Cortical and Striatal Functional Connectivity in Juvenile-Onset Huntington’s Disease


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample
2.2. Magnetic Resonance Imaging
2.3. Measures
2.4. Statistical Analyses
3. Results
3.1. Participant Demographics
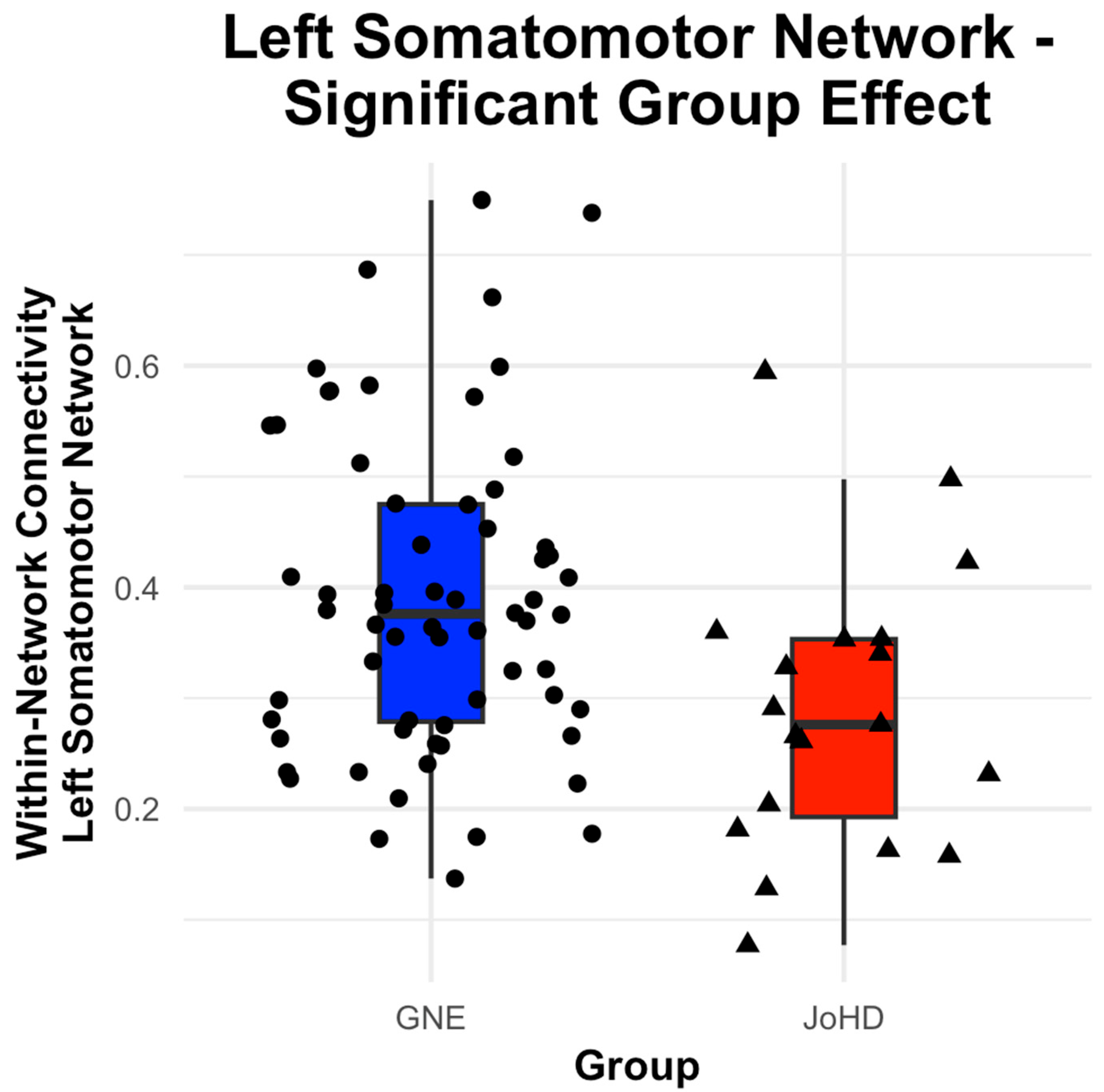
3.2. Cortical Network Group Differences
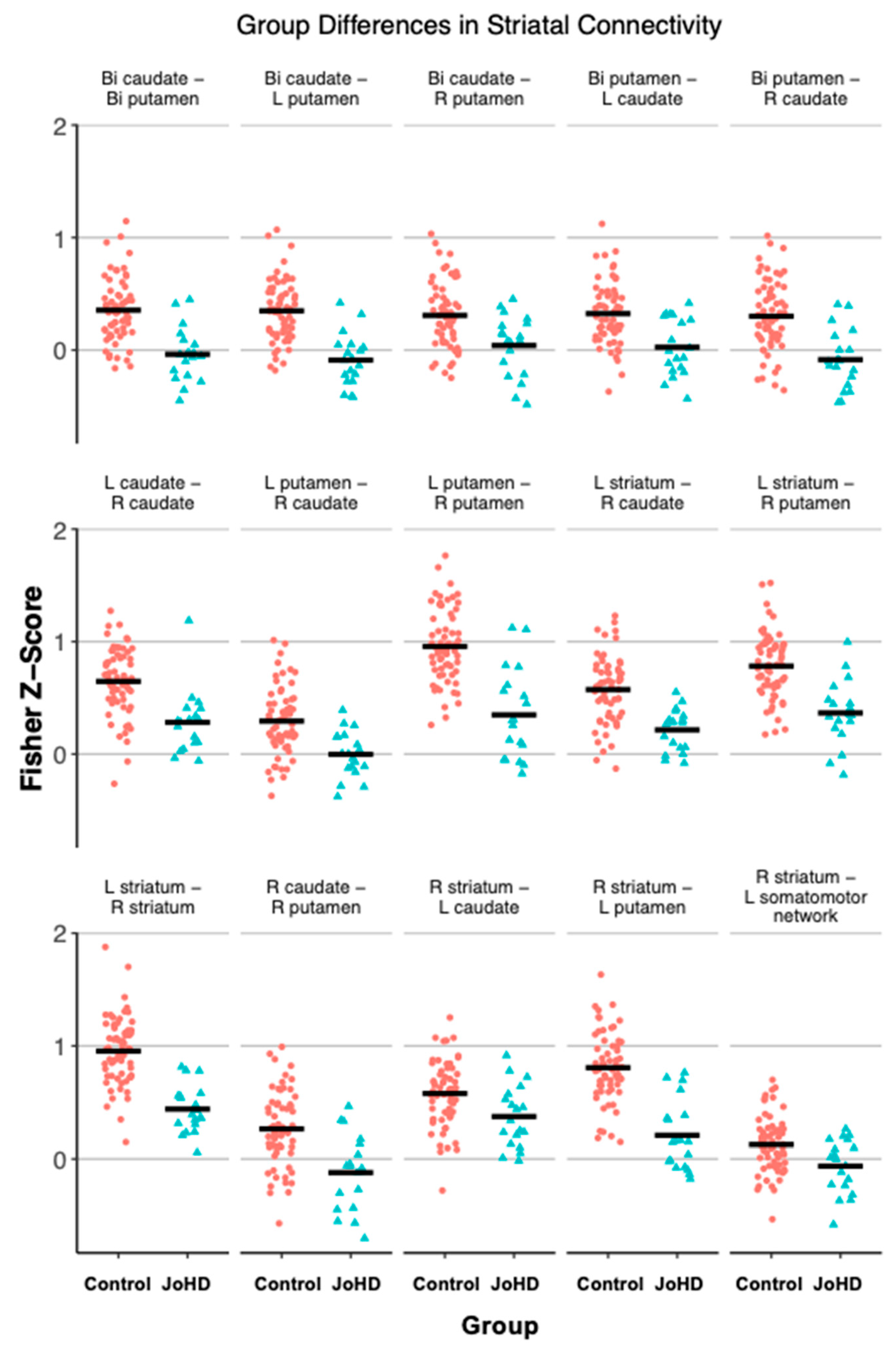
3.3. Striatal Group Differences
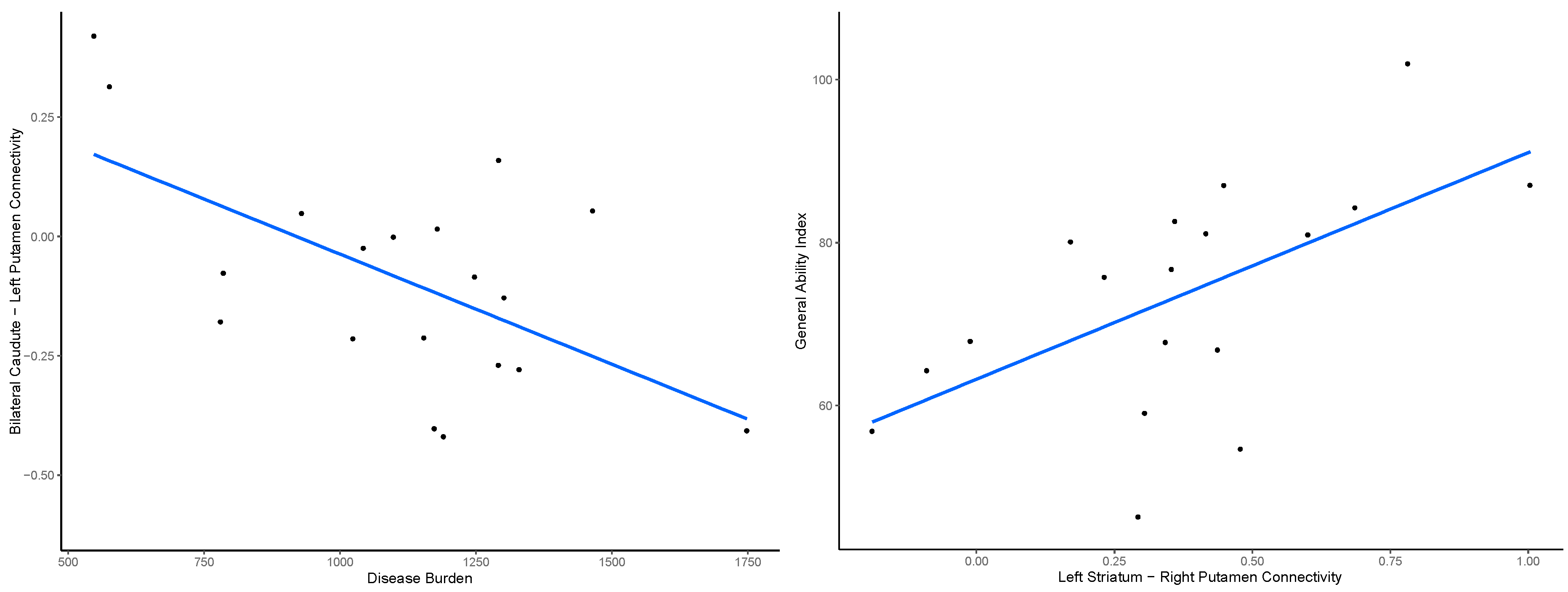
3.4. Outcome Measures
3.5. Scanner and Nonlinear Age Effects
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| JoHD | Juvenile-onset Huntington’s Disease |
| HD | Huntington’s Disease |
| GE | Gene-expanded |
| GNE | Gene-non-expanded |
| CAG | Cytosine-adenine-guanine |
| MRI | Magnetic resonance imaging |
| GAI | General ability index |
| ROI | Region of interest |
| UHDRS | Unified Huntington’s Disease Rating Scale |
| TMS | Total motor score |
| LME | Linear Mixed-Effects |
References
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntington’s Disease Chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Langbehn, D.R.; Brinkman, R.R.; Falush, D.; Paulsen, J.S.; Hayden, M.R. International Huntington’s Disease Collaborative Group A New Model for Prediction of the Age of Onset and Penetrance for Huntington’s Disease Based on CAG Length. Clin. Genet. 2004, 65, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Roos, R.A.C. Huntington’s Disease: A Clinical Review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Douglas, I.; Evans, S.; Rawlins, M.D.; Smeeth, L.; Tabrizi, S.J.; Wexler, N.S. Juvenile Huntington’s Disease: A Population-Based Study Using the General Practice Research Database. BMJ Open 2013, 3, e002085. [Google Scholar] [CrossRef]
- Roos, R. Orpha.net: Juvenile Huntington Disease. Available online: https://www.orpha.net (accessed on 5 March 2025).
- Achenbach, J.; Thiels, C.; Lücke, T.; Saft, C. Clinical Manifestation of Juvenile and Pediatric HD Patients: A Retrospective Case Series. Brain Sci. 2020, 10, 340. [Google Scholar] [CrossRef]
- Tereshchenko, A.V.; Schultz, J.L.; Bruss, J.E.; Magnotta, V.A.; Epping, E.A.; Nopoulos, P.C. Abnormal Development of Cerebellar-Striatal Circuitry in Huntington Disease. Neurology 2020, 94, e1908–e1915. [Google Scholar] [CrossRef]
- Schultz, J.L.; Langbehn, D.R.; Al-Kaylani, H.M.; van der Plas, E.; Koscik, T.R.; Epping, E.A.; Espe-Pfeifer, P.B.; Martin, E.P.; Moser, D.J.; Magnotta, V.A.; et al. Longitudinal Clinical and Biological Characteristics in Juvenile-Onset Huntington’s Disease. Mov. Disord. 2023, 38, 113–122. [Google Scholar] [CrossRef]
- White, N.; Roddey, C.; Shankaranarayanan, A.; Han, E.; Rettmann, D.; Santos, J.; Kuperman, J.; Dale, A. PROMO: Real-Time Prospective Motion Correction in MRI Using Image-Based Tracking. Magn. Reson. Med. 2010, 63, 91–105. [Google Scholar] [CrossRef]
- Tustison, N.J.; Cook, P.A.; Holbrook, A.J.; Johnson, H.J.; Muschelli, J.; Devenyi, G.A.; Duda, J.T.; Das, S.R.; Cullen, N.C.; Gillen, D.L.; et al. The ANTsX Ecosystem for Quantitative Biological and Medical Imaging. Sci. Rep. 2021, 11, 9068. [Google Scholar] [CrossRef]
- Behzadi, Y.; Restom, K.; Liau, J.; Liu, T.T. A Component Based Noise Correction Method (CompCor) for BOLD and Perfusion Based FMRI. Neuroimage 2007, 37, 90–101. [Google Scholar] [CrossRef]
- Schaefer, A.; Kong, R.; Gordon, E.M.; Laumann, T.O.; Zuo, X.-N.; Holmes, A.J.; Eickhoff, S.B.; Yeo, B.T.T. Local-Global Parcellation of the Human Cerebral Cortex from Intrinsic Functional Connectivity MRI. Cereb. Cortex 2018, 28, 3095–3114. [Google Scholar] [CrossRef] [PubMed]
- Young Kim, E.; Johnson, H.J. Robust Multi-Site MR Data Processing: Iterative Optimization of Bias Correction, Tissue Classification, and Registration. Front. Neuroinform. 2013, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Jenkinson, M.; Woolrich, M.W.; Beckmann, C.F.; Behrens, T.E.J.; Johansen-Berg, H.; Bannister, P.R.; De Luca, M.; Drobnjak, I.; Flitney, D.E.; et al. Advances in Functional and Structural MR Image Analysis and Implementation as FSL. Neuroimage 2004, 23 (Suppl. 1), S208–S219. [Google Scholar] [CrossRef] [PubMed]
- Huntington Study Group. Unified Huntington’s Disease Rating Scale: Reliability and Consistency. Mov. Disord. 1996, 11, 136–142. [Google Scholar] [CrossRef]
- Wechsler, D. Wechsler Intelligence Scale for Children, 5th ed.; Pearson: London, UK, 2014. [Google Scholar]
- Wechsler, D. Wechsler Adult Intelligence Scale, 4th ed.; Pearson: London, UK, 2008. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling False Discovery Rate—Practical Powerful Approach Multiple Testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Tereshchenko, A.; Magnotta, V.; Epping, E.; Mathews, K.; Espe-Pfeifer, P.; Martin, E.; Dawson, J.; Duan, W.; Nopoulos, P. Brain Structure in Juvenile-Onset Huntington Disease. Neurology 2019, 92, e1939–e1947. [Google Scholar] [CrossRef]
- Neema, M.; Schultz, J.L.; Langbehn, D.R.; Conrad, A.L.; Epping, E.A.; Magnotta, V.A.; Nopoulos, P.C. Mutant Huntingtin Drives Development of an Advantageous Brain Early in Life: Evidence in Support of Antagonistic Pleiotropy. Ann. Neurol. 2024, 96, 1006–1019. [Google Scholar] [CrossRef]
- Jiang, A.; You, L.; Handley, R.R.; Hawkins, V.; Reid, S.J.; Jacobsen, J.C.; Patassini, S.; Rudiger, S.R.; Mclaughlan, C.J.; Kelly, J.M.; et al. Single Nuclei RNA-Seq Reveals a Medium Spiny Neuron Glutamate Excitotoxicity Signature Prior to the Onset of Neuronal Death in an Ovine Huntington’s Disease Model. Hum. Mol. Genet. 2024, 33, 1524–1539. [Google Scholar] [CrossRef]
- Estevez-Fraga, C.; Altmann, A.; Parker, C.S.; Scahill, R.I.; Costa, B.; Chen, Z.; Manzoni, C.; Zarkali, A.; Durr, A.; Roos, R.A.C.; et al. Genetic Topography and Cortical Cell Loss in Huntington’s Disease Link Development and Neurodegeneration. Brain 2023, 146, 4532–4546. [Google Scholar] [CrossRef]
- Stocksdale, J.T.; Leventhal, M.J.; Lam, S.; Xu, Y.-X.; Wang, Y.O.; Wang, K.Q.; Thomas, R.; Faghihmonzavi, Z.; Raghav, Y.; Smith, C.; et al. Intersecting Impact of CAG Repeat and Huntingtin Knockout in Stem Cell-Derived Cortical Neurons. Neurobiol. Dis. 2025, 210, 106914. [Google Scholar] [CrossRef]
- Kuhlmann, N.; Wagner Valladolid, M.; Quesada-Ramírez, L.; Farrer, M.J.; Milnerwood, A.J. Chronic and Acute Manipulation of Cortical Glutamate Transmission Induces Structural and Synaptic Changes in Co-Cultured Striatal Neurons. Front. Cell. Neurosci. 2021, 15, 569031. [Google Scholar] [CrossRef] [PubMed]
- Lila, E.; Hunt, D.; Child, D.D.; Latimer, C.; Le, B.; Davis, M.; Jayadev, S.; Bird, T.D.; Shojaie, A.; Mac Donald, C.L. Asymmetric Brain Atrophy in Huntington’s Disease: A Postmortem MRI Study. J. Huntingt. Dis. 2025, 18796397251333334. [Google Scholar] [CrossRef] [PubMed]
- Byun, S.; Lee, M.; Kim, M. Gene Therapy for Huntington’s Disease: The Final Strategy for a Cure? J. Mov. Disord. 2022, 15, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Alegre, P.; Afifi, A.K. Clinical Characteristics of Childhood-Onset (Juvenile) Huntington Disease: Report of 12 Patients and Review of the Literature. J. Child Neurol. 2006, 21, 223–229. [Google Scholar] [CrossRef]
- Oosterloo, M.; Touze, A.; Byrne, L.M.; Achenbach, J.; Aksoy, H.; Coleman, A.; Lammert, D.; Nance, M.; Nopoulos, P.; Reilmann, R.; et al. Clinical Review of Juvenile Huntington’s Disease. J. Huntingt. Dis. 2024, 13, 149–161. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Control (n = 64) | JoHD (n = 19) | |||
|---|---|---|---|---|
| n (%) | n (%) | X2 | p | |
| Age (mean ± standard deviation) | 14.33 ± 3.25 | 17.35 ± 5.75 | t = −2.19 | 0.04 |
| Sex | X2 = 0 | 1 | ||
| Female | 38 (59%) | 11 (58%) | ||
| Male | 26 (41%) | 8 (42%) | ||
| CAG Repeats | 20.31 ± 4.16 | 66.84 ± 10.97 | −18.11 | <0.001 |
| Age at Diagnosis | NA | 13.67 ± 4.86 | NA | NA |
| Disease Duration | NA | 4.08 ± 2.92 | NA | NA |
| Network | Predictor | Estimate | SE | df | t | p-adj | 95% CI |
|---|---|---|---|---|---|---|---|
| Left Visual | Group | −0.01 | 0.03 | 79.00 | −0.41 | 0.87 | [−0.067, 0.043] |
| Age | −0.001 | 0.003 | 79.00 | −0.42 | 0.87 | [−0.007, 0.004] | |
| Sex | −0.004 | 0.02 | 79.00 | −0.17 | 0.87 | [−0.049, 0.041] | |
| Left Somatomotor | Group | −0.09 | 0.04 | 78.24 | −2.50 | 0.03 * | [−0.163, −0.018] |
| Age | −0.01 | 0.004 | 78.98 | −2.22 | 0.04 * | [−0.016, 0.000] | |
| Sex | 0.02 | 0.03 | 78.06 | 0.75 | 0.45 | [−0.035, 0.081] | |
| Left Dorsal Attention | Group | 0.03 | 0.04 | 78.27 | 0.78 | 0.58 | [−0.052, 0.115] |
| Age | −0.01 | 0.004 | 78.87 | −1.14 | 0.52 | [−0.015, 0.003] | |
| Sex | −0.01 | 0.03 | 78.07 | −0.15 | 0.88 | [−0.074, 0.062] | |
| Left Salience | Group | −0.04 | 0.03 | 78.34 | −1.32 | 0.25 | [−0.090, 0.015] |
| Age | 0.004 | 0.003 | 78.44 | 1.52 | 0.25 | [−0.002, 0.010] | |
| Sex | −0.02 | 0.02 | 78.09 | −0.87 | 0.39 | [−0.062, 0.023] | |
| Left Limbic | Group | 0.03 | 0.06 | 78.43 | 0.43 | 0.67 | [−0.091, 0.157] |
| Age | −0.01 | 0.01 | 77.35 | −0.84 | 0.54 | [−0.018, 0.010] | |
| Sex | −0.08 | 0.05 | 78.11 | −1.65 | 0.21 | [−0.182, 0.018] | |
| Left Control | Group | 0.05 | 0.04 | 78.33 | 1.35 | 0.28 | [−0.026, 0.124] |
| Age | −0.01 | 0.004 | 78.50 | −1.26 | 0.28 | [−0.014, 0.002] | |
| Sex | −0.03 | 0.03 | 78.09 | −0.88 | 0.38 | [−0.088, 0.032] | |
| Left Default Mode | Group | −0.03 | 0.02 | 78.52 | −1.42 | 0.32 | [−0.071, 0.009] |
| Age | 0.0003 | 0.002 | 75.51 | 0.12 | 0.90 | [−0.005, 0.004] | |
| Sex | −0.01 | 0.02 | 78.14 | −0.77 | 0.59 | [−0.046, 0.019] | |
| Right Visual | Group | 0.02 | 0.03 | 78.67 | 0.55 | 0.58 | [−0.039, 0.078] |
| Age | 0.003 | 0.003 | 70.78 | 1.00 | 0.58 | [−0.002, 0.010] | |
| Sex | −0.02 | 0.02 | 78.19 | −0.71 | 0.58 | [−0.064, 0.031] | |
| Right Somatomotor | Group | 0.05 | 0.04 | 78.19 | 1.03 | 0.41 | [−0.042, 0.129] |
| Age | −0.01 | 0.005 | 78.99 | −1.18 | 0.41 | [−0.015, 0.003] | |
| Sex | 0.01 | 0.04 | 78.05 | 0.29 | 0.77 | [−0.060, 0.079] | |
| Right Dorsal Attention | Group | 0.02 | 0.05 | 78.29 | 0.47 | 0.64 | [−0.074, 0.114] |
| Age | −0.01 | 0.01 | 78.79 | −0.97 | 0.64 | [−0.016, 0.004] | |
| Sex | −0.03 | 0.04 | 78.07 | −0.67 | 0.64 | [−0.103, 0.049] | |
| Right Salience | Group | 0.004 | 0.03 | 78.26 | 0.11 | 0.98 | [−0.062, 0.067] |
| Age | 0.002 | 0.004 | 78.93 | 0.55 | 0.98 | [−0.006, 0.008] | |
| Sex | 0.0006 | 0.03 | 78.06 | 0.02 | 0.98 | [−0.052, 0.052] | |
| Right Limbic | Group | −0.01 | 0.10 | 79.00 | −0.10 | 0.92 | [−0.194, 0.174] |
| Age | −0.01 | 0.01 | 79.00 | −1.27 | 0.35 | [−0.031, 0.006] | |
| Sex | 0.09 | 0.08 | 79.00 | 1.13 | 0.35 | [−0.063, 0.237] | |
| Right Control | Group | 0.02 | 0.03 | 78.67 | 0.67 | 0.61 | [−0.032, 0.073] |
| Age | 0.002 | 0.003 | 70.73 | 0.54 | 0.61 | [−0.003, 0.008] | |
| Sex | −0.01 | 0.02 | 78.19 | −0.51 | 0.61 | [−0.053, 0.032] | |
| Right Default Mode | Group | 0.02 | 0.03 | 78.67 | 0.55 | 0.58 | [−0.039, 0.078] |
| Age | 0.003 | 0.003 | 70.78 | 1.00 | 0.58 | [−0.002, 0.010] | |
| Sex | −0.02 | 0.02 | 78.19 | −0.71 | 0.58 | [−0.064, 0.031] |
| Roi Pair | Estimate | SE | df | t | p-adj | 95% CI |
|---|---|---|---|---|---|---|
| Left striatum–right striatum | −0.48 | 0.08 | 78.56 | −6.21 | <0.001 * | [−0.63, −0.33] |
| Left striatum–right caudate | −0.35 | 0.08 | 78.71 | −4.64 | <0.001 * | [−0.51, −0.21] |
| Left striatum–right putamen | −0.36 | 0.08 | 78.87 | −4.61 | <0.001 * | [−0.52, −0.21] |
| Right striatum–left caudate | −0.18 | 0.08 | 79 | −2.34 | 0.04 * | [−0.33, −0.03] |
| Right striatum–left putamen | −0.54 | 0.08 | 78.74 | −6.59 | <0.001 * | [−0.71, −0.39] |
| Bilateral caudate–bilateral putamen | −0.36 | 0.07 | 79 | −4.79 | <0.001 * | [−0.50, −0.21] |
| Bilateral caudate–left putamen | −0.40 | 0.07 | 79 | −5.62 | <0.001 * | [−0.53, −0.26] |
| Bilateral caudate–right putamen | −0.24 | 0.08 | 79 | −3.04 | 0.01 * | [−0.39, −0.09] |
| Left caudate–right caudate | −0.35 | 0.08 | 79 | −4.36 | <0.001 * | [−0.50, −0.19] |
| Left putamen–right caudate | −0.28 | 0.08 | 78.56 | −3.69 | <0.001 * | [−0.44, −0.14] |
| Right caudate–right putamen | −0.36 | 0.09 | 78.96 | −3.98 | <0.001 * | [−0.53, −0.19] |
| Bilateral putamen–right caudate | −0.37 | 0.08 | 78.7 | −4.37 | <0.001 * | [−0.54, −0.21] |
| Bilateral putamen–left caudate | −0.25 | 0.07 | 79 | −3.43 | <0.001 * | [−0.40, −0.11] |
| Left putamen–right putamen | −0.50 | 0.09 | 78.78 | −5.69 | <0.001 * | [−0.44, −0.14] |
| Bilateral striatum–bilateral somatomotor network | −0.13 | 0.07 | 79 | −1.83 | 0.20 | [−0.68, −0.34] |
| Left striatum–bilateral somatomotor network | −0.09 | 0.07 | 79 | −1.15 | 0.51 | [−0.27, 0.01] |
| Right striatum–bilateral somatomotor network | −0.17 | 0.07 | 78.9 | −2.51 | 0.06 | [−0.23, 0.06] |
| Bilateral caudate–bilateral somatomotor network | −0.05 | 0.06 | 79 | −0.84 | 0.54 | [−0.30, −0.04] |
| Left caudate–bilateral somatomotor network | 0.02 | 0.07 | 79 | 0.25 | 0.81 | [−0.17, 0.07] |
| Right caudate–bilateral somatomotor network | −0.11 | 0.06 | 79 | −1.79 | 0.22 | [−0.11, 0.14] |
| Bilateral putamen–bilateral somatomotor network | −0.14 | 0.08 | 78.96 | −1.79 | 0.31 | [−0.23, 0.01] |
| Left putamen–bilateral somatomotor network | −0.14 | 0.08 | 79 | −1.89 | 0.15 | [−0.29, 0.01] |
| Right putamen–bilateral somatomotor network | −0.13 | 0.07 | 78.62 | −1.75 | 0.34 | [−0.29, 0.003] |
| Bilateral striatum–left somatomotor network | −0.17 | 0.07 | 78.96 | −2.35 | 0.09 | [−0.28, 0.01] |
| Left striatum–left somatomotor network | −0.09 | 0.08 | 79 | −1.14 | 0.55 | [−0.31, −0.03] |
| Right striatum–left somatomotor network | −0.21 | 0.07 | 78.8 | −3.17 | 0.01 * | [−0.24, 0.06] |
| Bilateral caudate–left somatomotor network | −0.13 | 0.07 | 79 | −1.78 | 0.31 | [−0.34, −0.08] |
| Left caudate–left somatomotor network | −0.05 | 0.07 | 79 | −0.75 | 0.77 | [−0.27, 0.01] |
| Right caudate–left somatomotor network | −0.15 | 0.07 | 79 | −2.31 | 0.09 | [−0.20, 0.09] |
| Bilateral putamen–left somatomotor network | −0.12 | 0.07 | 79 | −1.69 | 0.38 | [−0.28, −0.03] |
| Left putamen–left somatomotor network | −0.11 | 0.08 | 79 | −1.41 | 0.66 | [−0.26, 0.02] |
| Right putamen–left somatomotor network | −0.12 | 0.07 | 79 | −1.77 | 0.33 | [−0.26, 0.04] |
| Bilateral striatum–right somatomotor network | −0.08 | 0.07 | 78.51 | −1.23 | 0.61 | [−0.26, 0.01] |
| Left striatum–right somatomotor network | −0.1 | 0.06 | 79 | −1.61 | 0.22 | [−0.22, 0.04] |
| Right striatum–right somatomotor network | −0.07 | 0.07 | 78.28 | −0.98 | 0.92 | [−0.23, 0.02] |
| Bilateral caudate–right somatomotor network | 0.02 | 0.06 | 79 | 0.41 | 0.68 | [−0.20, 0.06] |
| Left caudate–right somatomotor network | 0.04 | 0.06 | 78.81 | 0.64 | 0.67 | [−0.09, 0.13] |
| Right caudate–right somatomotor network | −0.02 | 0.06 | 78.71 | −0.3 | 0.82 | [−0.08, 0.16] |
| Bilateral putamen–right somatomotor network | −0.14 | 0.08 | 78.4 | −1.8 | 0.30 | [−0.13, 0.09] |
| Left putamen–right somatomotor network | −0.16 | 0.08 | 78.78 | −2.07 | 0.17 | [−0.30, 0.01] |
| Right putamen–right somatomotor network | −0.11 | 0.07 | 78.44 | −1.46 | 0.59 | [−0.32, −0.02] |
| Left somatomotor network–right somatomotor network | −0.03 | 0.09 | 78.99 | −0.34 | 0.74 | [−0.26, 0.03] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barry, A.; Nopoulos, P.C. Cortical and Striatal Functional Connectivity in Juvenile-Onset Huntington’s Disease. Brain Sci. 2025, 15, 663. https://doi.org/10.3390/brainsci15060663
Barry A, Nopoulos PC. Cortical and Striatal Functional Connectivity in Juvenile-Onset Huntington’s Disease. Brain Sciences. 2025; 15(6):663. https://doi.org/10.3390/brainsci15060663
Chicago/Turabian StyleBarry, Amy, and Peg C. Nopoulos. 2025. "Cortical and Striatal Functional Connectivity in Juvenile-Onset Huntington’s Disease" Brain Sciences 15, no. 6: 663. https://doi.org/10.3390/brainsci15060663
APA StyleBarry, A., & Nopoulos, P. C. (2025). Cortical and Striatal Functional Connectivity in Juvenile-Onset Huntington’s Disease. Brain Sciences, 15(6), 663. https://doi.org/10.3390/brainsci15060663