
Metabolic Syndrome and Parkinson’s Disease: Two Villains Join Forces
 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Study Assessments
- Obesity: body mass index (BMI) > 30 kg/m2.
- Hyperglycemia: fasting glucose ≥ 100 mg/dL, previously diagnosed diabetes mellitus, or current use of antidiabetic medication.
- Hypertriglyceridemia: triglycerides ≥ 150 mg/dL, or prior diagnosis/treatment for elevated triglycerides.
- Low HDL cholesterol: HDL < 40 mg/dL in men or <50 mg/dL in women, or current treatment for low HDL levels.
- Hypertension: blood pressure ≥ 130/85 mmHg, previously diagnosed hypertension, or use of antihypertensive therapy.
2.3. Genetic Assessment in PD Patients with or Without MetS
2.4. Statistical Analysis
3. Results
3.1. Baseline Demographic and Clinical Characteristics
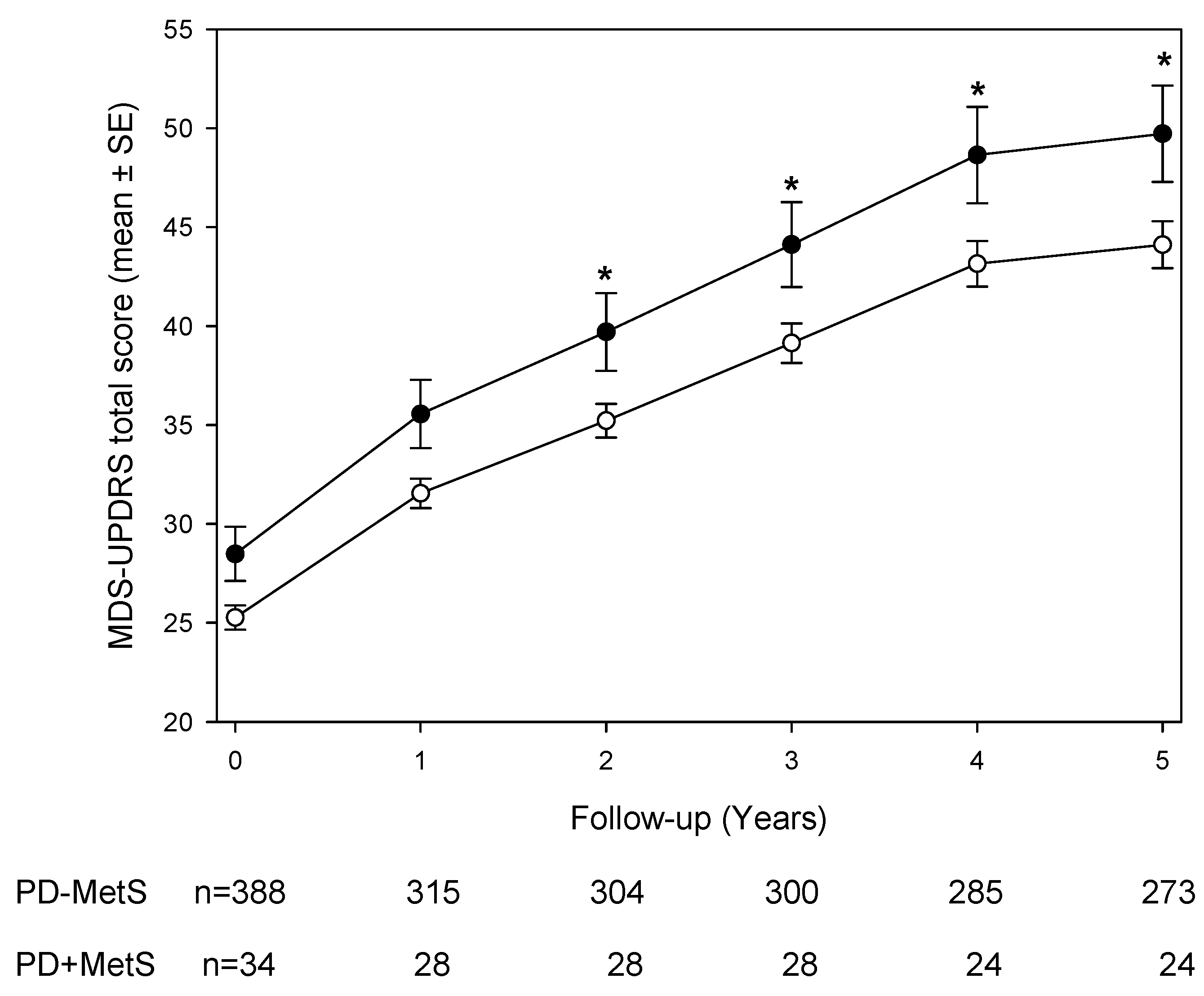
3.2. Longitudinal MDS-UPDRS Scores and Subscales
3.3. Genetic Association Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The Parkinson Progression Marker Initiative. The Parkinson Progression Marker Initiative (PPMI). Prog. Neurobiol. 2010, 95, 629–635. [Google Scholar]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E.; Espay, A.J. Disease Modification in Parkinson’s Disease: Current Approaches, Challenges, and Future Considerations. Mov. Disord. 2018, 33, 660–677. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Brewer, H.B., Jr.; Cleeman, J.I.; Smith, S.C., Jr.; Lenfant, C. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 2004, 109, 433–438. [Google Scholar] [CrossRef]
- Otero-Losada, M.; Cao, G.; Gómez Llambí, H.; Nobile, M.H.; Azzato, F.; Milei, J. Oxidative Stress and Antioxidants in Experimental Metabolic Syndrome. In Biochemistry of Oxidative Stress; Gelpi, R.J., Boveris, A., Poderoso, J.J., Eds.; Springer: Cham, Switzerland, 2016; pp. 375–390. [Google Scholar]
- Xia, X.; Wang, Y.; Zheng, J.C. Extracellular vesicles, from the pathogenesis to the therapy of neurodegenerative diseases. Transl. Neurodegener. 2022, 11, 53. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, M.A.; Labandeira, C.M.; Lago-Baameiro, N.; Valenzuela, R.; Pardo, M.; Labandeira-Garcia, J.L.; Rodriguez-Perez, A.I. Extracellular vesicles and their renin-angiotensin cargo as a link between metabolic syndrome and Parkinson’s disease. Antioxidants 2023, 12, 12. [Google Scholar] [CrossRef]
- Meléndez-Flores, J.D.; Castillo-Torres, S.A.; Cerda-Contreras, C.; Chávez-Luévanos, B.; Estrada-Bellmann, I. Clinical features of metabolic syndrome in patients with Parkinson’s disease. Rev. Neurol. 2021, 72, 9–15. [Google Scholar]
- Peng, Z.; Dong, S.; Tao, Y.; Huo, Y.; Zhou, Z.; Huang, W.; Qu, H.; Liu, J.; Chen, Y.; Xu, Z.; et al. Metabolic syndrome contributes to cognitive impairment in patients with Parkinson’s disease. Park. Relat. Disord. 2018, 55, 68–74. [Google Scholar] [CrossRef]
- Leehey, M.; Luo, S.; Sharma, S.; Wills, A.A.; Bainbridge, J.L.; Wong, P.S.; Simon, D.K.; Schneider, J.; Zhang, Y.; Perez, A.; et al. Association of metabolic syndrome and change in Unified Parkinson’s Disease Rating Scale scores. Neurology 2017, 89, 1789–1794. [Google Scholar] [CrossRef]
- Li, L.Y.; Liu, S.F.; Zhuang, J.L.; Li, M.M.; Huang, Z.P.; Chen, Y.H.; Chen, X.R.; Chen, C.N.; Lin, S.; Ye, L.C. Recent research progress on metabolic syndrome and risk of Parkinson’s disease. Rev. Neurosci. 2023, 34, 719–735. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Goetz, C.G.; Tilley, B.C.; Shaftman, S.R.; Stebbins, G.T.; Fahn, S.; Martinez-Martin, P.; Poewe, W.; Sampaio, C.; Stern, M.B.; Dodel, R.; et al. Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Scale presentation and clinimetric testing results. Mov. Disord. 2008, 23, 2129–2170. [Google Scholar] [CrossRef] [PubMed]
- Wang, M. Generalized estimating equations in longitudinal data analysis: A review and recent developments. Adv. Stat. 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Nam, G.E.; Kim, S.M.; Han, K.; Kim, N.H.; Chung, H.S.; Kim, J.W.; Han, B.; Cho, S.J.; Yu, J.H.; Park, Y.G.; et al. Metabolic syndrome and risk of Parkinson disease: A nationwide cohort study. PLoS Med. 2018, 15, e1002640. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.H.; Lee, S.; Yoon, J.-H. Metabolic syndrome and Parkinson’s disease incidence: A nationwide study using propensity score matching. Metab. Syndr. Relat. Disord. 2021, 19, 1–7. [Google Scholar] [CrossRef]
- Souza, A.; Barros, W.M.A.; Silva, J.M.L.; Silva, M.R.M.; Silva, A.B.J.; Fernandes, M.S.d.S.; dos Santos, M.E.R.A.; da Silva, M.L.; Carmo, T.S.D.; Silva, R.K.P.; et al. Effect of metabolic syndrome on Parkinson’s disease: A systematic review. Clinics 2021, 76, e3379. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Zhou, R.; Liu, D.; Cui, M.; Yu, K.; Yang, H.; Li, L.; Liu, J.; Chen, Y.; Hong, W.; et al. Association between metabolic syndrome and mild parkinsonian signs progression in the elderly. Front. Aging Neurosci. 2021, 13, 722836. [Google Scholar]
- Athauda, D.; Maclagan, K.; Skene, S.S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Budnik, N.; Zampedri, L.; Dickson, J.; et al. Exenatide once weekly versus placebo in Parkinson’s disease: A randomized, double-blind, placebo-controlled trial. Lancet 2017, 390, 1664–1675. [Google Scholar] [CrossRef]
- Perez-Lloret, S.; Otero-Losada, M.; Toblli, J.E.; Capani, F. Renin-angiotensin system as a potential target for new therapeutic approaches in Parkinson’s disease. Expert. Opin. Investig. Drugs 2017, 26, 1163–1173. [Google Scholar] [CrossRef]
- de Kloet, A.D.; Krause, E.G.; Woods, S.C. The renin angiotensin system and the metabolic syndrome. Physiol. Behav. 2010, 100, 525–534. [Google Scholar] [CrossRef]
- Auluck, P.K.; Chan, H.Y.; Trojanowski, J.Q.; Lee, V.M.; Bonini, N.M. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model. Science 2002, 295, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Wolfer, D.P.; Lipp, H.P.; Büeler, H. Hsp70 gene transfer inhibits MPTP-induced degeneration in mouse PD model. Mol. Ther. 2005, 11, 80–88. [Google Scholar] [CrossRef]
- Klucken, J.; Shin, Y.; Masliah, E.; Hyman, B.T.; McLean, P.J. Hsp70 reduces alpha-synuclein aggregation. J. Biol. Chem. 2004, 279, 25497–25502. [Google Scholar] [CrossRef]
- Roodveldt, C.; Bertoncini, C.W.; Andersson, A.; van der Goot, A.T.; Hsu, S.T.; Fernández-Montesinos, R.; de Jong, J.; van Ham, T.J.; Nollen, E.A.; Pozo, D.; et al. Stabilization of Hsp70/α-synuclein complex by Hip. EMBO J. 2009, 28, 3758–3770. [Google Scholar] [CrossRef] [PubMed]
- Østvold, A.C.; Grundt, K.; Wiese, C. NUCKS1 is a highly modified, chromatin-associated protein. Biochem. J. 2022, 479, 1205–1220. [Google Scholar] [CrossRef] [PubMed]
- Hock, R.; Furusawa, T.; Ueda, T.; Bustin, M. HMG chromosomal proteins in development and disease. Trends Cell Biol. 2007, 17, 72–79. [Google Scholar] [CrossRef]
- Stoka, V.; Turk, V.; Turk, B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res. Rev. 2016, 32, 22–37. [Google Scholar] [CrossRef]
- Drobny, A.; Boros, F.A.; Balta, D.; Huarcaya, S.P.; Caylioglu, D.; Qazi, N.; Vandrey, J.; Schneider, Y.; Dobert, J.P.; Pitcairn, C.; et al. Reciprocal effects of alpha-synuclein aggregation and lysosomal homeostasis. Transl. Neurodegener. 2023, 12, 31. [Google Scholar] [CrossRef]
- Araujo, T.F.; Cordeiro, A.V.; Vasconcelos, D.A.; Vitzel, K.F.; Silva, V.R. Role of cathepsin B in autophagy during obesity: A systematic review. Life Sci. 2018, 209, 274–281. [Google Scholar] [CrossRef]
- Grenier-Larouche, T.; Kwee, L.C.; Deleye, Y.; Leon-Mimila, P.; Walejko, J.M.; McGarrah, R.W.; Marceau, S.; Trahan, S.; Racine, C.; Carpentier, A.C.; et al. Altered branched-chain alpha-keto acid metabolism is a feature of NAFLD in individuals with severe obesity. JCI Insight 2022, 7, e159204. [Google Scholar] [CrossRef] [PubMed]
- Jones-Tabah, J.; He, K.; Senkevich, K.; Karpilovsky, N.; Deyab, G.; Cousineau, Y.; Nikanorova, D.; Goldsmith, T.; Del Cid Pellitero, E.; Chen, C.X.; et al. The Parkinson’s disease risk gene cathepsin B promotes fibrillar alpha-synuclein clearance, lysosomal function and glucocerebrosidase activity in dopaminergic neurons. Mol. Neurodegener. 2024, 19, 88. [Google Scholar] [CrossRef] [PubMed]
- Burrell, J.A.; Stephens, J.M. KAT8, lysine acetyltransferase 8, is required for adipocyte differentiation in vitro. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166103. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Gupta, S.; Dinsmore, I.; Kollu, A.; Cawley, A.M.; Anwar, M.Y.; Chen, H.H.; Petty, L.E.; Seshadri, S.; Graff, M.; et al. Integrating Genetic and Transcriptomic Data to Identify Genes Underlying Obesity Risk Loci. medRxiv 2024. [Google Scholar] [CrossRef]
- Jishi, A.; Hu, D.; Shang, Y.; Wang, R.; Gunzler, S.A.; Qi, X. BCKDK loss impairs mitochondrial Complex I activity and drives alpha-synuclein aggregation in models of Parkinson’s disease. Acta Neuropathol. Commun. 2024, 12, 198. [Google Scholar] [CrossRef]

{kind=link}
| PD + MetS (n = 34) | PD–MetS (n = 389) | p-Value | |
|---|---|---|---|
| Patients’ characteristics | |||
| Male sex | 32 (94%) | 246 (63%) | <0.01 |
| Age | 64.9 ± 9.4 | 61.4 ± 9.7 | 0.04 |
| PD family history | 8 (24%) | 126 (32%) | 0.38 |
| Hoehn and Yahr score | |||
| I | 8 (24%) | 177 (46%) | 0.01 |
| II | 26 (76%) | 212 (54%) | |
| MDS-UPDRS Total score | 30.7 ± 9.6 | 26.5 ± 11.2 | 0.03 |
| MetS components | |||
| Body mass Index | 29 (85%) | 241 (62%) | <0.01 |
| Hyperglycemia | 17 (50%) | 5 (1%) | <0.01 |
| Low HDL-C | 13 (38%) | 23 (6%) | <0.01 |
| Hypertriglyceridemia | 19 (56%) | 11 (3%) | <0.01 |
| Hypertension | 28 (83%) | 255 (66%) | 0.02 |
| Year | Part I | Part II | Part III | Part IV | ||||
|---|---|---|---|---|---|---|---|---|
| PD–MetS | PD + MetS | PD–MetS | PD + MetS | PD–MetS | PD + MetS | PD–MetS | PD + MetS | |
| 0 | 1.2 ± 1.6 | 1.1 ± 1.2 | 5.8 ± 4.1 | 7.1 ± 4.7 ** | 20.7 ± 9.0 | 23.6 ± 7.2 ** | - | - |
| 1 | 1.5 ± 1.8 | 1.8 ± 1.7 | 7.2 ± 5.0 | 9.4 ± 4.5 * | 24.2 ± 10.4 | 28.4 ± 10.0 * | 0.2 ± 0.9 | 0.5 ± 1.3 |
| 2 | 1.9 ± 2.3 | 1.9 ± 2.0 | 7.8 ± 5.3 | 9.4 ± 5.2 | 26.4 ± 11.2 | 34.8 ± 10.7 ** | 0.5 ± 1.4 | 0.6 ± 1.7 |
| 3 | 1.9 ± 2.3 | 2.3 ± 1.9 | 8.7 ± 5.7 | 10.4 ± 5.5 | 27.7 ± 12.1 | 35.6 ± 9.9 ** | 0.7 ± 1.7 | 1.1 ± 2.1 |
| 4 | 2.2 ± 2.5 | 2.5 ± 3.6 | 9.7 ± 6.7 | 11.8 ± 5.5 | 30.4 ± 12.8 | 35.2 ± 9.7 * | 1.4 ± 2.5 | 1.4 ± 2.0 |
| 5 | 2.3 ± 3.0 | 2.3 ± 2.0 | 9.9 ± 6.8 | 11.7 ± 5.9 | 29.9 ± 13.6 | 35.7 ± 9.5 * | 2.0 ± 2.8 | 1.8 ± 2.0 |
| Number of Risk Alleles for Each SNP | Overall | No MetS | MetS | Chi-Sq | Additive Model | Dominant Model | Recessive Model | Full Model |
|---|---|---|---|---|---|---|---|---|
| (N = 409) | (N = 376) | (N = 33) | p-Value | OR (95% CI) | OR (95% CI) | OR (95% CI) | OR (95% CI) | |
| p-Value (AIC) | p-Value (AIC) | p-Value (AIC) | p-Value | |||||
| GBA_N370S_rs76763715 | ||||||||
| 0 | 401 (98.0%) | 370 (98.4%) | 31 (93.9%) | 0.003 | 4.22 (0.99, 16.19) | 3.98 (0.57, 18.12) | - | 2.91 (0.61, 12.70) |
| 1 | 7 (1.7%) | 6 (1.6%) | 1 (3.0%) | 0.03 (229.6) | 0.10 (231.25) | 0.141 | ||
| 2 | 1 (0.2%) | 0 (0%) | 1 (3.0%) | SELECTED | ||||
| NUCKS1_rs823118 | ||||||||
| 0 | 135 (33.0%) | 128 (34.0%) | 7 (21.2%) | 0.011 | 0.98 (0.59, 1.62) | 1.92 (0.85, 4.90) | 0.26 (0.04, 0.88) | 0.21 (0.03, 0.77) |
| 1 | 197 (48.2%) | 173 (46.0%) | 24 (72.7%) | 0.93 (233.29) | 0.14 (203.97) | 0.05 (228.57) | 0.043 | |
| 2 | 77 (18.8%) | 75 (19.9%) | 2 (6.1%) | SELECTED | ||||
| CTSB_rs1293298 | ||||||||
| 0 | 237 (57.9%) | 211 (56.1%) | 26 (78.8%) | 0.027 | 0.62 (0.38, 1.01) | 0.34 (0.13, 0.77) | - | 0.35 (0.13, 0.84) |
| 1 | 143 (35.0%) | 136 (36.2%) | 7 (21.2%) | 0.06 (229.75) | 0.01 (226.49) | 0.025 | ||
| 2 | 29 (7.1%) | 29 (7.7%) | 0 (0%) | SELECTED | ||||
| ZNF646.KAT8.BCKDK_rs14235 | ||||||||
| 0 | 147 (35.9%) | 134 (35.6%) | 13 (39.4%) | 0.023 | 1.29 (0.77, 2.14) | 0.85 (0.41, 1.81) | 2.59 (1.12, 5.62) | 3.06 (1.24, 7.29) |
| 1 | 198 (48.4%) | 188 (50.0%) | 10 (30.3%) | 0.33 (232.46) | 0.67 (233.21) | 0.02 (228.48) | 0.012 | |
| 2 | 64 (15.6%) | 54 (14.4%) | 10 (30.3%) | SELECTED | ||||
| COMT_rs4633 | ||||||||
| 0 | 116 (28.4%) | 104 (27.7%) | 12 (36.4%) | 0.105 | 0.62 (0.38, 1.01) | 0.67 (0.32, 1.45) | 0.33 (0.10, 0.87) | 0.40 (0.11, 1.10) |
| 1 | 179 (43.8%) | 162 (43.1%) | 17 (51.5%) | 0.06 (229.75) | 0.29 (232.22) | 0.04 (228.25) | 0.107 | |
| 2 | 114 (27.9%) | 110 (29.3%) | 4 (12.1%) | SELECTED |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udovin, L.; Bordet, S.; Barbar, H.; Otero-Losada, M.; Pérez-Lloret, S.; Capani, F. Metabolic Syndrome and Parkinson’s Disease: Two Villains Join Forces. Brain Sci. 2025, 15, 706. https://doi.org/10.3390/brainsci15070706
Udovin L, Bordet S, Barbar H, Otero-Losada M, Pérez-Lloret S, Capani F. Metabolic Syndrome and Parkinson’s Disease: Two Villains Join Forces. Brain Sciences. 2025; 15(7):706. https://doi.org/10.3390/brainsci15070706
Chicago/Turabian StyleUdovin, Lucas, Sofía Bordet, Hanny Barbar, Matilde Otero-Losada, Santiago Pérez-Lloret, and Francisco Capani. 2025. "Metabolic Syndrome and Parkinson’s Disease: Two Villains Join Forces" Brain Sciences 15, no. 7: 706. https://doi.org/10.3390/brainsci15070706
APA StyleUdovin, L., Bordet, S., Barbar, H., Otero-Losada, M., Pérez-Lloret, S., & Capani, F. (2025). Metabolic Syndrome and Parkinson’s Disease: Two Villains Join Forces. Brain Sciences, 15(7), 706. https://doi.org/10.3390/brainsci15070706