
VEXAS Syndrome and Alzheimer’s Disease—Are There Connections?



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. What Is VEXAS Syndrome?
2. Alzheimer’s Disease—Brief Reminder
2.1. Clinical Features of Alzheimer’s Disease
2.2. Diagnosis and Current Treatment
2.3. Neuropathology of Alzheimer’s Disease
3. Inflammatory Process and Its Role in Alzheimer’s Disease
3.1. Role of Microglia and Astrocytes in Neuroinflammation
3.2. Inflammatory Mediators and Their Impact on Neuronal Function
3.3. The Role of Inflammation in Disease Progression and Potential Therapeutic Implications
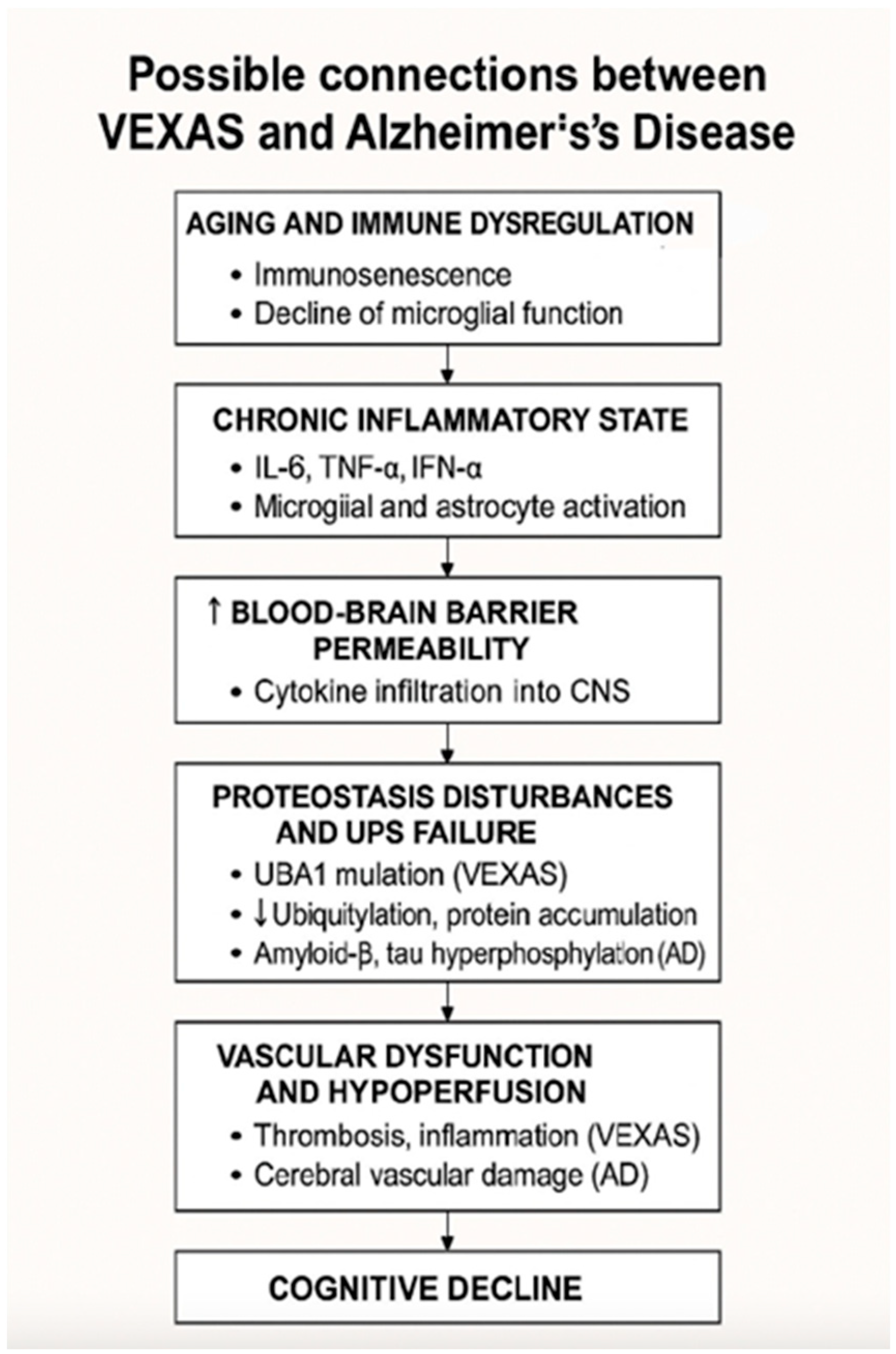
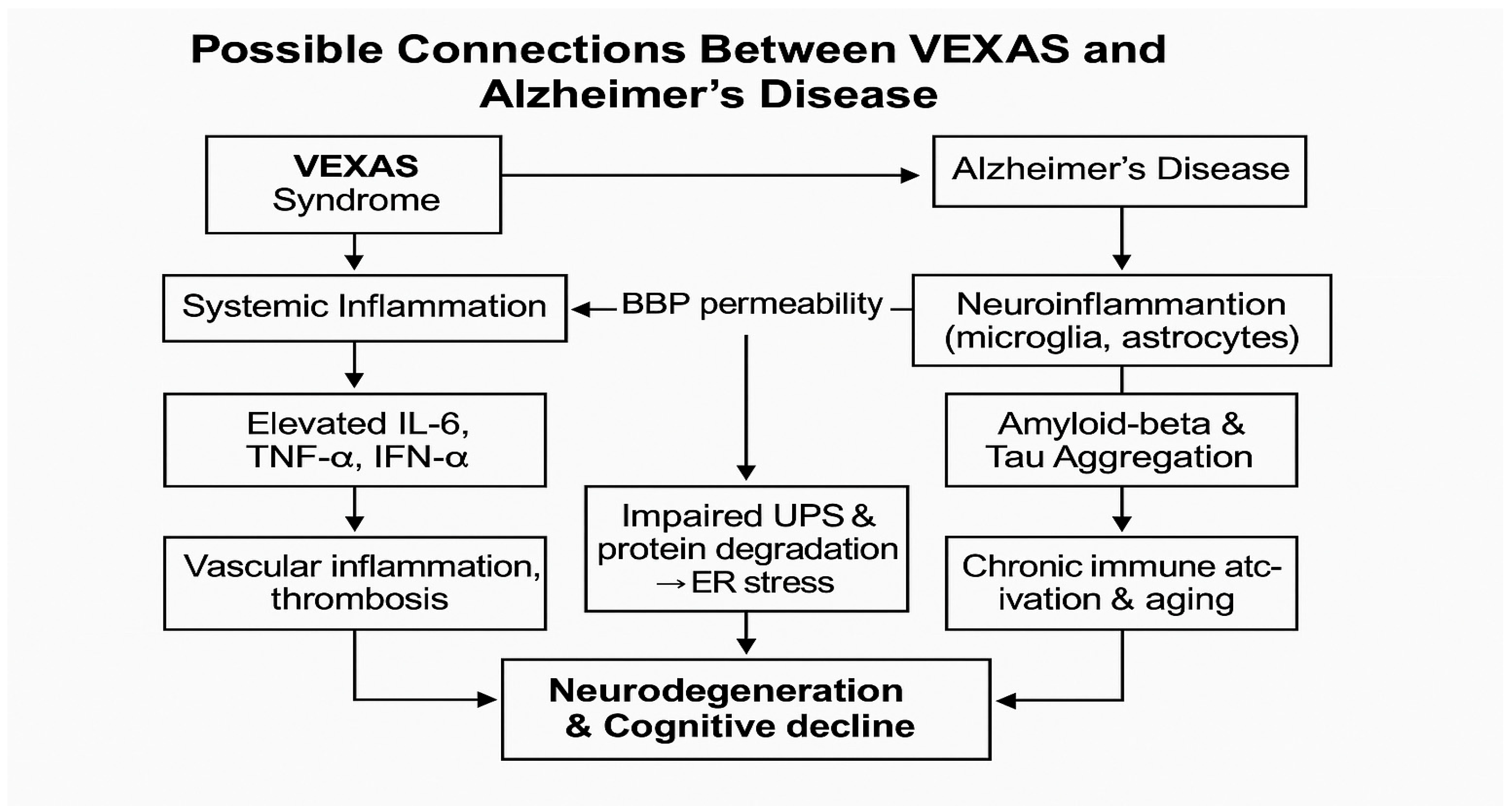
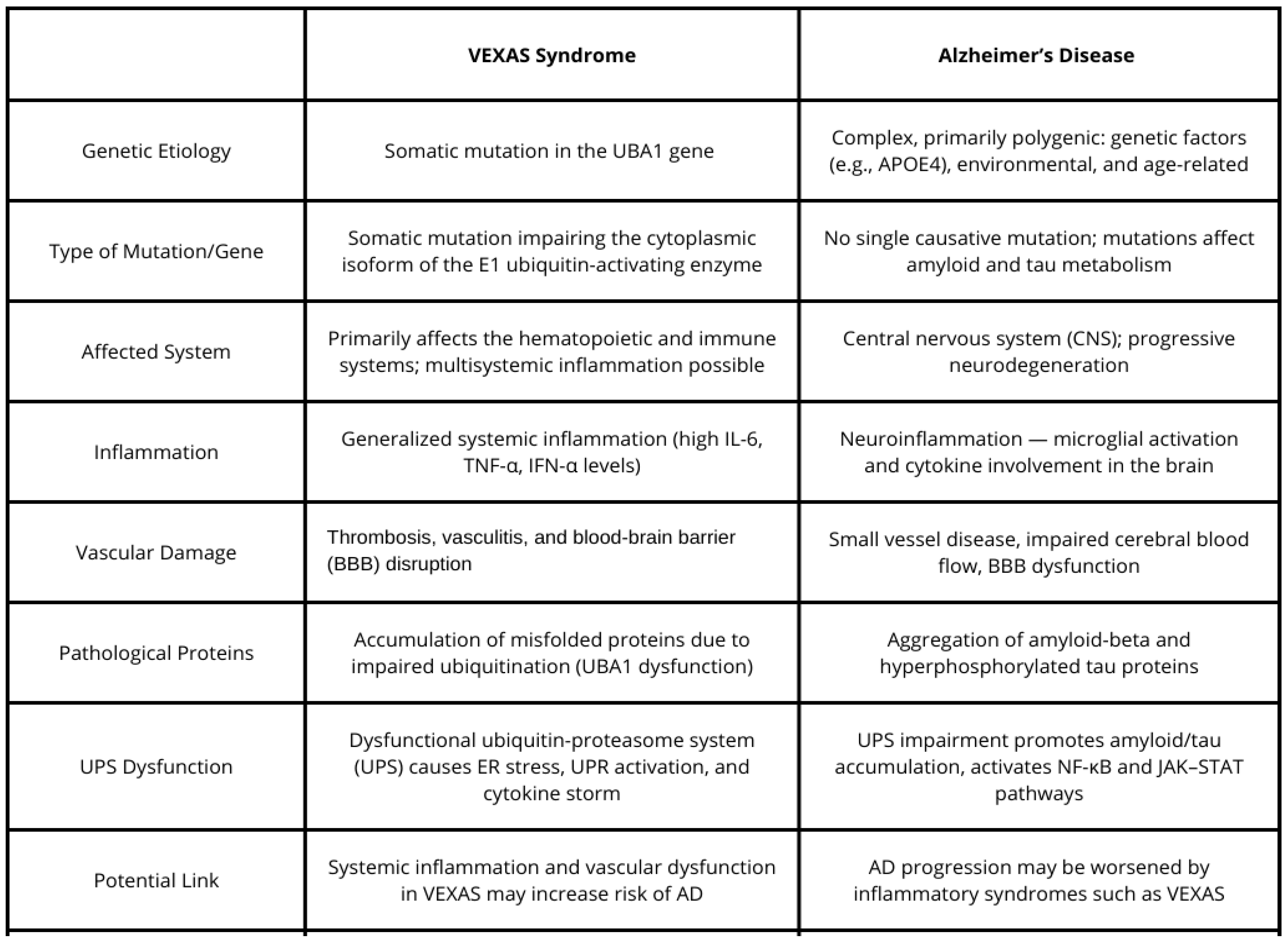
4. Possible Connections Between VEXAS and Alzheimer’s Disease
4.1. Inflammation and Neuroinflammation
4.2. Vascular Dysfunction and Cognitive Decline
4.3. Genetic Mechanisms and Protein Misfolding
4.4. Age-Related Immune Dysfunction and Cognitive Decline
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beck, D.B.; Ferrada, M.A.; Sikora, K.A.; Ombrello, A.K.; Collins, J.C.; Pei, W.; Balanda, N.; Ross, D.L.; Cardona, D.O.; Wu, Z.; et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N. Engl. J. Med. 2020, 383, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Prevalence of VEXAS Syndrome Higher than Expected, Genome-First Study Finds. Available online: https://dceg.cancer.gov (accessed on 26 March 2025).
- Obiorah, I.E.; Patel, B.A.; Groarke, E.M.; Wang, W.; Trick, M.; Ombrello, A.K.; Ferrada, M.A.; Wu, Z.; Gutierrez-Rodrigues, F.; Lotter, J.; et al. Benign and malignant hematologic manifestations in patients with VEXAS syndrome due to somatic mutations in UBA1. Blood Adv. 2021, 5, 3203–3215. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.L.; Ferrada, M.A.; Beck, D.B. Clinical features and management of VEXAS syndrome: A comprehensive review. J. Autoimmun. 2023, 130, 102865. [Google Scholar]
- Collins, J.C.; Balanda, N.; Magaziner, S.J.; English, M.; Ospina Cardona, D.; Patnaik, M.M.; Terrier, B.; Kosmider, O.; Werner, A.; Beck, D.B. Novel Disease-Causing Mutations in UBA1 Reveal Disease Mechanisms in Bone Marrow Failure and Inflammation. Blood 2022, 140 (Suppl. S1), 2914–2915. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Bertoni, D.; Magana, P.; Paramval, U.; Pidruchna, I.; Radhakrishnan, M.; Tsenkov, M.; Nair, S.; Mirdita, M.; Yeo, J.; et al. AlphaFold Protein Structure Database in 2024: Providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 2024, 52, D368–D375. [Google Scholar] [CrossRef]
- Georgin-Lavialle, S.; Terrier, B.; Olivier, K.; Mekinian, A. OP0049: Further characterization of clinical and laboratory features occurring in VEXAS syndrome in a large-scale analysis of multicenter case-series of 116 French patients. Ann. Rheum. Dis. 2022, 81, 33. [Google Scholar] [CrossRef]
- Battipaglia, G.; Vincenzi, A.; Falconi, G.; Fiore, A.; D’Agostino, F.; Iannotta, R.; Grimaldi, F.; Gurnari, C.; Galossi, E.; Crisà, E.; et al. New scenarios in Vacuoles, E1 enzyme, X linked, Autoinflammatory, Somatic (VEXAS) syndrome: Evolution from myelodysplastic syndrome to acute myeloid leukemia. Curr. Res. Transl. Med. 2023, 71, 103386. [Google Scholar] [CrossRef]
- Ferrada, M.A.; Sikora, K.A.; Luo, Y.; Wells, K.V.; Patel, B.; Groarke, E.M.; Cardona, D.O.; Rominger, E.; Hoffmann, P.; Le, M.T.; et al. Somatic Mutations in UBA1 Define a Distinct Subset of Relapsing Polychondritis Patients With VEXAS. Arthritis Rheumatol. 2021, 73, 1886–1895. [Google Scholar] [CrossRef]
- Al-Hakim, A.; Savic, S. An update on VEXAS syndrome. Expert Rev. Clin. Immunol. 2022, 19, 203–215. [Google Scholar] [CrossRef]
- Beck, D.B.; Bodian, D.L.; Shah, V.; Mirshahi, U.L.; Kim, J.; Ding, Y.; Magaziner, S.J.; Strande, N.T.; Cantor, A.; Haley, J.S.; et al. Estimated prevalence and clinical manifestations of UBA1 variants associated with VEXAS syndrome in a clinical population. JAMA 2023, 329, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Rodrigues, F.; Kusne, Y.; Fernandez, J.; Lasho, T.; Shalhoub, R.; Ma, X.; Alessi, H.; Finke, C.; Koster, M.J.; Mangaonkar, A.; et al. Spectrum of clonal hematopoiesis in VEXAS syndrome. Blood 2023, 142, 244–259. [Google Scholar] [CrossRef] [PubMed]
- Ferrada, M.A.; Savic, S.; Ospina Cardona, D.; Collins, J.C.; Alessi, H.; Gutierrez-Rodrigues, F.; Uthaya Kumar, D.B.; Wilson, L.; Goodspeed, W.; Topilow, J.S.; et al. Translation of cytoplasmic UBA1 contributes to VEXAS syndrome pathogenesis. Blood 2022, 140, 1496–1506. [Google Scholar] [CrossRef]
- Oliviero, F.; Pompilio, G.; de Bonis, M. A comprehensive overview of VEXAS syndrome and its vascular implications. Vasc. Med. 2020, 25, 417–425. [Google Scholar]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent advancements in pathogenesis, diagnostics and treatment of Alzheimer’s disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef]
- Marrero, J.A.; Beck, D.B.; Kastner, D.L. VEXAS syndrome: An inflammatory disorder with hematologic manifestations. Hematol. Oncol. Clin. N. Am. 2022, 36, 313–326. [Google Scholar]
- Lyketsos, C.G.; Carrillo, M.C.; Ryan, J.M.; Khachaturian, A.S.; Trzepacz, P.; Amatniek, J.; Cedarbaum, J.; Brashear, R.; Miller, D.S. Neuropsychiatric symptoms in Alzheimer’s disease: Phenomenology, epidemiology, and treatment. J. Clin. Psychiatry 2000, 61 (Suppl. S1), 5–13. [Google Scholar]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Birks, J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst. Rev. 2006, 2006, CD005593. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau pathology in Alzheimer’s disease and other tauopathies. Biochim. Biophys. Acta 2010, 1802, 1316–1335. [Google Scholar]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Frost Georgia, R.; Li, Y.-M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Wyss-Coray, T. Inflammation in Alzheimer disease: A brief review of the literature. J. Alzheimers Dis. 2006, 9, 303–312. [Google Scholar]
- Sharo, C.; Zhai, T.; Huang, Z. Investigation of Potential Drug Targets Involved in Inflammation Contributing to Alzheimer’s Disease Progression. Pharmaceuticals 2024, 17, 137. [Google Scholar] [CrossRef]
- Townsend, M.; Laver, J. Inflammation and Alzheimer’s disease: The potential of anti-inflammatory therapies. J. Clin. Immunol. 2009, 29, 770–778. [Google Scholar]
- Gérard, L.; Frolow, M.; Jourdain, R. VEXAS syndrome: A review of clinical and genetic findings. Autoimmun. Rev. 2022, 21, 102723. [Google Scholar]
- Poulter, J.A.; Collins, J.C.; Cargo, C.; De Tute, R.M.; Evans, P.; Ospina Cardona, D.; Bowen, D.T.; Cunnington, J.R.; Baguley, E.; Quinn, M.; et al. Novel somatic mutations in UBA1 as a cause of VEXAS syndrome. Blood 2021, 137, 3676–3681. [Google Scholar] [CrossRef] [PubMed]
- Sakata, K.; Fujii, H.; Ueno, Y. VEXAS syndrome: Pathogenesis, clinical features, and treatment. Front. Immunol. 2021, 12, 732611. [Google Scholar]
- Wu, Z.; Gao, S.; Gao, Q.; Patel, B.A.; Groarke, E.M.; Feng, X.; Manley, A.L.; Li, H.; Ospina Cardona, D.; Kajigaya, S.; et al. Early activation of inflammatory pathways in UBA1-mutated hematopoietic stem and progenitor cells in VEXAS. Cell Rep. Med. 2023, 4, 101160. [Google Scholar] [CrossRef]
- Foulongne, V.; Guillot, L. The X-linked UBA1 mutation in VEXAS syndrome: Implications for pathogenesis and treatment. Rheumatology 2021, 60, 585–590. [Google Scholar]
- Loeza-Uribe, M.P.; Hinojosa-Azaola, A.; Sánchez-Hernández, B.E.; Crispín, J.C.; Apodaca-Chávez, E.; Ferrada, M.A.; Martín-Nares, E. VEXAS syndrome: Clinical manifestations, diagnosis, and treatment. Reumatol. Clin. 2024, 20, 47–56. [Google Scholar] [CrossRef]
- Xie, J.; Van Hoecke, L.; Vandenbroucke, R.E. The Impact of Systemic Inflammation on Alzheimer’s Disease Pathology. Front. Immunol. 2022, 12, 796867. [Google Scholar] [CrossRef]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Targeting neuroinflammation in Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8, 704–712. [Google Scholar]
- Roy, E.R.; Chiu, G.; Li, S.; Propson, N.E.; Kanchi, R.; Wang, B.; Coarfa, C.; Zheng, H.; Cao, W. Concerted type I interferon signaling in microglia and neural cells promotes memory impairment associated with amyloid β plaques. Immunity 2022, 55, 879–894.e6. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Gao, W.; Zhang, M.; Zhang, F.; Sun, X.; Wu, B.; Liu, Y.; Li, X.; Li, H. Diagnostic implications of ubiquitination-related gene signatures in Alzheimer’s disease. Sci. Rep. 2024, 14, 10728. [Google Scholar] [CrossRef]
- Galea, I. The blood-brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef] [PubMed]
- McManus, R.M.; Higgins, S.C.; Mills, K.H.; Lynch, M.A. Respiratory infection promotes T cell infiltration and amyloid-β deposition in APP/PS1 mice. Neurobiol. Aging 2014, 35, 109–121. [Google Scholar] [CrossRef]
- Moore, Z.; Mobilio, F.; Walker, F.R.; Taylor, J.M.; Crack, P.J. Abrogation of type-I interferon signalling alters the microglial response to Aβ1-42. Sci. Rep. 2020, 10, 3153. [Google Scholar] [CrossRef]
- Chen, L.; Ruan, G.; Cheng, Y.; Yi, A.; Chen, D.; Wei, Y. The role of Th17 cells in inflammatory bowel disease and the research progress. Front. Immunol. 2023, 13, 1055914. [Google Scholar] [CrossRef]
- Roy, E.R.; Wang, B.; Wan, Y.-W.; Chiu, G.; Cole, A.; Yin, Z.; Propson, N.E.; Xu, Y.; Jankowsky, J.L.; Liu, Z.; et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Investig. 2020, 130, 1912–1930. [Google Scholar] [CrossRef]
- Minter, M.R.; Moore, Z.; Zhang, M.; Brody, K.M.; Jones, N.C.; Shultz, S.R.; Taylor, J.M.; Crack, P.J. Deletion of the type-1 interferon receptor in APPSWE/PS1ΔE9 mice preserves cognitive function and alters glial phenotype. Acta Neuropathol. Commun. 2016, 4, 72. [Google Scholar] [CrossRef]
- Chen, Z.; Lu, Y.; Wang, Y.; Wang, Q.; Yu, L.; Liu, J. Natural products targeting tau protein phosphorylation: A promising therapeutic avenue for Alzheimer’s disease. Planta Med. 2025, in press. [Google Scholar] [CrossRef]
- Langlois, V.; Curie, A.; Demas, A.; Etancelin, P.; Sauvètre, G.; Leclancher, A.; Mekinian, A. Central nervous system vasculitis in VEXAS syndrome: A rare involvement. Clin. Neurol. Neurosurg. 2024, 242, 108351. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Song, W.; Xu, Z.; Danziger, M.M.; Karavani, E.; Zang, C.; Chen, X.; Li, Y.; Paz, I.M.R.; Gohel, D.; et al. Single-microglia transcriptomic transition network-based prediction and real-world patient data validation identifies ketorolac as a repurposable drug for Alzheimer’s disease. Alzheimers Dement. 2025, 21, e14373. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sowa, A.; Malicka, M.; Biernacka, M.; Beszłej, J.A.; Leszek, J. VEXAS Syndrome and Alzheimer’s Disease—Are There Connections? Brain Sci. 2025, 15, 573. https://doi.org/10.3390/brainsci15060573
Sowa A, Malicka M, Biernacka M, Beszłej JA, Leszek J. VEXAS Syndrome and Alzheimer’s Disease—Are There Connections? Brain Sciences. 2025; 15(6):573. https://doi.org/10.3390/brainsci15060573
Chicago/Turabian StyleSowa, Aleksandra, Marta Malicka, Magdalena Biernacka, Jan Aleksander Beszłej, and Jerzy Leszek. 2025. "VEXAS Syndrome and Alzheimer’s Disease—Are There Connections?" Brain Sciences 15, no. 6: 573. https://doi.org/10.3390/brainsci15060573
APA StyleSowa, A., Malicka, M., Biernacka, M., Beszłej, J. A., & Leszek, J. (2025). VEXAS Syndrome and Alzheimer’s Disease—Are There Connections? Brain Sciences, 15(6), 573. https://doi.org/10.3390/brainsci15060573