennow, K.; Marcusson, J.; Brisby, H. Increased CSF levels of phosphorylated neurofilament heavy protein following bout in amateur boxers. PLoS ONE 2013, 8, e81249. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Smith, D.H.; Blennow, K. Biomarkers of mild traumatic brain injury in cerebrospinal fluid and blood. Nat. Rev. Neurol. 2013, 9, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.M.; Jones, M.T.; Kirk, K.M.; Gable, D.A.; Repshas, J.T.; Johnson, T.A.; Andreasson, U.; Norgren, N.; Blennow, K.; Zetterberg, H. Serum Neurofilament Light in American Football Athletes over the Course of a Season. J. Neurotrauma 2016, 33, 1784–1789. [Google Scholar] [CrossRef] [PubMed]
- Nizamutdinov, D.; Shapiro, L.A. Overview of Traumatic Brain Injury: An Immunological Context. Brain Sci. 2017, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.G.; Satapathy, S.S.; Dagro, A.M.; McKee, P.J. Numerical Study of Head/helmet interaction due to blast loading. In Proceedings of the ASME International Mechanical Engineering Congress & Exposition, San Diego, CA, USA, 15–21 November 2013. [Google Scholar]
- Roth, T.L.; Nayak, D.; Atanasijevic, T.; Koretsky, A.P.; Latour, L.L.; McGavern, D.B. Transcranial amelioration of inflammation and cell death after brain injury. Nature 2014, 505, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.K.; Yang, Z.; Yue, J.K.; Zhang, Z.; Winkler, E.A.; Puccio, A.M.; Diaz-Arrastia, R.; Lingsma, H.F.; Yuh, E.L.; Mukherjee, P. Plasma anti-glial fibrillary acidic protein autoantibody levels during the acute and chronic phases of traumatic brain injury: A transforming research and clinical knowledge in traumatic brain injury pilot study. J. Neurotrauma 2016, 33, 1270–1277. [Google Scholar] [CrossRef] [PubMed]
- Bargerstock, E.; Puvenna, V.; Iffland, P.; Falcone, T.; Hossain, M.; Vetter, S.; Man, S.; Dickstein, L.; Marchi, N.; Ghosh, C. Is peripheral immunity regulated by blood-brain barrier permeability changes? PLoS ONE 2014, 9, e101477. [Google Scholar] [CrossRef] [PubMed]
- Marchi, N.; Bazarian, J.J.; Puvenna, V.; Janigro, M.; Ghosh, C.; Zhong, J.; Zhu, T.; Blackman, E.; Stewart, D.; Ellis, J. Consequences of repeated blood-brain barrier disruption in football players. PLoS ONE 2013, 8, e56805. [Google Scholar] [CrossRef] [PubMed]
- Grunwald, G.B.; Klein, R.; Simmonds, M.A.; Kornguth, S.E. Autoimmune basis for visual paraneoplastic syndrome in patients with small-cell lung carcinoma. Lancet 1985, 1, 658–661. [Google Scholar] [CrossRef]
- Kornguth, S.E.; Kalinke, T.; Grunwald, G.B.; Schutta, H.; Dahl, D. Anti-neurofilament antibodies in the sera of patients with small cell carcinoma of the lung and with visual paraneoplastic syndrome. Cancer Res. 1986, 46, 2588–2595. [Google Scholar] [PubMed]
- Spear, P.D.; Miller, S.; Vielhuber, K.; Kornguth, S.E. Visual field defects in cats with neonatal or adult immunological loss of retinal ganglion cells. Brain Res. 1986, 368, 154–157. [Google Scholar] [CrossRef]
- Williams, R.W.; Crabtree, J.W.; Chalupa, L.M.; Spear, P.D.; Kornguth, S.E. Selectivity of antibody-mediated destruction of axons in the cat’s optic nerve. Brain Res. 1985, 336, 57–66. [Google Scholar] [CrossRef]
- Crabtree, J.W.; Spear, P.D.; McCall, M.A.; Tong, L.; Jones, K.R.; Kornguth, S.E. Dose-response analysis of effects of antibodies to large ganglion cells on the cat’s retinogeniculate pathways. J. Neurosci. 1986, 6, 1199–1210. [Google Scholar] [PubMed]
- Reuter, M.; Schmansky, N.J.; Rosas, H.D.; Fischl, B. Within-subject template estimation for unbiased longitudinal image analysis. Neuroimage 2012, 61, 1402–1418. [Google Scholar] [CrossRef] [PubMed]
- Poldrack, R.A.; Laumann, T.O.; Koyejo, O.; Gregory, B.; Hover, A.; Chen, M.Y.; Gorgolewski, K.J.; Luci, J.; Joo, S.J.; Boyd, R.L.; et al. Long-term neural and physiological phenotyping of a single human. Nat. Commun. 2015, 6, 8885. [Google Scholar] [CrossRef] [PubMed]
- Hedman, A.M.; van Haren, N.E.; Schnack, H.G.; Kahn, R.S.; Hulshoff Pol, H.E. Human brain changes across the life span: A review of 56 longitudinal magnetic resonance imaging studies. Hum. Brain Mapp. 2012, 33, 1987–2002. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, M.; Tam, R.; Hernandez-Torres, E.; Martin, N.; Perera, W.; Zhao, Y.; Shahinfard, E.; Dadachanji, S.; Taunton, J.; Li, D.K.; et al. A Prospective Pilot Investigation of Brain Volume, White Matter Hyperintensities, and Hemorrhagic Lesions after Mild Traumatic Brain Injury. Front. Neurol. 2016, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Ghidaoui, M.; Zhao, M.; McInnis, D.; Axworth, D. A Review of Water Hammer Theory and Practice. Appl. Mech. Rev. 2005, 58, 49–75. [Google Scholar] [CrossRef]
- Budday, S.; Nay, R.; de Rooij, R.; Steinmann, P.; Wyrobek, T.; Ovaert, T.C.; Kuhl, E. Mechanical properties of gray and white matter brain tissue by indentation. J. Mech. Behav. Biomed. Mater. 2015, 46, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Puvenna, V.; Janigro, D. Biomarkers of Traumatic Brain Injury and Their Relationship to Pathology. In Translational Research in Traumatic Brain Injury; Laskowitz, D., Grant, G., Eds.; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Puvenna, V.; Engeler, M.; Banjara, M.; Brennan, C.; Schreiber, P.; Dadas, A.; Bahrami, A.; Solanki, J.; Bandyopadhyay, A.; Morris, J.K. Is phosphorylated tau unique to chronic traumatic encephalopathy? Phosphorylated tau in epileptic brain and chronic traumatic encephalopathy. Brain Res. 2016, 1630, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.L.; Partington, C.; Robbins, M.; Graziano, F.; Turski, P.; Kornguth, S. Magnetic resonance imaging of central nervous system lesions in patients with lupus erythematosus. Correlation with clinical remission and antineurofilament and anticardiolipin antibody titers. Arthritis Rheum. 1991, 34, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Robbins, M.L.; Kornguth, S.E.; Bell, C.L.; Kalinke, T.; England, D.; Turski, P.; Graziano, F.M. Antineurofilament antibody evaluation in neuropsychiatric systemic lupus erythematosus. Combination with anticardiolipin antibody assay and magnetic resonance imaging. Arthritis Rheum. 1988, 31, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Ontiveros, D.G.; Tajiri, N.; Acosta, S.; Giunta, B.; Tan, J.; Borlongan, C.V. Microglia activation as a biomarker for traumatic brain injury. Front. Neurol. 2013, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F. Immune function of microglia. Glia 2001, 36, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Kornguth, S.; Bersu, E.; Mack, K. MHC class I gene expression. Science 1995, 270, 720–721. [Google Scholar] [PubMed]
- Kornguth, S.; Mack, K.J.; Bersu, E. Relationship between the neural dysgenesis and increased production of class I MHC H-2Kk mRNA and protein in neurons of murine trisomy 16 fetuses. Biochem. Biophys. Res. Commun. 1991, 179, 102–107. [Google Scholar] [CrossRef]
- Neumann, H.; Cavalie, A.; Jenne, D.E.; Wekerle, H. Induction of MHC class I genes in neurons. Science 1995, 269, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.; Boucraut, J.; Hahnel, C.; Misgeld, T.; Wekerle, H. Neuronal control of MHC class II inducibility in rat astrocytes and microglia. Eur. J. Neurosci. 1996, 8, 2582–2590. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.; Schmidt, H.; Cavalie, A.; Jenne, D.; Wekerle, H. Major histocompatibility complex (MHC) class I gene expression in single neurons of the central nervous system: Differential regulation by interferon (IFN)-γ and tumor necrosis factor (TNF)-α. J. Exp. Med. 1997, 185, 305–316. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, L.P.; Lampson, L.A. Local neurochemicals and site-specific immune regulation in the CNS. J. Neuropathol. Exp. Neurol. 2000, 59, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Byrne, L.M.; Rodrigues, F.B.; Blennow, K.; Durr, A.; Leavitt, B.R.; Roos, R.A.C.; Scahill, R.I.; Tabrizi, S.J.; Zetterberg, H.; Langbehn, D.; et al. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington’s disease: A retrospective cohort analysis. Lancet Neurol. 2017, 16, 601–609. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Kochanek, P.M.; Clark, R.S.; Ciallella, J.R.; Dixon, C.E. Secondary Injury after Head Trauma: Subacute and Long-term Mechanisms. Semin. Clin. Neuropsychiatry 1998, 3, 176–185. [Google Scholar] [PubMed]
- Lu, D.; Goussev, A.; Chen, J.; Pannu, P.; Li, Y.; Mahmood, A.; Chopp, M. Atorvastatin reduces neurological deficit and increases synaptogenesis, angiogenesis, and neuronal survival in rats subjected to traumatic brain injury. J. Neurotrauma 2004, 21, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lu, D.; Jiang, H.; Xiong, Y.; Qu, C.; Li, B.; Mahmood, A.; Zhou, D.; Chopp, M. Increase in phosphorylation of Akt and its downstream signaling targets and suppression of apoptosis by simvastatin after traumatic brain injury. J. Neurosurg. 2008, 109, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.C.; Lucke-Wold, B.; Lucke-Wold, N.; Elliott, A.S.; Logsdon, A.F.; Rosen, C.L.; Huber, J.D. Neuroprotection for ischemic stroke: Moving past shortcomings and identifying promising directions. Int. J. Mol. Sci. 2013, 14, 1890–1917. [Google Scholar] [CrossRef] [PubMed]
- Elewa, H.F.; Hilali, H.; Hess, D.C.; Machado, L.S.; Fagan, S.C. Minocycline for short-term neuroprotection. Pharmacotherapy 2006, 26, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Fagan, S.C.; Waller, J.L.; Nichols, F.T.; Edwards, D.J.; Pettigrew, L.C.; Clark, W.M.; Hall, C.E.; Switzer, J.A.; Ergul, A.; Hess, D.C. Minocycline to improve neurologic outcome in stroke (MINOS): A dose-finding study. Stroke 2010, 41, 2283–2287. [Google Scholar] [CrossRef] [PubMed]
- Seifert, H.; Collier, L.; Chapman, C.; Benkovic, S.; Willing, A.; Pennypacker, K. Proinflammatory Interferon gamma signaling is directly associated with stroke induced neurodegeneration. J. Neuroimmune Pharmacol. 2014, 9, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Sironi, L.; Cimino, M.; Guerrini, U.; Calvio, A.M.; Lodetti, B.; Asdente, M.; Balduini, W.; Paoletti, R.; Tremoli, E. Treatment with statins after induction of focal ischemia in rats reduces the extent of brain damage. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Vedantam, S.; Moller, A. Minocycline: A Novel Stroke Therapy. J. Neurol. Stroke 2015, 2, 73. [Google Scholar] [CrossRef]
- Kantarci, O.H.; Hebrink, D.D.; Schaefer-Klein, J.; Sun, Y.; Achenbach, S.; Atkinson, E.J.; Heggarty, S.; Cotleur, A.C.; de Andrade, M.; Vandenbroeck, K.; et al. Interferon gamma allelic variants: Sex-biased multiple sclerosis susceptibility and gene expression. Arch. Neurol. 2008, 65, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Harbour, S.N.; Kolde, R.; Latorre, I.J.; Tun, H.M.; Schoeb, T.R.; Turner, H.; Moon, J.J.; Khafipour, E.; Xavier, R.J.; et al. Selective Induction of Homeostatic Th17 Cells in the Murine Intestine by Cholera Toxin Interacting with the Microbiota. J. Immunol. 2017, 199, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Ottum, P.A.; Arellano, G.; Reyes, L.I.; Iruretagoyena, M.; Naves, R. Opposing Roles of Interferon-Gamma on Cells of the Central Nervous System in Autoimmune Neuroinflammation. Front. Immunol. 2015, 6, 539. [Google Scholar] [CrossRef] [PubMed]
- Dretsch, M.N.; Williams, K.; Emmerich, T.; Crynen, G.; Ait-Ghezala, G.; Chaytow, H.; Mathura, V.; Crawford, F.C.; Iverson, G.L. Brain-derived neurotropic factor polymorphisms, traumatic stress, mild traumatic brain injury, and combat exposure contribute to postdeployment traumatic stress. Brain Behav. 2016, 6, e00392. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Player | Memory Verbal | Memory Visual | Motor | Reaction Time | Impulse Control | Total Symptoms | Cognitive Efficiency |
|---|---|---|---|---|---|---|---|
| 10 | 80 | 83 | 48 | 0.47 | 8 | 10 | 0.28 |
| 11 | 100 | 88 | 51 | 0.6 | 7 | 3 | 0.37 |
| 12 | 88 | 77 | 38 | 0.54 | 14 | 5 | 0.37 |
| 13 | 85 | 58 | 33 | 0.72 | 1 | 3 | 0.37 |
| 14 | 85 | 70 | 43 | 0.68 | 3 | 28 | 0.24 |
| 15 | 77 | 73 | 42 | 0.54 | 7 | 6 | 0.2 |
| 15 * | 62 | 73 | 22 | 1.17 | 2 | 18 | 0.08 |
| 15 ** | 88 | 83 | 41 | 0.6 | 7 | 1 | 0.49 |
| 16 | 100 | 85 | 37 | 0.56 | 2 | 5 | 0.35 |
| 17 | 100 | 94 | 42 | 0.64 | 3 | 1 | 0.39 |
| 29 | 85 | 57 | 41 | 0.5 | 6 | 6 | 0.47 |
| 29 * | 88 | 74 | 38 | 0.49 | 6 | 0 | 0.37 |
| 30 | 100 | 80 | 46 | 0.7 | 7 | 13 | 0.02 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kornguth, S.; Rutledge, N.; Perlaza, G.; Bray, J.; Hardin, A. A Proposed Mechanism for Development of CTE Following Concussive Events: Head Impact, Water Hammer Injury, Neurofilament Release, and Autoimmune Processes. Brain Sci. 2017, 7, 164. https://doi.org/10.3390/brainsci7120164
Kornguth S, Rutledge N, Perlaza G, Bray J, Hardin A. A Proposed Mechanism for Development of CTE Following Concussive Events: Head Impact, Water Hammer Injury, Neurofilament Release, and Autoimmune Processes. Brain Sciences. 2017; 7(12):164. https://doi.org/10.3390/brainsci7120164
Chicago/Turabian StyleKornguth, Steven, Neal Rutledge, Gabe Perlaza, James Bray, and Allen Hardin. 2017. "A Proposed Mechanism for Development of CTE Following Concussive Events: Head Impact, Water Hammer Injury, Neurofilament Release, and Autoimmune Processes" Brain Sciences 7, no. 12: 164. https://doi.org/10.3390/brainsci7120164
APA StyleKornguth, S., Rutledge, N., Perlaza, G., Bray, J., & Hardin, A. (2017). A Proposed Mechanism for Development of CTE Following Concussive Events: Head Impact, Water Hammer Injury, Neurofilament Release, and Autoimmune Processes. Brain Sciences, 7(12), 164. https://doi.org/10.3390/brainsci7120164