The APOE–Microglia Axis in Alzheimer’s Disease: Functional Divergence and Therapeutic Perspectives—A Narrative Review


Abstract
1. Introduction
2. The Role of APOEε4 in Microglia Function Associated with AD
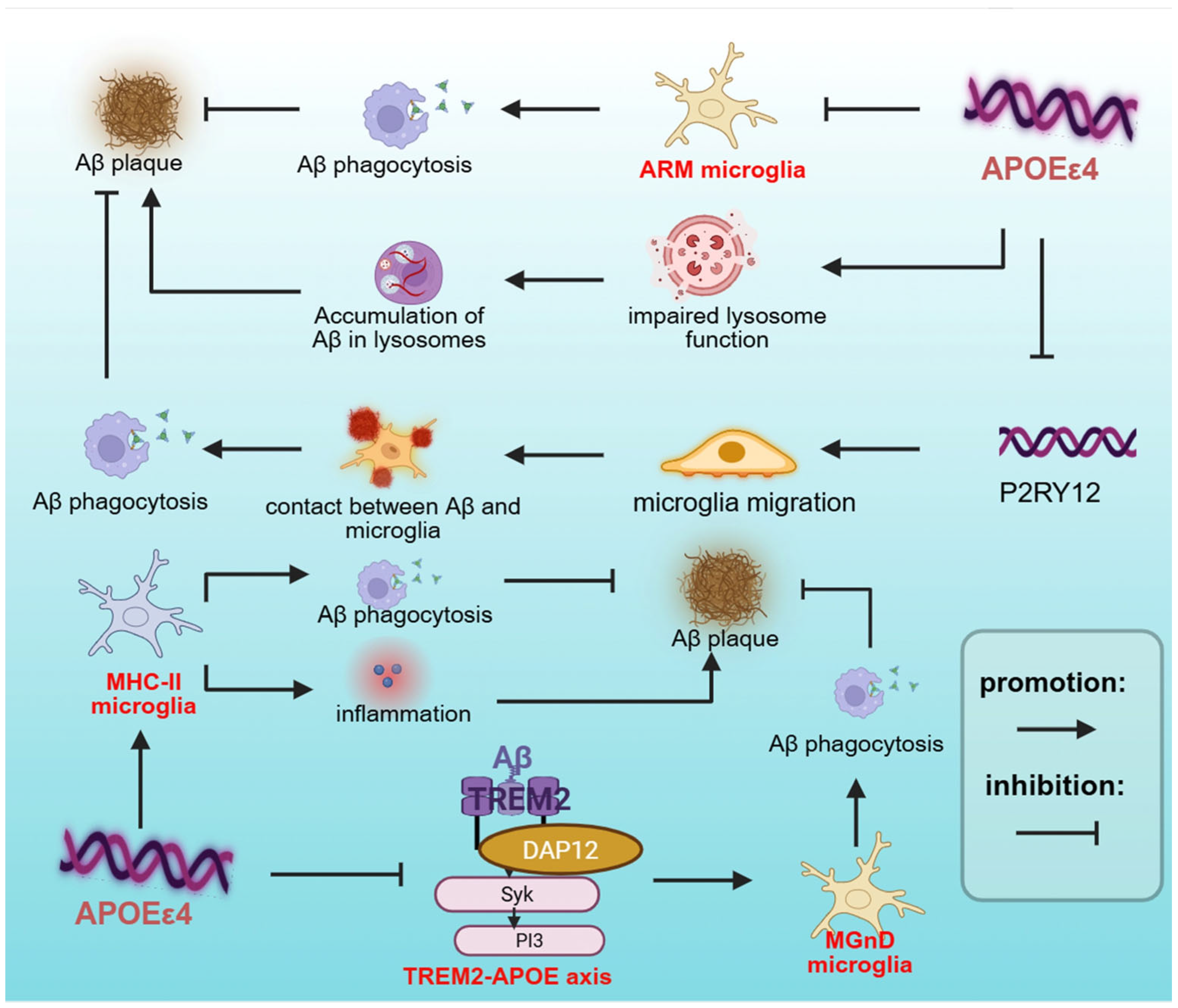
2.1. APOEε4 Regulates Microglial Activation States in AD Pathogenesis
2.2. APOΕε4 Regulates Microglial Inflammatory Responses
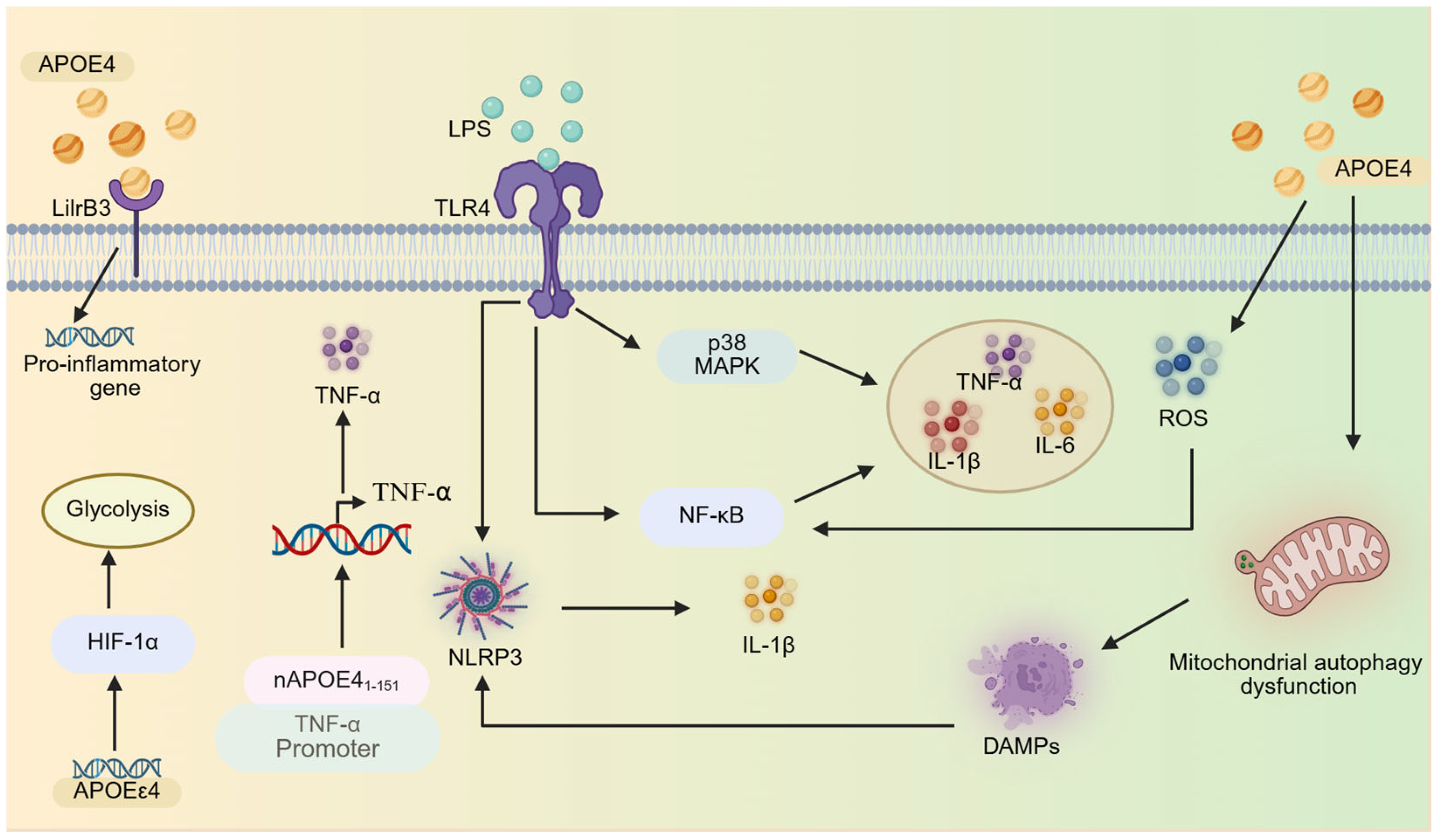
2.2.1. Inflammatory Pathway Activation
2.2.2. Metabolic Reprogramming
2.3. APOEε4 Regulates Microglial Phagocytosis in AD
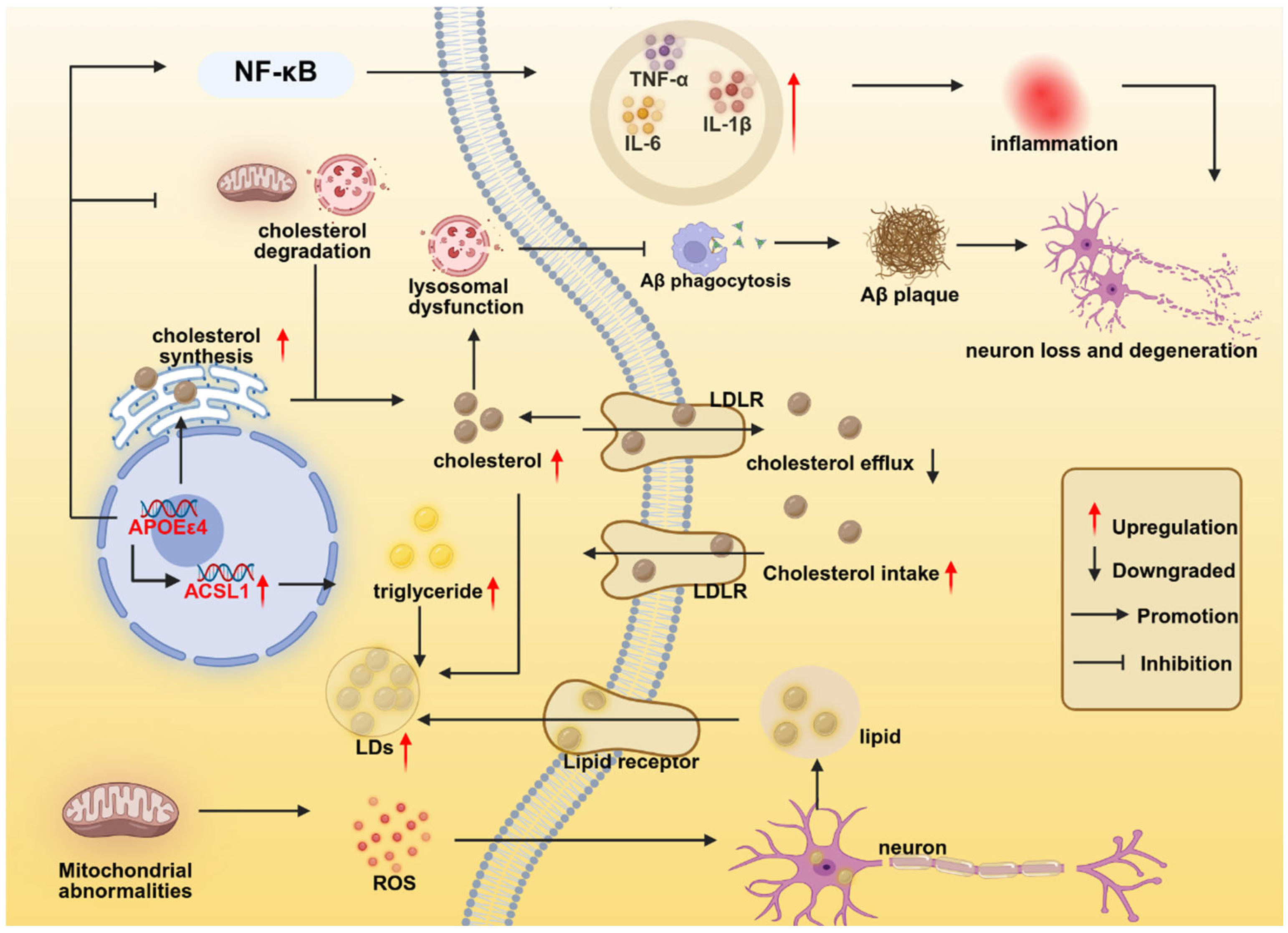
2.4. APOEε4 Regulates Microglial Lipid Metabolism
3. The Role of APOEε2 in Microglia Function Associated with AD
4. The Role of APOEε3 in Microglia Function Associated with AD
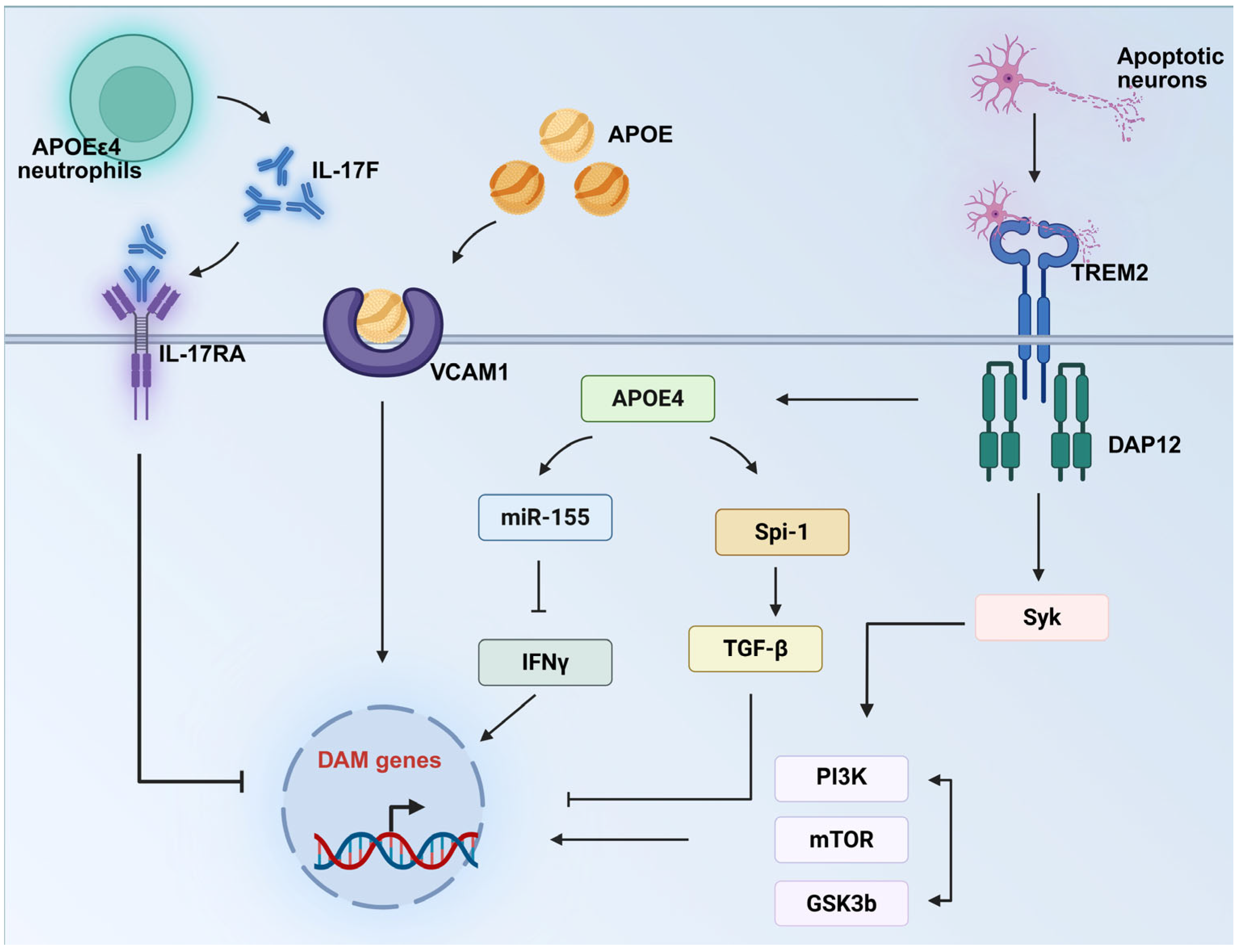
5. The APOE–Microglia Axis: A Novel Concept in Alzheimer’s Disease Pathogenesis and Therapy
6. Clinical Therapeutic Prospects
7. Future Research Directions
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rogus-Pulia, N.; Foundas, A.L.; Mueller, K.D. Alzheimer’s Disease. In Neurologic and Neurodegenerative Diseases of the Larynx; Weissbrod, P.A., Francis, D.O., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 177–189. [Google Scholar] [CrossRef]
- Tahami Monfared, A.A.; Byrnes, M.J.; White, L.A.; Zhang, Q. Alzheimer’s Disease: Epidemiology and Clinical Progression. Neurol. Ther. 2022, 11, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Dumurgier, J.; Sabia, S. Epidemiology of Alzheimer’s disease: Latest trends. Rev. Prat. 2020, 70, 149–151. [Google Scholar] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- AmeliMojarad, M.; AmeliMojarad, M. The neuroinflammatory role of microglia in Alzheimer’s disease and their associated therapeutic targets. CNS Neurosci. Ther. 2024, 30, e14856. [Google Scholar] [CrossRef]
- Al-Ghraiybah, N.F.; Wang, J.; Alkhalifa, A.E.; Roberts, A.B.; Raj, R.; Yang, E.; Kaddoumi, A. Glial Cell-Mediated Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 10572. [Google Scholar] [CrossRef]
- Li, Y.; Chen, X.; Zhou, M.; Feng, S.; Peng, X.; Wang, Y. Microglial TLR4/NLRP3 Inflammasome Signaling in Alzheimer’s Disease. J. Alzheimer’s Dis. 2024, 97, 75–88. [Google Scholar] [CrossRef]
- Bird, T.D. Genetic aspects of Alzheimer disease. Genet. Med. 2008, 10, 231–239. [Google Scholar] [CrossRef]
- Laws, S.M.; Hone, E.; Gandy, S.; Martins, R.N. Expanding the association between the APOE gene and the risk of Alzheimer’s disease: Possible roles for APOE promoter polymorphisms and alterations in APOE transcription. J. Neurochem. 2003, 84, 1215–1236. [Google Scholar] [CrossRef]
- Wang, C.; Xiong, M.; Gratuze, M.; Bao, X.; Shi, Y.; Andhey, P.S.; Manis, M.; Schroeder, C.; Yin, Z.; Madore, C.; et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 2021, 109, 1657–1674.e1657. [Google Scholar] [CrossRef]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019, 27, 1293–1306.e1296. [Google Scholar] [CrossRef]
- Kang, S.S.; Ebbert, M.T.W.; Baker, K.E.; Cook, C.; Wang, X.; Sens, J.P.; Kocher, J.P.; Petrucelli, L.; Fryer, J.D. Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J. Exp. Med. 2018, 215, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Liu, C.C.; Qiao, W.; Bu, G. Apolipoprotein E, Receptors, and Modulation of Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Raulin, A.C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurodegener. 2022, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Weisgraber, K.H.; Rall, S.C., Jr.; Mahley, R.W. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J. Biol. Chem. 1981, 256, 9077–9083. [Google Scholar] [CrossRef]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. J. Am. Med. Assoc. 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Risch, N.J.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Rimmler, J.B., Jr.; Locke, P.A.; Conneally, P.M.; Schmader, K.E.; et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef]
- Genin, E.; Hannequin, D.; Wallon, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Bullido, M.J.; Engelborghs, S.; De Deyn, P.; Berr, C.; et al. APOE and Alzheimer disease: A major gene with semi-dominant inheritance. Mol. Psychiatry 2011, 16, 903–907. [Google Scholar] [CrossRef]
- Huebbe, P.; Rimbach, G. Evolution of human apolipoprotein E (APOE) isoforms: Gene structure, protein function and interaction with dietary factors. Ageing Res. Rev. 2017, 37, 146–161. [Google Scholar] [CrossRef]
- Therriault, J.; Benedet, A.L.; Pascoal, T.A.; Mathotaarachchi, S.; Savard, M.; Chamoun, M.; Thomas, E.; Kang, M.S.; Lussier, F.; Tissot, C.; et al. APOEε4 potentiates the relationship between amyloid-β and tau pathologies. Mol. Psychiatry 2021, 26, 5977–5988. [Google Scholar] [CrossRef] [PubMed]
- Kloske, C.M.; Dugan, A.; Woolums, A.E.; Lee, T.; Anderson, S.; Patel, E.; Abner, E.L.; Nelson, P.T.; Fardo, D.W.; Wilcock, D.M. Impaired neuroinflammatory response of ApoE4 in Alzheimer’s disease patients. Alzheimer’s Dement. 2020, 16, e041052. [Google Scholar] [CrossRef]
- Friedberg, J.S.; Aytan, N.; Cherry, J.D.; Xia, W.; Standring, O.J.; Alvarez, V.E.; Nicks, R.; Svirsky, S.; Meng, G.; Jun, G.; et al. Associations between brain inflammatory profiles and human neuropathology are altered based on apolipoprotein E ε4 genotype. Sci. Rep. 2020, 10, 2924. [Google Scholar] [CrossRef]
- Lopresti, B.J.; Campbell, E.M.; Yu, Z.; Anderson, S.J.; Cohen, A.D.; Minhas, D.S.; Snitz, B.E.; Royse, S.K.; Becker, C.R.; Aizenstein, H.J.; et al. Influence of apolipoprotein-E genotype on brain amyloid load and longitudinal trajectories. Neurobiol. Aging 2020, 94, 111–120. [Google Scholar] [CrossRef]
- Wang, N.; Cai, L.; Pei, X.; Lin, Z.; Huang, L.; Liang, C.; Wei, M.; Shao, L.; Guo, T.; Huang, F.; et al. Microglial apolipoprotein E particles contribute to neuronal senescence and synaptotoxicity. iScience 2024, 27, 110006. [Google Scholar] [CrossRef]
- Jackson, R.J.; Keiser, M.S.; Meltzer, J.C.; Fykstra, D.P.; Dierksmeier, S.E.; Hajizadeh, S.; Kreuzer, J.; Morris, R.; Melloni, A.; Nakajima, T.; et al. APOE2 gene therapy reduces amyloid deposition and improves markers of neuroinflammation and neurodegeneration in a mouse model of Alzheimer disease. Mol. Ther. 2024, 32, 1373–1386. [Google Scholar] [CrossRef]
- Groot, C. Multimodal imaging correlates of the protective APOE2 allele. Alzheimer’s Dement. 2020, 16, e039421. [Google Scholar] [CrossRef]
- Li, Z.; Shue, F.; Zhao, N.; Shinohara, M.; Bu, G. APOE2: Protective mechanism and therapeutic implications for Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 63. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Qian, J.; Monsell, S.E.; Betensky, R.A.; Hyman, B.T. APOEε2 is associated with milder clinical and pathological Alzheimer disease. Ann. Neurol. 2015, 77, 917–929. [Google Scholar] [CrossRef]
- Goldberg, T.E.; Huey, E.D.; Devanand, D.P. Association of APOE e2 genotype with Alzheimer’s and non-Alzheimer’s neurodegenerative pathologies. Nat. Commun. 2020, 11, 4727. [Google Scholar] [CrossRef]
- Roda, A.R.; Montoliu-Gaya, L.; Villegas, S. The Role of Apolipoprotein E Isoforms in Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 68, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Medway, C.W.; Abdul-Hay, S.; Mims, T.; Ma, L.; Bisceglio, G.; Zou, F.; Pankratz, S.; Sando, S.B.; Aasly, J.O.; Barcikowska, M.; et al. ApoE variant p.V236E is associated with markedly reduced risk of Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Arboleda-Velasquez, J.F.; Lopera, F.; O’Hare, M.; Delgado-Tirado, S.; Marino, C.; Chmielewska, N.; Saez-Torres, K.L.; Amarnani, D.; Schultz, A.P.; Sperling, R.A.; et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: A case report. Nat. Med. 2019, 25, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Uddin, M.S.; Lim, L.W. Glial cells in Alzheimer’s disease: From neuropathological changes to therapeutic implications. Ageing Res. Rev. 2022, 78, 101622. [Google Scholar] [CrossRef]
- Bolós, M.; Llorens-Martín, M.; Jurado-Arjona, J.; Hernández, F.; Rábano, A.; Avila, J. Direct Evidence of Internalization of Tau by Microglia In Vitro and In Vivo. J. Alzheimer’s Dis. 2016, 50, 77–87. [Google Scholar] [CrossRef]
- Skuljec, J.; Sun, H.; Pul, R.; Bénardais, K.; Ragancokova, D.; Moharregh-Khiabani, D.; Kotsiari, A.; Trebst, C.; Stangel, M. CCL5 induces a pro-inflammatory profile in microglia in vitro. Cell. Immunol. 2011, 270, 164–171. [Google Scholar] [CrossRef]
- Doens, D.; Fernández, P.L. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflammation 2014, 11, 48. [Google Scholar] [CrossRef]
- Xie, Z.; Meng, J.; Wu, Z.; Nakanishi, H.; Hayashi, Y.; Kong, W.; Lan, F.; Narengaowa Yang, Q.; Qing, H.; Ni, J. The Dual Nature of Microglia in Alzheimer’s Disease: A Microglia-Neuron Crosstalk Perspective. Neuroscientist 2023, 29, 616–638. [Google Scholar] [CrossRef]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015, 138 Pt 6, 1738–1755. [Google Scholar] [CrossRef]
- Miao, J.; Ma, H.; Yang, Y.; Liao, Y.; Lin, C.; Zheng, J.; Yu, M.; Lan, J. Microglia in Alzheimer’s disease: Pathogenesis, mechanisms, and therapeutic potentials. Front. Aging Neurosci. 2023, 15, 1201982. [Google Scholar] [CrossRef] [PubMed]
- Botella Lucena, P.; Heneka, M.T. Inflammatory aspects of Alzheimer’s disease. Acta Neuropathol. 2024, 148, 31. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wei, M.; Li, X.; Jia, L.; Li, J.; Xu, H.; Zhang, Y.-w.; Zheng, H. Apolipoprotein E4 expressed by microglia impairs microglial functions and enhances neurotoxicity. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Liu, C.C.; Wang, N.; Chen, Y.; Inoue, Y.; Shue, F.; Ren, Y.; Wang, M.; Qiao, W.; Ikezu, T.C.; Li, Z.; et al. Cell-autonomous effects of APOE4 in restricting microglial response in brain homeostasis and Alzheimer’s disease. Nat. Immunol. 2023, 24, 1854–1866. [Google Scholar] [CrossRef]
- Rao, A.; Chen, N.; Kim, M.J.; Blumenfeld, J.; Yip, O.; Liang, Z.; Shostak, D.; Hao, Y.; Nelson, M.R.; Koutsodendris, N. Microglia depletion reduces human neuronal APOE4-related pathologies in a chimeric Alzheimer’s disease model. Cell Stem Cell 2025, 32, 86–104.e107. [Google Scholar] [CrossRef]
- Najm, R.; Zalocusky, K.A.; Zilberter, M.; Yoon, S.Y.; Hao, Y.; Koutsodendris, N.; Nelson, M.; Rao, A.; Taubes, A.; Jones, E.A.; et al. In Vivo Chimeric Alzheimer’s Disease Modeling of Apolipoprotein E4 Toxicity in Human Neurons. Cell Rep. 2020, 32, 107962. [Google Scholar] [CrossRef]
- Fitz, N.F.; Nam, K.N.; Wolfe, C.M.; Letronne, F.; Playso, B.E.; Iordanova, B.E.; Kozai, T.D.Y.; Biedrzycki, R.J.; Kagan, V.E.; Tyurina, Y.Y.; et al. Phospholipids of APOE lipoproteins activate microglia in an isoform-specific manner in preclinical models of Alzheimer’s disease. Nat. Commun. 2021, 12, 3416. [Google Scholar] [CrossRef]
- Sepulveda, J.; Kim, J.Y.; Binder, J.; Vicini, S.; Rebeck, G.W. APOE4 genotype and aging impair injury-induced microglial behavior in brain slices, including toward Aβ, through P2RY12. Mol. Neurodegener. 2024, 19, 24. [Google Scholar] [CrossRef]
- Minett, T.; Classey, J.; Matthews, F.E.; Fahrenhold, M.; Taga, M.; Brayne, C.; Ince, P.G.; Nicoll, J.A.; Boche, D. Microglial immunophenotype in dementia with Alzheimer’s pathology. J. Neuroinflammation 2016, 13, 135. [Google Scholar] [CrossRef]
- Maezawa, I.; Nivison, M.; Montine, K.S.; Maeda, N.; Montine, T.J. Neurotoxicity from innate immune response is greatest with targeted replacement of E4 allele of apolipoprotein E gene and is mediated by microglial p38MAPK. Faseb J. 2006, 20, 797–799. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wang, M.; Jeevaratnam, S.; Rosenberg, C.; Ikezu, T.C.; Shue, F.; Doss, S.V.; Alnobani, A.; Martens, Y.A.; Wren, M.; et al. Opposing effects of apoE2 and apoE4 on microglial activation and lipid metabolism in response to demyelination. Mol. Neurodegener. 2022, 17, 75. [Google Scholar] [CrossRef] [PubMed]
- Iannucci, J.; Sen, A.; Grammas, P. Isoform-Specific Effects of Apolipoprotein E on Markers of Inflammation and Toxicity in Brain Glia and Neuronal Cells In Vitro. Curr Issues Mol. Biol. 2021, 43, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Lee, C.Y.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T.M.; Collins, J.L.; et al. ApoE promotes the proteolytic degradation of Abeta. Neuron 2008, 58, 681–693. [Google Scholar] [CrossRef]
- Yin, Z.; Rosenzweig, N.; Kleemann, K.L.; Zhang, X.; Brandão, W.; Margeta, M.A.; Schroeder, C.; Sivanathan, K.N.; Silveira, S.; Gauthier, C.; et al. APOE4 impairs the microglial response in Alzheimer’s disease by inducing TGFβ-mediated checkpoints. Nat. Immunol. 2023, 24, 1839–1853. [Google Scholar] [CrossRef]
- Bohlen, C.J.; Friedman, B.A.; Dejanovic, B.; Sheng, M. Microglia in Brain Development, Homeostasis, and Neurodegeneration. Annu. Rev. Genet. 2019, 53, 263–288. [Google Scholar] [CrossRef]
- Benarroch, E.E. Microglia: Multiple roles in surveillance, circuit shaping, and response to injury. Neurology 2013, 81, 1079–1088. [Google Scholar] [CrossRef]
- Moser, V.A.; Workman, M.J.; Hurwitz, S.J.; Lipman, R.M.; Pike, C.J.; Svendsen, C.N. Microglial transcription profiles in mouse and human are driven by APOE4 and sex. iScience 2021, 24, 103238. [Google Scholar] [CrossRef]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e1147. [Google Scholar] [CrossRef]
- Muth, C.; Hartmann, A.; Sepulveda-Falla, D.; Glatzel, M.; Krasemann, S. Phagocytosis of Apoptotic Cells Is Specifically Upregulated in ApoE4 Expressing Microglia in vitro. Front. Cell. Neurosci. 2019, 13, 181. [Google Scholar] [CrossRef]
- Egensperger, R.; Kösel, S.; von Eitzen, U.; Graeber, M.B. Microglial activation in Alzheimer disease: Association with APOE genotype. Brain Pathol. 1998, 8, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Ferrari-Souza, J.P.; Lussier, F.Z.; Leffa, D.T.; Therriault, J.; Tissot, C.; Bellaver, B.; Ferreira, P.C.L.; Malpetti, M.; Wang, Y.T.; Povala, G.; et al. APOEε4 associates with microglial activation independently of Aβ plaques and tau tangles. Sci. Adv. 2023, 9, eade1474. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e569. [Google Scholar] [CrossRef]
- Prinz, M.; Masuda, T.; Wheeler, M.A.; Quintana, F.J. Microglia and Central Nervous System-Associated Macrophages-From Origin to Disease Modulation. Annu. Rev. Immunol. 2021, 39, 251–277. [Google Scholar] [CrossRef]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [CrossRef]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Millet, A.; Ledo, J.H.; Tavazoie, S.F. An exhausted-like microglial population accumulates in aged and APOE4 genotype Alzheimer’s brains. Immunity 2024, 57, 153–170.e156. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Wang, K.; Hu, G.; Wang, X.; Miao, Z.; Azevedo, J.A.; Suh, E.; Van Deerlin, V.M.; Choi, D.; Roeder, K.; et al. APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol. 2020, 140, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef]
- Haney, M.S.; Pálovics, R.; Munson, C.N.; Long, C.; Johansson, P.K.; Yip, O.; Dong, W.; Rawat, E.; West, E.; Schlachetzki, J.C.M.; et al. APOE4/4 is linked to damaging lipid droplets in Alzheimer’s disease microglia. Nature 2024, 628, 154–161. [Google Scholar] [CrossRef]
- Lee, S.; Devanney, N.A.; Golden, L.R.; Smith, C.T.; Schwartz, J.L.; Walsh, A.E.; Clarke, H.A.; Goulding, D.S.; Allenger, E.J.; Morillo-Segovia, G.; et al. APOE modulates microglial immunometabolism in response to age, amyloid pathology, and inflammatory challenge. Cell Rep. 2023, 42, 112196. [Google Scholar] [CrossRef]
- Litvinchuk, A.; Suh, J.H.; Guo, J.L.; Lin, K.; Davis, S.S.; Bien-Ly, N.; Tycksen, E.; Tabor, G.T.; Remolina Serrano, J.; Manis, M.; et al. Amelioration of Tau and ApoE4-linked glial lipid accumulation and neurodegeneration with an LXR agonist. Neuron 2024, 112, 384–403.e388. [Google Scholar] [CrossRef]
- Koutsodendris, N.; Blumenfeld, J.; Agrawal, A.; Traglia, M.; Grone, B.; Zilberter, M.; Yip, O.; Rao, A.; Nelson, M.R.; Hao, Y.; et al. Neuronal APOE4 removal protects against tau-mediated gliosis, neurodegeneration and myelin deficits. Nat. Aging 2023, 3, 275–296. [Google Scholar] [CrossRef]
- Wang, S.; Sudan, R.; Peng, V.; Zhou, Y.; Du, S.; Yuede, C.M.; Lei, T.; Hou, J.; Cai, Z.; Cella, M.; et al. TREM2 drives microglia response to amyloid-β via SYK-dependent and -independent pathways. Cell 2022, 185, 4153–4169.e4119. [Google Scholar] [CrossRef]
- Yin, Z.; Herron, S.; Silveira, S.; Kleemann, K.; Gauthier, C.; Mallah, D.; Cheng, Y.; Margeta, M.A.; Pitts, K.M.; Barry, J.L.; et al. Identification of a protective microglial state mediated by miR-155 and interferon-γ signaling in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2023, 26, 1196–1207. [Google Scholar] [CrossRef]
- Rosenzweig, N.; Kleemann, K.L.; Rust, T.; Carpenter, M.; Grucci, M.; Aronchik, M.; Brouwer, N.; Valenbreder, I.; Cooper-Hohn, J.; Iyer, M.; et al. Sex-dependent APOE4 neutrophil-microglia interactions drive cognitive impairment in Alzheimer’s disease. Nat. Med. 2024, 30, 2990–3003. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.F.; Wu, W.; Wong, H.Y.; Ouyang, L.; Qiao, Y.; Xu, J.; Lau, J.H.; Wong, C.; Jiang, Y.; Holtzman, D.M.; et al. The VCAM1-ApoE pathway directs microglial chemotaxis and alleviates Alzheimer’s disease pathology. Nat. Aging 2023, 3, 1219–1236. [Google Scholar] [CrossRef] [PubMed]
- Machlovi, S.I.; Neuner, S.M.; Hemmer, B.M.; Khan, R.; Liu, Y.; Huang, M.; Zhu, J.D.; Castellano, J.M.; Cai, D.; Marcora, E.; et al. APOE4 confers transcriptomic and functional alterations to primary mouse microglia. Neurobiol. Dis. 2022, 164, 105615. [Google Scholar] [CrossRef] [PubMed]
- Mhatre-Winters, I.; Eid, A.; Han, Y.; Tieu, K.; Richardson, J.R. Sex and APOE Genotype Alter the Basal and Induced Inflammatory States of Primary Microglia from APOE Targeted Replacement Mice. Int. J. Mol. Sci. 2022, 23, 9829. [Google Scholar] [CrossRef]
- Lanfranco, M.F.; Sepulveda, J.; Kopetsky, G.; Rebeck, G.W. Expression and secretion of apoE isoforms in astrocytes and microglia during inflammation. Glia 2021, 69, 1478–1493. [Google Scholar] [CrossRef]
- Zhu, Y.; Nwabuisi-Heath, E.; Dumanis, S.B.; Tai, L.M.; Yu, C.; Rebeck, G.W.; LaDu, M.J. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia 2012, 60, 559–569. [Google Scholar] [CrossRef]
- Li, Z.; Martens, Y.A.; Ren, Y.; Jin, Y.; Sekiya, H.; Doss, S.V.; Kouri, N.; Castanedes-Casey, M.; Christensen, T.A.; Miller Nevalainen, L.B.; et al. APOE genotype determines cell-type-specific pathological landscape of Alzheimer’s disease. Neuron 2025, 113, 1380–1397. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Li, Z.; Noori, A.; Nguyen, H.N.; Mezlini, A.; Li, L.; Hudry, E.; Jackson, R.J.; Hyman, B.T.; Das, S. Effect of APOE alleles on the glial transcriptome in normal aging and Alzheimer’s disease. Nat. Aging 2021, 1, 919–931. [Google Scholar] [CrossRef]
- Kloske, C.M.; Dugan, A.J.; Weekman, E.M.; Winder, Z.; Patel, E.; Nelson, P.T.; Fardo, D.W.; Wilcock, D.M. Inflammatory Pathways Are Impaired in Alzheimer Disease and Differentially Associated With Apolipoprotein E Status. J. Neuropathol. Exp. Neurol. 2021, 80, 922–932. [Google Scholar] [CrossRef]
- Liu, X.T.; Chen, X.; Zhao, N.; Geng, F.; Zhu, M.M.; Ren, Q.G. Synergism of ApoE4 and systemic infectious burden is mediated by the APOE-NLRP3 axis in Alzheimer’s disease. Psychiatry Clin. Neurosci. 2024, 78, 517–526. [Google Scholar] [CrossRef]
- Yao, Z.; Liu, N.; Zhu, X.; Wang, L.; Zhao, Y.; Liu, Q.; Gao, C.; Li, J. Subanesthetic isoflurane abates ROS-activated MAPK/NF-κB signaling to repress ischemia-induced microglia inflammation and brain injury. Aging 2020, 12, 26121–26139. [Google Scholar] [CrossRef] [PubMed]
- Schoonbroodt, S.; Piette, J. Oxidative stress interference with the nuclear factor-kappa B activation pathways. Biochem. Pharmacol. 2000, 60, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- LaDu, M.; Wohlgenant, S.; Van Eldik, L.; Tai, L. TLR4-p38 pathway as a mechanism of APOE-modulated neuroinflammation in Alzheimer disease. FASEB J. 2014, 28, 1056–1060. [Google Scholar] [CrossRef]
- Balu, D.; Valencia-Olvera, A.C.; Nguyen, A.; Patnam, M.; York, J.; Peri, F.; Neumann, F.; LaDu, M.J.; Tai, L.M. A small-molecule TLR4 antagonist reduced neuroinflammation in female E4FAD mice. Alzheimers Res. Ther. 2023, 15, 181. [Google Scholar] [CrossRef]
- Love, J.E.; Day, R.J.; Gause, J.W.; Brown, R.J.; Pu, X.; Theis, D.I.; Caraway, C.A.; Poon, W.W.; Rahman, A.A.; Morrison, B.E.; et al. Nuclear uptake of an amino-terminal fragment of apolipoprotein E4 promotes cell death and localizes within microglia of the Alzheimer’s disease brain. Int. J. Physiol. Pathophysiol. Pharmacol. 2017, 9, 40–57. [Google Scholar]
- Pollock, T.B.; Mack, J.M.; Day, R.J.; Isho, N.F.; Brown, R.J.; Oxford, A.E.; Morrison, B.E.; Hayden, E.J.; Rohn, T.T. A Fragment of Apolipoprotein E4 Leads to the Downregulation of a CXorf56 Homologue, a Novel ER-Associated Protein, and Activation of BV2 Microglial Cells. Oxidative Med. Cell. Longev. 2019, 2019, 5123565. [Google Scholar] [CrossRef]
- Pollock, T.B.; Cholico, G.N.; Isho, N.F.; Day, R.J.; Suresh, T.; Stewart, E.S.; McCarthy, M.M.; Rohn, T.T. Transcriptome Analyses in BV2 Microglial Cells Following Treatment with Amino-Terminal Fragments of Apolipoprotein E. Front. Aging Neurosci. 2020, 12, 256. [Google Scholar] [CrossRef]
- Rohn, T.T.; Beck, J.D.; Galla, S.J.; Isho, N.F.; Pollock, T.B.; Suresh, T.; Kulkarni, A.; Sanghal, T.; Hayden, E.J. Fragmentation of Apolipoprotein E4 is Required for Differential Expression of Inflammation and Activation Related Genes in Microglia Cells. Int. J. Neurodegener. Dis. 2021, 4, 20. [Google Scholar] [CrossRef]
- Theendakara, V.; Peters-Libeu, C.A.; Spilman, P.; Poksay, K.S.; Bredesen, D.E.; Rao, R.V. Direct Transcriptional Effects of Apolipoprotein E. J. Neurosci. 2016, 36, 685–700. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, Y.; Huang, G.; Yang, M.; Zhu, Y.; Jin, C.; Jing, D.; Ji, K.; Shi, Y. LilrB3 is a putative cell surface receptor of APOE4. Cell Res. 2023, 33, 116–130. [Google Scholar] [CrossRef]
- Wong, M.Y.; Lewis, M.; Doherty, J.J.; Shi, Y.; Cashikar, A.G.; Amelianchik, A.; Tymchuk, S.; Sullivan, P.M.; Qian, M.; Covey, D.F.; et al. 25-Hydroxycholesterol amplifies microglial IL-1β production in an apoE isoform-dependent manner. J. Neuroinflammation 2020, 17, 192. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Mai, W.; Chen, L.; Cao, K.; Zhang, B.; Zhang, Z.; Liu, Y.; Lou, H.; Duan, S.; Gao, Z. mTOR-mediated metabolic reprogramming shapes distinct microglia functions in response to lipopolysaccharide and ATP. Glia 2020, 68, 1031–1045. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Sobotka, K.S.; Joshi, P.; Gressens, P.; Fleiss, B.; Thornton, C.; Mallard, C.; Hagberg, H. Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia 2019, 67, 1047–1061. [Google Scholar] [CrossRef]
- Victor, M.B.; Leary, N.; Luna, X.; Meharena, H.S.; Scannail, A.N.; Bozzelli, P.L.; Samaan, G.; Murdock, M.H.; von Maydell, D.; Effenberger, A.H.; et al. Lipid accumulation induced by APOE4 impairs microglial surveillance of neuronal-network activity. Cell Stem Cell 2022, 29, 1197–1212.e1198. [Google Scholar] [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef]
- Stephen, T.L.; Cacciottolo, M.; Balu, D.; Morgan, T.E.; LaDu, M.J.; Finch, C.E.; Pike, C.J. APOE genotype and sex affect microglial interactions with plaques in Alzheimer’s disease mice. Acta Neuropathol. Commun. 2019, 7, 82. [Google Scholar] [CrossRef]
- Fitz, N.F.; Wolfe, C.M.; Playso, B.E.; Biedrzycki, R.J.; Lu, Y.; Nam, K.N.; Lefterov, I.; Koldamova, R. Trem2 deficiency differentially affects phenotype and transcriptome of human APOE3 and APOE4 mice. Mol. Neurodegener. 2020, 15, 41. [Google Scholar] [CrossRef]
- McQuade, A.; Kang, Y.J.; Hasselmann, J.; Jairaman, A.; Sotelo, A.; Coburn, M.; Shabestari, S.K.; Chadarevian, J.P.; Fote, G.; Tu, C.H.; et al. Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat. Commun. 2020, 11, 5370. [Google Scholar] [CrossRef]
- Pey, P.; Pearce, R.K.; Kalaitzakis, M.E.; Griffin, W.S.; Gentleman, S.M. Phenotypic profile of alternative activation marker CD163 is different in Alzheimer’s and Parkinson’s disease. Acta Neuropathol. Commun. 2014, 2, 21. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Gómez-Isla, T.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. A phenotypic change but not proliferation underlies glial responses in Alzheimer disease. Am. J. Pathol. 2013, 182, 2332–2344. [Google Scholar] [CrossRef]
- Perlmutter, L.S.; Scott, S.A.; Barrón, E.; Chui, H.C. MHC class II-positive microglia in human brain: Association with Alzheimer lesions. J. Neurosci. Res. 1992, 33, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Mittal, K.; Eremenko, E.; Berner, O.; Elyahu, Y.; Strominger, I.; Apelblat, D.; Nemirovsky, A.; Spiegel, I.; Monsonego, A. CD4 T Cells Induce A Subset of MHCII-Expressing Microglia that Attenuates Alzheimer Pathology. iScience 2019, 16, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Bassal, R.; Rivkin-Natan, M.; Rabinovich, A.; Michaelson, D.M.; Frenkel, D.; Pinkas-Kramarski, R. APOE4 impairs autophagy and Aβ clearance by microglial cells. Inflamm. Res. 2025, 74, 61. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.A.; Tai, L.M.; LaDu, M.J.; Rebeck, G.W. Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition. J. Neuroinflammation 2014, 11, 111. [Google Scholar] [CrossRef]
- Fote, G.M.; Geller, N.R.; Efstathiou, N.E.; Hendricks, N.; Vavvas, D.G.; Reidling, J.C.; Thompson, L.M.; Steffan, J.S. Isoform-dependent lysosomal degradation and internalization of apolipoprotein E requires autophagy proteins. J. Cell Sci. 2022, 135, jcs258687. [Google Scholar] [CrossRef]
- Solé-Domènech, S.; Cruz, D.L.; Capetillo-Zarate, E.; Maxfield, F.R. The endocytic pathway in microglia during health, aging and Alzheimer’s disease. Ageing Res. Rev. 2016, 32, 89–103. [Google Scholar] [CrossRef]
- Hao, Y.; Su, C.; Liu, X.; Sui, H.; Shi, Y.; Zhao, L. Bioengineered microglia-targeted exosomes facilitate Aβ clearance via enhancing activity of microglial lysosome for promoting cognitive recovery in Alzheimer’s disease. Biomater. Adv. 2022, 136, 212770. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kanekiyo, T.; Shinohara, M.; Zhang, Y.; LaDu, M.J.; Xu, H.; Bu, G. Differential regulation of amyloid-β endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J. Biol. Chem. 2012, 287, 44593–44601. [Google Scholar] [CrossRef]
- Kim, Y.; Ha, T.Y.; Lee, M.S.; Chang, K.A. Regulatory Mechanisms and Therapeutic Implications of Lysosomal Dysfunction in Alzheimer’s Disease. Int. J. Biol. Sci. 2025, 21, 1014–1031. [Google Scholar] [CrossRef]
- Hopp, S.C.; Lin, Y.; Oakley, D.; Roe, A.D.; DeVos, S.L.; Hanlon, D.; Hyman, B.T. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflammation 2018, 15, 269. [Google Scholar] [CrossRef]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Gratuze, M.; Schlachetzki, J.C.M.; D’Oliveira Albanus, R.; Jain, N.; Novotny, B.; Brase, L.; Rodriguez, L.; Mansel, C.; Kipnis, M.; O’Brien, S.; et al. TREM2-independent microgliosis promotes tau-mediated neurodegeneration in the presence of ApoE4. Neuron 2023, 111, 202–219.e207. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.F.; Cao, H.; Fu, A.K.Y.; Ip, N.Y. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 25800–25809. [Google Scholar] [CrossRef] [PubMed]
- Carmeli, C.; Donati, A.; Antille, V.; Viceic, D.; Ghika, J.; von Gunten, A.; Clarke, S.; Meuli, R.; Frackowiak, R.S.; Knyazeva, M.G. Demyelination in mild cognitive impairment suggests progression path to Alzheimer’s disease. PLoS ONE 2013, 8, e72759. [Google Scholar] [CrossRef]
- Wang, C.; Lu, J.; Sha, X.; Qiu, Y.; Chen, H.; Yu, Z. TRPV1 regulates ApoE4-disrupted intracellular lipid homeostasis and decreases synaptic phagocytosis by microglia. Exp. Mol. Med. 2023, 55, 347–363. [Google Scholar] [CrossRef]
- Martenka, J.; Soszyńska, K. Predictive value of apolipoproteine E genotypes. Wiad. Lek. 2016, 69, 569–575. [Google Scholar]
- Yen, J.J.; Yu, I.I. The role of ApoE-mediated microglial lipid metabolism in brain aging and disease. Immunometabolism 2023, 5, e00018. [Google Scholar] [CrossRef]
- Sienski, G.; Narayan, P.; Bonner, J.M.; Kory, N.; Boland, S.; Arczewska, A.A.; Ralvenius, W.T.; Akay, L.; Lockshin, E.; He, L.; et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci. Transl. Med. 2021, 13, eaaz4564. [Google Scholar] [CrossRef]
- Tcw, J.; Qian, L.; Pipalia, N.H.; Chao, M.J.; Liang, S.A.; Shi, Y.; Jain, B.R.; Bertelsen, S.E.; Kapoor, M.; Marcora, E.; et al. Cholesterol and matrisome pathways dysregulated in astrocytes and microglia. Cell 2022, 185, 2213–2233.e2225. [Google Scholar] [CrossRef]
- Lee, C.Y.; Tse, W.; Smith, J.D.; Landreth, G.E. Apolipoprotein E promotes β-amyloid trafficking and degradation by modulating microglial cholesterol levels. J. Biol. Chem. 2012, 287, 2032–2044. [Google Scholar] [CrossRef]
- Kaji, S.; Berghoff, S.A.; Spieth, L.; Schlaphoff, L.; Sasmita, A.O.; Vitale, S.; Büschgens, L.; Kedia, S.; Zirngibl, M.; Nazarenko, T.; et al. Apolipoprotein E aggregation in microglia initiates Alzheimer’s disease pathology by seeding β-amyloidosis. Immunity 2024, 57, 2651–2668.e12. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Braun, D.; Fitzgerald, G.A.; Hsieh, Y.T.; Rougé, L.; Litvinchuk, A.; Steffek, M.; Propson, N.E.; Heffner, C.M.; Discenza, C.; et al. Decreased lipidated ApoE-receptor interactions confer protection against pathogenicity of ApoE and its lipid cargoes in lysosomes. Cell 2024, 188, 187–206.e26. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lin, S.; Bales, K.R.; Gelfanova, V.; Koger, D.; Delong, C.; Hale, J.; Liu, F.; Hunter, J.M.; Paul, S.M. Macrophage-mediated degradation of beta-amyloid via an apolipoprotein E isoform-dependent mechanism. J. Neurosci. 2009, 29, 3603–3612. [Google Scholar] [CrossRef]
- Pankiewicz, J.E.; Guridi, M.; Kim, J.; Asuni, A.A.; Sanchez, S.; Sullivan, P.M.; Holtzman, D.M.; Sadowski, M.J. Blocking the apoE/Aβ interaction ameliorates Aβ-related pathology in APOE ε2 and ε4 targeted replacement Alzheimer model mice. Acta Neuropathol. Commun. 2014, 2, 75. [Google Scholar] [CrossRef]
- Olah, M.; Patrick, E.; Villani, A.C.; Xu, J.; White, C.C.; Ryan, K.J.; Piehowski, P.; Kapasi, A.; Nejad, P.; Cimpean, M.; et al. A transcriptomic atlas of aged human microglia. Nat. Commun. 2018, 9, 539. [Google Scholar] [CrossRef]
- Chen, Y.; Song, S.; Parhizkar, S.; Lord, J.; Zhu, Y.; Strickland, M.R.; Wang, C.; Park, J.; Tabor, G.T.; Jiang, H.; et al. APOE3ch alters microglial response and suppresses Aβ-induced tau seeding and spread. Cell 2024, 187, 428–445.e420. [Google Scholar] [CrossRef]
- Chen, G.; Wang, M.; Zhang, Z.; Hong, D.K.; Ahn, E.H.; Liu, X.; Kang, S.S.; Ye, K. ApoE3 R136S binds to Tau and blocks its propagation, suppressing neurodegeneration in mice with Alzheimer’s disease. Neuron 2025, 113, 719–736.e5. [Google Scholar] [CrossRef]
- Nelson, M.R.; Liu, P.; Agrawal, A.; Yip, O.; Blumenfeld, J.; Traglia, M.; Kim, M.J.; Koutsodendris, N.; Rao, A.; Grone, B.; et al. The APOE-R136S mutation protects against APOE4-driven Tau pathology, neurodegeneration and neuroinflammation. Nat. Neurosci. 2023, 26, 2104–2121. [Google Scholar] [CrossRef]
- Liu, C.C.; Murray, M.E.; Li, X.; Zhao, N.; Wang, N.; Heckman, M.G.; Shue, F.; Martens, Y.; Li, Y.; Raulin, A.C.; et al. APOE3-Jacksonville (V236E) variant reduces self-aggregation and risk of dementia. Sci. Transl. Med. 2021, 13, eabc9375. [Google Scholar] [CrossRef]
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5000-person neuropathological study. Nat. Commun. 2020, 11, 667. [Google Scholar] [CrossRef]
- Jackson, R.J.; Hyman, B.T.; Serrano-Pozo, A. Multifaceted roles of APOE in Alzheimer disease. Nat. Rev. Neurol. 2024, 20, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Macyczko, J.R.; Liu, C.C.; Bu, G. ApoE4 reduction: An emerging and promising therapeutic strategy for Alzheimer’s disease. Neurobiol. Aging 2022, 115, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Mouzat, K.; Chudinova, A.; Polge, A.; Kantar, J.; Camu, W.; Raoul, C.; Lumbroso, S. Regulation of Brain Cholesterol: What Role Do Liver X Receptors Play in Neurodegenerative Diseases? Int. J. Mol. Sci. 2019, 20, 3858. [Google Scholar] [CrossRef]
- Zhang, R.; Wuerch, E.; Yong, V.W.; Xue, M. LXR agonism for CNS diseases: Promises and challenges. J. Neuroinflamm. 2024, 21, 97. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Huang, Z.T.; Yuan, M.H.; Jing, F.; Cai, R.L.; Zou, Q.; Pu, Y.S.; Wang, S.Y.; Chen, F.; Yi, W.M.; et al. Role of Hypoxia Inducible Factor-1α in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 80, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Belaidi, A.A.; Bush, A.I.; Ayton, S. Apolipoprotein E in Alzheimer’s disease: Molecular insights and therapeutic opportunities. Mol. Neurodegener. 2025, 20, 47. [Google Scholar] [CrossRef]
- Tashima, T. Smart Strategies for Therapeutic Agent Delivery into Brain across the Blood-Brain Barrier Using Receptor-Mediated Transcytosis. Chem. Pharm. Bull. 2020, 68, 316–325. [Google Scholar] [CrossRef]
- Jones, A.R.; Shusta, E.V. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm. Res. 2007, 24, 1759–1771. [Google Scholar] [CrossRef]
- Kimura, S.; Harashima, H. Non-invasive gene delivery across the blood-brain barrier: Present and future perspectives. Neural Regen. Res. 2022, 17, 785–787. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef]
- Wu, Y.; Eisel, U.L.M. Microglia-Astrocyte Communication in Alzheimer’s Disease. J. Alzheimer’s Dis. 2023, 95, 785–803. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Nigro, M.; Travagli, A.; Gessi, S. Microglia and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 12990. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Aylward, A.J.; Pearse, R.V., 2nd; Lish, A.M.; Hsieh, Y.C.; Augur, Z.M.; Benoit, C.R.; Chou, V.; Knupp, A.; Pan, C.; et al. Cell-type-specific regulation of APOE and CLU levels in human neurons by the Alzheimer’s disease risk gene SORL1. Cell Rep. 2023, 42, 112994. [Google Scholar] [CrossRef] [PubMed]
- Chausse, B.; Kakimoto, P.A.; Kann, O. Microglia and lipids: How metabolism controls brain innate immunity. Semin. Cell Dev. Biol. 2021, 112, 137–144. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microglia Function | APOEε2 | APOEε3 | APOEε4 |
|---|---|---|---|
| Activation states | Mild activation | Homeostatic | Hyperactive |
| Inflammation | Suppressed | Neutral | Elevated |
| Aβ clearance | Efficient | Moderate | Impaired |
| Tau pathology | Protective | Mild | deteriorating |
| Lipid metabolism | Balanced | Balanced | Disrupted |
| Migration ability | Normal | Normal | Impaired |
| Type | Promoting Factors | Inhibiting Factors | Related Genes | Functional Features | Refs. |
|---|---|---|---|---|---|
| DAM (MGnD) | APOE-TREM2 axis, Aβ plaques, apoptotic neurons | —— | TREM2, APOE, TYROBP, LPL | Increased phagocytosis | [64,65] |
| TIMs | Neuroinflammation, aging, APOEε4, cellular stress | —— | NF-κB, C/EBP, AP-1 | Impaired phagocytosis/ inflammation | [73] |
| ARM | Aβ plaques, tau pathology | APOEε4 TREM2-R47H | CD163 | Increased phagocytosis | [74] |
| MHC-II microglia | Aβ plaques, tau pathology, Neuronal APOE4 | —— | MHC-II | Increased inflammation/ phagocytosis | [47] |
| LDAM | Inflammation, APOEε4, Aβ plaques | —— | ACSL1 | Increased inflammation, impaired phagocytosis | [75,76] |
| Axis | Therapeutic Target | Intervention | Mechanisms | Refs. |
|---|---|---|---|---|
| LXR-APOE axis | LXR | LXR agonists (GW3965) | Activation of LXR promotes APOE upregulation, enhancing cholesterol transport and metabolism, and preventing intracellular cholesterol accumulation. | [144,145] |
| APOE—HIF-1α axis | HIF-1α | HIF-1α inhibitors | APOEε4 upregulates HIF-1α, driving microglia toward a phenotype resembling DAM/MGnD with pro-inflammatory and glycolytic metabolic shifts. | [77,146] |
| VCAM1-APOE axis | VCAM1 | VCAM1 agonists | Microglial VCAM1 expression promoting migration toward APOE-containing Aβ plaques. VCAM1-APOE interaction induces DAM/MGnD transformation, enhancing Aβ clearance. | [83] |
| APOE4—ITGB8-TGFβ axis | TGFβ | TGFβ inhibitor | The microglial APOE4-ITGB8-TGFβ pathway serves as a negative regulator of microglial response to AD pathology, and restoring the MGnD phenotype via blocking ITGB8-TGFβ signaling provides a promising therapeutic intervention for AD. | [56,65] |
| TREM2-APOE axis | TREM2, SYK, miR155 | TREM2/SYK agonists, miR-15 inhibitor | TREM2-SYK signaling axis activation is essential for the DAM/MGnD phenotype. miR-155 suppresses the downstream IFNγ signaling pathway, thereby blocking the DAM/MGnD response. | [65,80,81] |
| IL-17F—IL-17RA axis | IL-17F | IL-17F inhibitor | APOEε4-associated neutrophils exhibit elevated IL-17F expression, which engages microglial IL-17RA to inhibit the DAM/MGnD phenotype. Disrupting this IL-17F/IL-17RA axis improved cognitive function in a mouse model of AD. | [82] |
| APOE—NF-κB/NLRP3 axis | NF-κB, NLRP3 | NF-κB/NLRP3 inhibitor, | APOE4 can significantly induce the activation of NF-κB and more effectively activate the NLRP3 inflammasome, enhancing the neuroinflammatory response of microglia. | [91] |
| APOE—NF-κB ACSL1 axis | ACSL1 | ACSL1 inhibitor (Triacin C) | APOEε4 promotes microglial lipid droplet accumulation (LDAM phenotype) through NF-κB-mediated transcriptional activation of ACSL1. | [76] |
| APOE4- LilrB3 axis | LilrB3 | LilrB3 antagonist | The specific interaction between APOE4 protein and LilrB3(an immune checkpoint receptor protein expressed on the surface of microglia) activates microglia, driving their transition into a pro-inflammatory state. | [101] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, A.; Wang, T.; Yang, L.; Zhou, Y. The APOE–Microglia Axis in Alzheimer’s Disease: Functional Divergence and Therapeutic Perspectives—A Narrative Review. Brain Sci. 2025, 15, 675. https://doi.org/10.3390/brainsci15070675
Liu A, Wang T, Yang L, Zhou Y. The APOE–Microglia Axis in Alzheimer’s Disease: Functional Divergence and Therapeutic Perspectives—A Narrative Review. Brain Sciences. 2025; 15(7):675. https://doi.org/10.3390/brainsci15070675
Chicago/Turabian StyleLiu, Aiwei, Tingxu Wang, Liu Yang, and Yu Zhou. 2025. "The APOE–Microglia Axis in Alzheimer’s Disease: Functional Divergence and Therapeutic Perspectives—A Narrative Review" Brain Sciences 15, no. 7: 675. https://doi.org/10.3390/brainsci15070675
APA StyleLiu, A., Wang, T., Yang, L., & Zhou, Y. (2025). The APOE–Microglia Axis in Alzheimer’s Disease: Functional Divergence and Therapeutic Perspectives—A Narrative Review. Brain Sciences, 15(7), 675. https://doi.org/10.3390/brainsci15070675