



Extracellular Vesicles and Purinergic Signaling in Alzheimer’s Disease—Joining Forces for Novel Therapeutic Approach



Abstract
1. Introduction
1.1. Alzheimer’s Disease
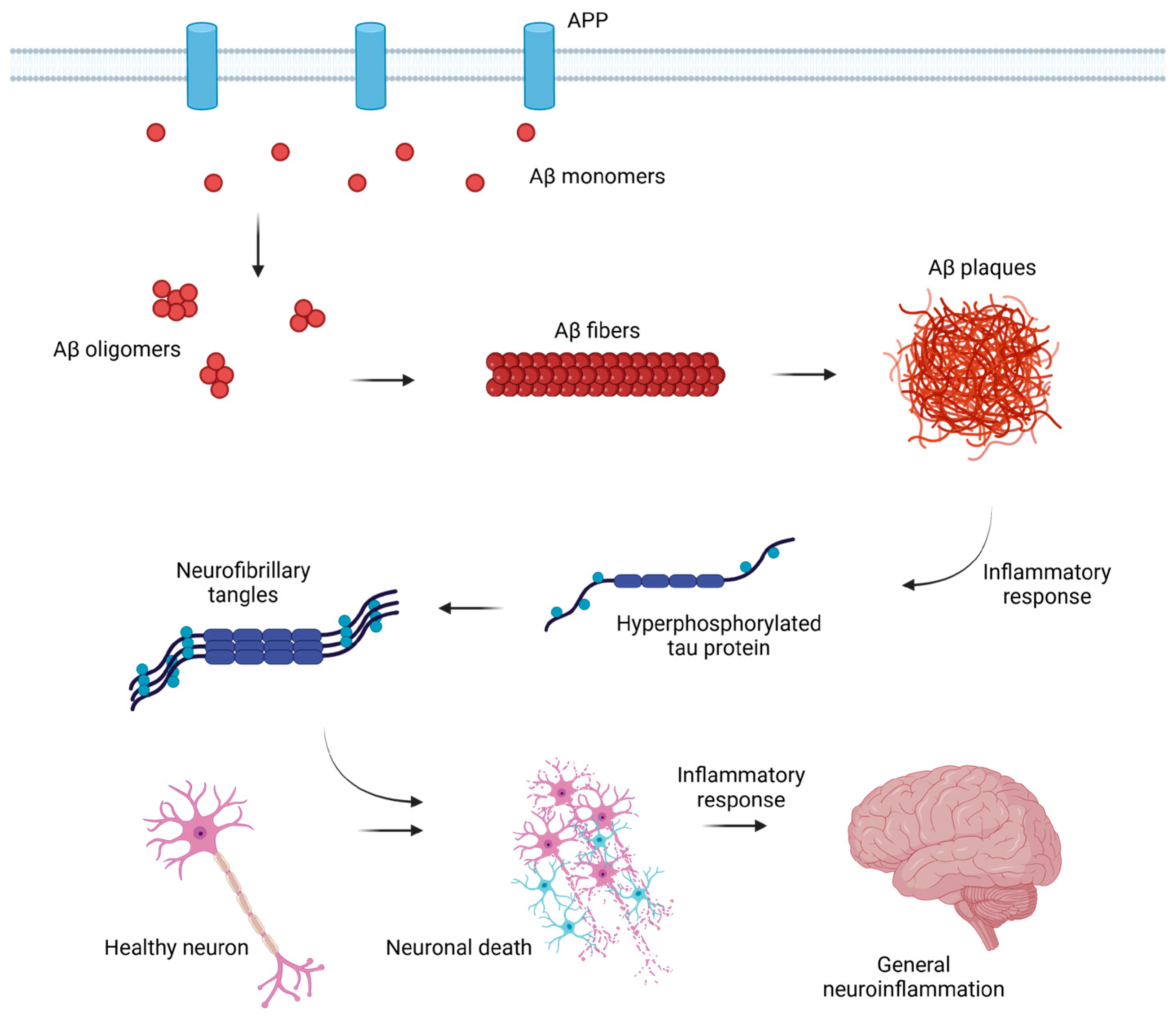
1.2. Mechanism of the Disorder Development
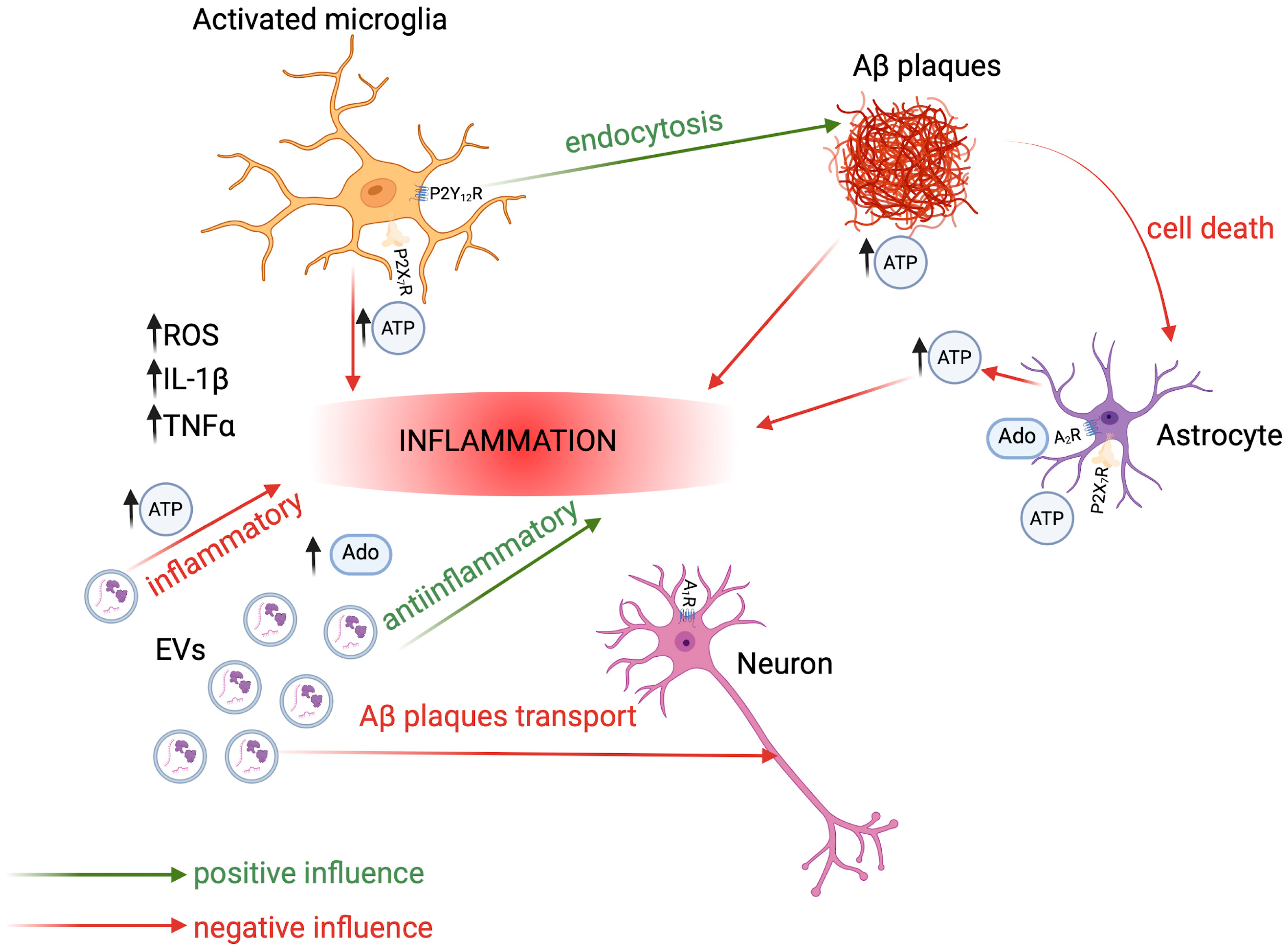
1.3. Basics of Neuroinflammation in AD
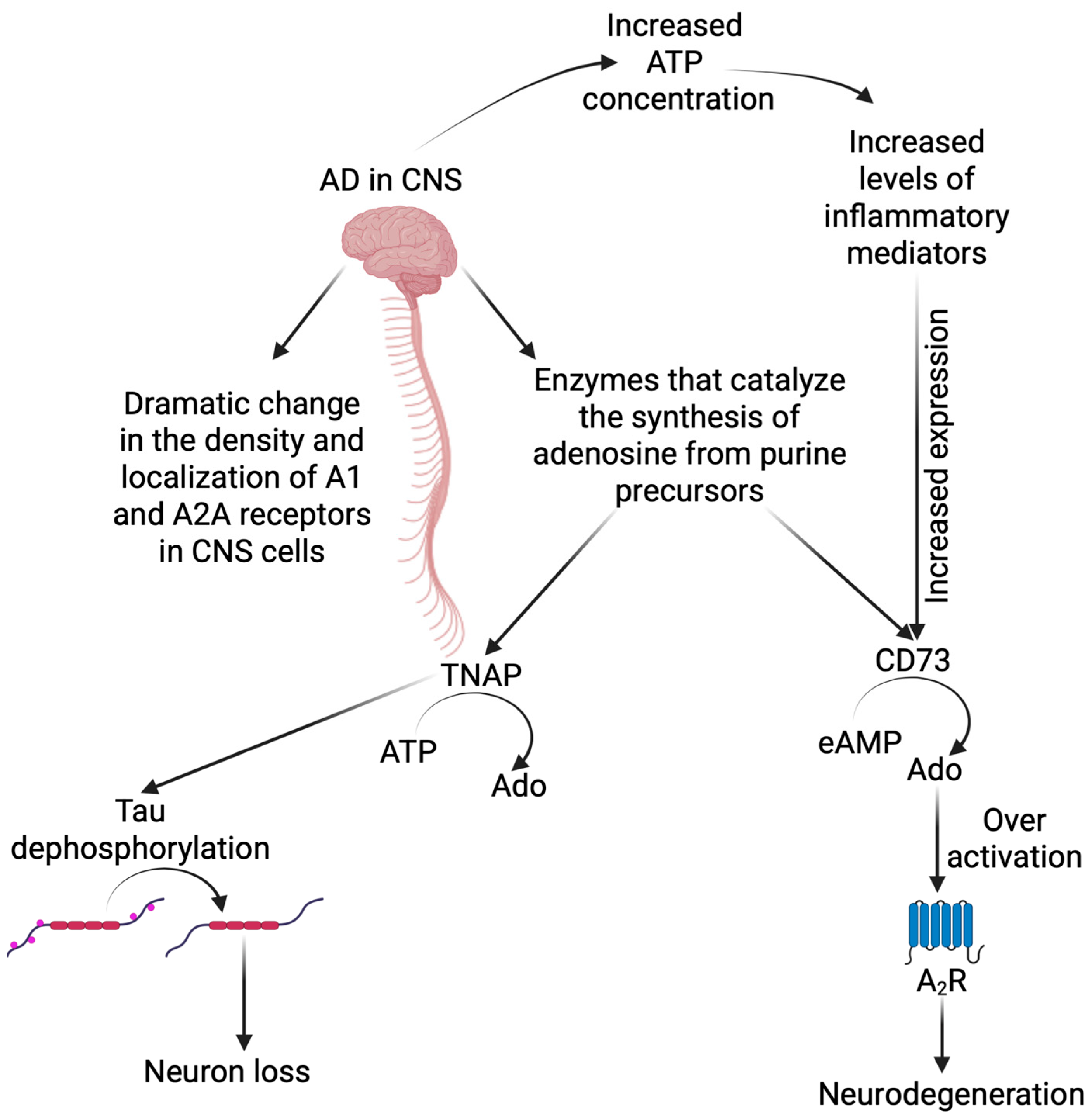
2. Purinergic Signaling in CNS and Its Role in Alzheimer’s Disease Development
2.1. Adenosine and Its Receptors
2.2. ATP and Its Receptors
2.2.1. P2X Receptors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Purinergic Receptor | Placement in the CNS | Receptor Activation Effect | Reference |
|---|---|---|---|
| P2X1 | Postsynaptic neurons | Interneuron depolarization | [102] |
| P2X2 | Presynaptic neurons | Increase in glutamate release | [102] |
| P2X3 | Presynaptic neurons | Interneuron depolarization | [103] |
| P2X4 | Microglia | Increase in migration and secretion | [104] |
| P2X7 | Microglia | Activation of inflammasome | [105,106] |
| Leukocytes | Necrotic cytolysis Apoptotic death | [99] | |
| Oligodendrocytes | Promotion of demyelination | [107] |
2.2.2. P2Y Receptors
2.3. Main Players in Purinergic Signaling in AD
2.3.1. Astrocytes
2.3.2. Microglia
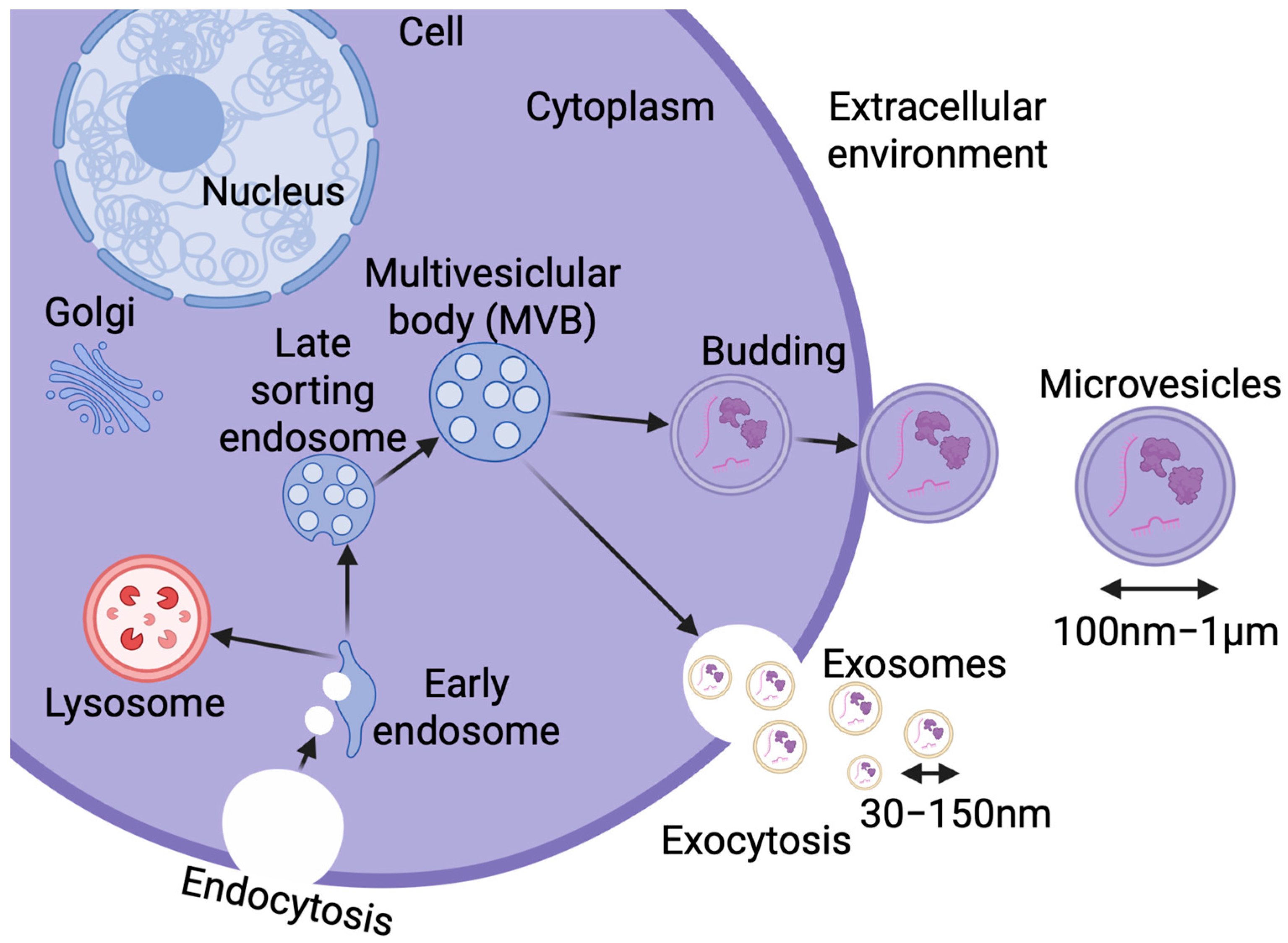
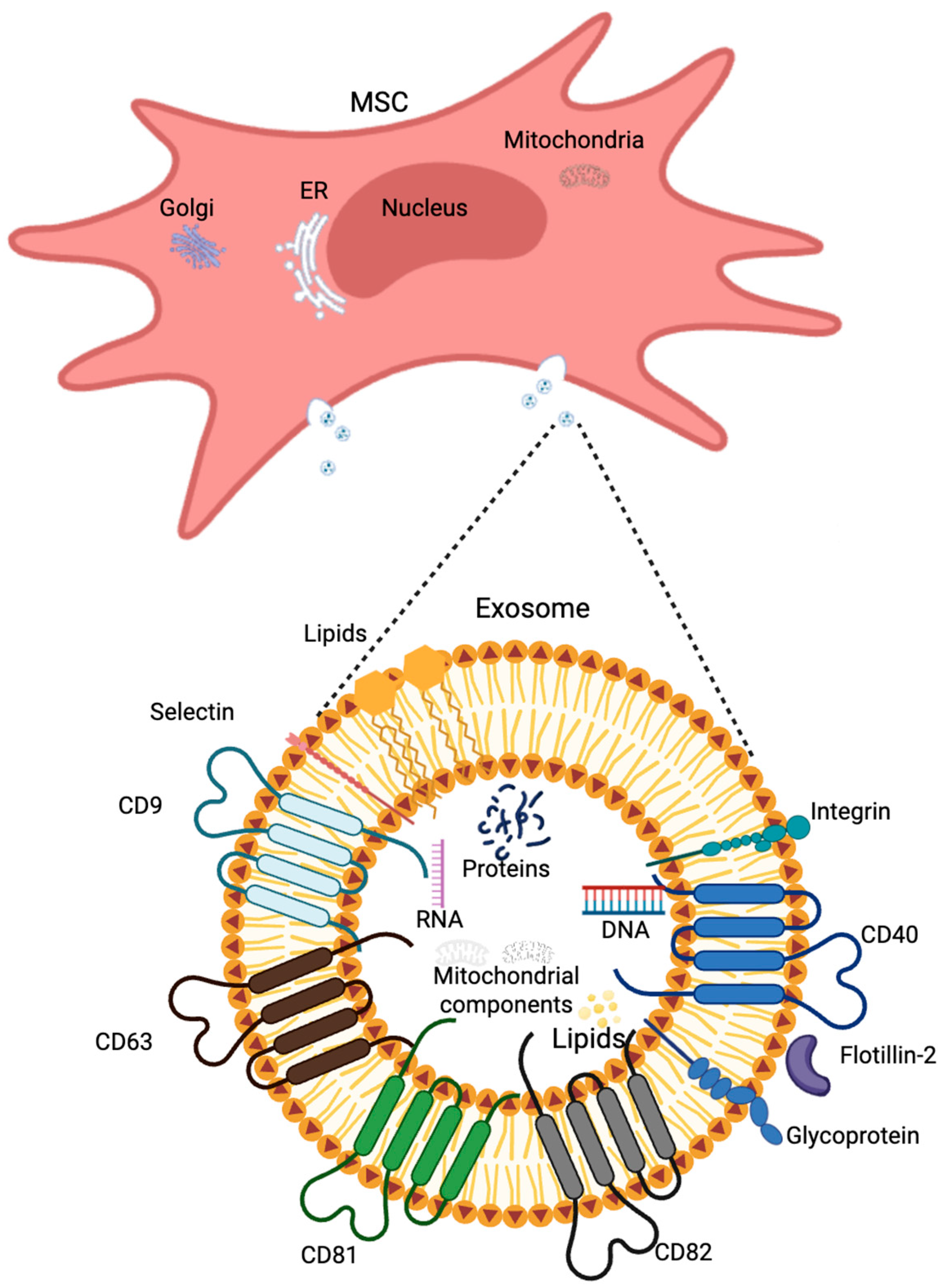
2.3.3. Extracellular Vesicles
3. Extracellular Vesicles Contribution to Alzheimer’s Disease
Therapeutic Potential of Extracellular Vesicles with Purinergic Compounds
4. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Serý, O.; Povová, J.; Míšek, I.; Pešák, L.; Janout, V. Molecular mechanisms of neuropathological changes in Alzheimer’s disease: A review. Folia Neuropathol. 2013, 51, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Small, D.H.; Cappai, R. Alois Alzheimer and Alzheimer’s disease: A centennial perspective. J Neurochem. 2006, 99, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Long, S.; Benoist, C.; Weidner, W. World Alzheimer Report 2023: Reducing Dementia Risk: Never Too Early, Never Too Late; Alzheimer’s Disease International: London, UK, 2023. [Google Scholar]
- World Health Organization. Global Action Plan on the Public Health Response to Dementia 2017–2025; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Yin, T.; Liu, Y.; Ji, W.; Zhuang, J.; Chen, X.; Gong, B.; Chu, J.; Liang, W.; Gao, J.; Yin, Y. Engineered mesenchymal stem cell-derived extracellular vesicles: A state-of-the-art multifunctional weapon against Alzheimer’s disease. Theranostics 2023, 13, 1264–1285. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lamptey, R.N.L.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Approaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vandendriessche, C.; Bruggeman, A.; Van Cauwenberghe, C.; Vandenbroucke, R.E. Extracellular Vesicles in Alzheimer’s and Parkinson’s Disease: Small Entities with Large Consequences. Cells 2020, 9, 2485. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, Z.; Wang, X.; Wang, X.; Yi, X.; Wong, Y.K.; Wu, J.; Xie, F.; Hu, D.; Wang, Q.; Wang, J.; et al. Research progress on the role of extracellular vesicles in neurodegenerative diseases. Transl. Neurodegener. 2023, 12, 43. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fox, N.C.; Scahill, R.I.; Crum, W.R.; Rossor, M.N. Correlation between rates of brain atrophy and cognitive decline in AD. Neurology 1999, 52, 1687–1689. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Petersen, R.C.; Xu, Y.C.; Waring, S.C.; O’Brien, P.C.; Tangalos, E.G.; Smith, G.E.; Ivnik, R.J.; Kokmen, E. Medial temporal atrophy on MRI in normal aging and very mild Alzheimer’s disease. Neurology 1997, 49, 786–794. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Piguet, O.; Double, K.L.; Kril, J.J.; Harasty, J.; Macdonald, V.; McRitchie, D.A.; Halliday, G.M. White matter loss in healthy ageing: A postmortem analysis. Neurobiol. Aging 2009, 30, 1288–1895. [Google Scholar] [CrossRef] [PubMed]
- Stout, J.C.; Jernigan, T.L.; Archibald, S.L.; Salmon, D.P. Association of dementia severity with cortical gray matter and abnormal white matter volumes in dementia of the Alzheimer type. Arch. Neurol. 1996, 53, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, S.; Montilli, C.; Finocchi, P.; Amadio, S. Membrane compartments and purinergic signalling: P2X receptors in neurodegenerative and neuroinflammatory events. FEBS J. 2009, 276, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gouras, G.K.; Olsson, T.T.; Hansson, O. β-Amyloid peptides and amyloid plaques in Alzheimer’s disease. Neurotherapeutics 2015, 12, 3–11. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Iwatsubo, T.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Nukina, N.; Ihara, Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: Evidence that an initially deposited species is A beta 42(43). Neuron 1994, 13, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Papayannopoulos, I.A.; Styles, J.; Bobin, S.A.; Lin, Y.Y.; Biemann, K.; Iqbal, K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Arch Biochem Biophys. 1993, 301, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rich, J.B.; Rasmusson, D.X.; Folstein, M.F.; Carson, K.A.; Kawas, C.; Brandt, J. Nonsteroidal anti-inflammatory drugs in Alzheimer’s disease. Neurology 1995, 45, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Lace, G.L.; Wharton, S.B.; Ince, P.G. A brief history of tau: The evolving view of the microtubule-associated protein tau in neurodegenerative diseases. Clin. Neuropathol. 2007, 26, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 17, 2181–2192. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, S.-L.; Wang, C.; Jiang, T.; Tan, L.; Xing, A. The Role of Cdk5 in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 4328–4342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: Challenges, successes and future. Signal Transduct Target Ther. 2023, 8, 248. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fà, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.P.; Honig, L.S.; et al. Role of Amyloid-β and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimers Dis. 2018, 64, S611–S631, Erratum in J. Alzheimers Dis. 2019, 68, 415. https://doi.org/10.3233/JAD-189015. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cieślak, M.; Wojtczak, A. Role of purinergic receptors in the Alzheimer’s disease. Purinergic Signal. 2018, 14, 331–344. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- McGeer, P.L.; McGeer, E.G. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: Implications for therapy. Acta Neuropathol. 2013, 126, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tarkowski, E.; Andreasen, N.; Tarkowski, A.; Blennow, K. Intrathecal inflammation precedes development of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1200–1205. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Griffin, W.S. Inflammation and neurodegenerative diseases. Am. J. Clin. Nutr. 2006, 83, 470S–474S. [Google Scholar] [CrossRef] [PubMed]
- Linden, J. Adenosine in tissue protection and tissue regeneration. Mol. Pharmacol. 2005, 67, 1385–1387. [Google Scholar] [CrossRef] [PubMed]
- Abbracchio, M.P.; Ceruti, S. P1 receptors and cytokine secretion. Purinergic Signal. 2007, 3, 13–25. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Benzing, W.C.; Wujek, J.R.; Ward, E.K.; Shaffer, D.; Ashe, K.H.; Younkin, S.G.; Brunden, K.R. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol. Aging 1999, 20, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.J.; Lee, H.G.; Zhu, X.; Perry, G.; Smith, M.A. Alzheimer disease pathology as a host response. J. Neuropathol. Exp. Neurol. 2008, 67, 523–531. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Graeber, M.B.; Streit, W.J. Microglia: Biology and pathology. Acta Neuropathol. 2010, 119, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Filiou, M.D.; Arefin, A.S.; Moscato, P.; Graeber, M.B. ‘Neuroinflammation’ differs categorically from inflammation: Transcriptomes of Alzheimer’s disease, Parkinson’s disease, schizophrenia and inflammatory diseases compared. Neurogenetics 2014, 15, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Xue, Q.S.; Tischer, J.; Bechmann, I. Microglial pathology. Acta Neuropathol. Commun. 2014, 2, 142. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Erb, L.; Woods, L.T.; Khalafalla, M.G.; Weisman, G.A. Purinergic signaling in Alzheimer’s disease. Brain Res. Bull. 2019, 151, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Ajit, D.; Peterson, T.S.; Wang, Y.; Camden, J.M.; Gibson Wood, W.; Sun, G.Y.; Erb, L.; Petris, M.; Weisman, G.A. Nucleotides released from Aβ1–42-treated microglial cells increase cell migration and Aβ1–42 uptake through P2Y2 receptor activation. J. Neurochem. 2012, 121, 228–238. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; Whitem, C.L., 3rd; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fillit, H.; Ding, W.H.; Buee, L.; Kalman, J.; Altstiel, L.; Lawlor, B.; Wolf-Klein, G. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci. Lett. 1991, 129, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Kann, O.; Ohlemeyer, C.; Hanisch, U.K.; Kettenmann, H. Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): Suppression of receptor-evoked calcium signaling and control of release function. J. Neurosci. 2003, 23, 4410–4419. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Reed-Geaghan, E.G.; Savage, J.C.; Hise, A.G.; Landreth, G.E. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J. Neurosci. 2009, 29, 11982–11992. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Toescu, E.C.; Möller, T.; Kettenmann, H.; Verkhratsky, A. Long-term activation of capacitative Ca2+ entry in mouse microglial cells. Neuroscience 1998, 86, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Vom Berg, J.; Prokop, S.; Miller, K.R.; Obst, J.; Kälin, R.E.; Lopategui-Cabezas, I.; Wegner, A.; Mair, F.; Schipke, C.G.; Peters, O.; et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nat. Med. 2012, 18, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stalder, M.; Deller, T.; Staufenbiel, M.; Jucker, M. 3D-Reconstruction of microglia and amyloid in APP23 transgenic mice: No evidence of intracellular amyloid. Neurobiol. Aging 2001, 22, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Wang, K.C.; Imaki, H.; Rubenstein, R.; Wronska, A.; Osuchowski, M.; Lipinski, W.J.; Walker, L.C.; LeVine, H. The role of microglial cells and astrocytes in fibrillar plaque evolution in transgenic APP(SW) mice. Neurobiol. Aging 2001, 22, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chakfe, Y.; Seguin, R.; Antel, J.P.; Morissette, C.; Malo, D.; Henderson, D.; Séguéla, P. ADP and AMP induce interleukin-1beta release from microglial cells through activation of ATP-primed P2X7 receptor channels. J. Neurosci. 2002, 22, 3061–3069. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, S.; Liu, Y.; Hao, W.; Wolf, L.; Kiliaan, A.J.; Penke, B.; Rübe, C.E.; Walter, J.; Heneka, M.T.; Hartmann, T.; et al. TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J. Immunol. 2012, 188, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Hide, I.; Ido, K.; Kohsaka, S.; Inoue, K.; Nakata, Y. Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J. Neurosci. 2004, 24, 1–7. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- de Rivero Vaccari, J.P.; Bastien, D.; Yurcisin, G.; Pineau, I.; Dietrich, W.D.; De Koninck, Y.; Keane, R.W.; Lacroix, S. P2X4 receptors influence inflammasome activation after spinal cord injury. J. Neurosci. 2012, 32, 3058–3066. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Puchałowicz, K.; Tarnowski, M.; Baranowska-Bosiacka, I.; Chlubek, D.; Dziedziejko, V. P2X and P2Y receptors—Role in the pathophysiology of the nervous system. Int. J. Mol. Sci. 2014, 15, 23672–23704. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Erb, L.; Cao, C.; Ajit, D.; Weisman, G.A. P2Y receptors in Alzheimer’s disease. Biol. Cell 2015, 107, 1–21. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, T.; Ulrich, H.; Semyanov, A.; Illes, P.; Tang, Y. Optical control of purinergic signaling. Purinergic Signal. 2021, 17, 385–392. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, L.; Li, Y.J.; Yang, X.; Yang, B.; Zhang, X.; Zhang, J.; Zhang, Q.; Cheng, X.D.; Wang, J.H.; Yu, N.W. Purinergic signaling: A potential therapeutic target for ischemic stroke. Purinergic Signal. 2023, 19, 173–183. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huang, Z.; Xie, N.; Illes, P.; Di Virgilio, F.; Ulrich, H.; Semyanov, A.; Verkhratsky, A.; Sperlagh, B.; Yu, S.G.; Huang, C.; et al. From purines to purinergic signaling: Molecular functions and human diseases. Signal Transduct Target Ther. 2021, 6, 162. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Burnstock, G. Purinergic nerves. Pharmacol. Rev. 1972, 24, 509–581. [Google Scholar] [CrossRef]
- Mizumoto, N.; Kumamoto, T.; Robson, S.C.; Sévigny, J.; Matsue, H.; Enjyoji, K.; Takashima, A. CD39 is the dominant Langerhans cell-associated ecto-NTPDase: Modulatory roles in inflammation and immune responsiveness. Nat. Med. 2002, 8, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Romio, M.; Reinbeck, B.; Bongardt, S.; Huls, S.; Burghoff, S.; Schrader, J. Extracellular purine metabolism and signaling of CD73-derived adenosine in murine Treg and Teff cells. Am. J. Physiol. Cell Physiol. 2011, 301, C530–C539. [Google Scholar] [CrossRef]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, J.P.; Liu, S.C.; Hu, S.Q.; Lu, J.F.; Wu, C.L.; Hu, D.X.; Zhang, W.J. ATP ion channel P2X purinergic receptors in inflammation response. Biomed. Pharmacother. 2023, 158, 114205. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, D.E.; Petiz, L.L.; Glaser, T.; Oliveira-Giacomelli, Á.; Andrejew, R.; Saab, F.A.R.; Milanis, M.D.S.; Campos, H.C.; Sampaio, V.F.A.; La Banca, S.; et al. Purinergic signaling in cognitive impairment and neuropsychiatric symptoms of Alzheimer’s disease. Neuropharmacology 2023, 226, 109371. [Google Scholar] [CrossRef] [PubMed]
- Wardas, J. Neuroprotective role of adenosine in the CNS. Pol. J. Pharmacol. 2002, 54, 313–326. [Google Scholar] [PubMed]
- Zarrinmayeh, H.; Territo, P.R. Purinergic Receptors of the Central Nervous System: Biology, PET Ligands, and Their Applications. Mol. Imaging 2020, 19, 1536012120927609. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ribeiro, J.A.; Sebastião, A.M.; de Mendonça, A. Adenosine receptors in the nervous system: Pathophysiological implications. Prog. Neurobiol. 2002, 68, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Trinh, P.N.H.; Baltos, J.A.; Hellyer, S.D.; May, L.T.; Gregory, K.J. Adenosine receptor signalling in Alzheimer’s disease. Purinergic Signal. 2022, 18, 359–381, Erratum in Purinergic Signal. 2022, 18, 407. https://doi.org/10.1007/s11302-022-09906-x. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rebola, N.; Pinheiro, P.C.; Oliveira, C.R.; Malva, J.O.; Cunha, R.A. Subcellular localization of adenosine A(1) receptors in nerve terminals and synapses of the rat hippocampus. Brain Res. 2003, 987, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Cherchi, F.; Pugliese, A.M.; Coppi, E. Oligodendrocyte precursor cell maturation: Role of adenosine receptors. Neural Regen. Res. 2021, 16, 1686–1692. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Boison, D.; Chen, J.F.; Fredholm, B.B. Adenosine signaling and function in glial cells. Cell Death Differ. 2010, 17, 1071–1082. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, X.X.; Nomura, T.; Aihara, H.; Nishizaki, T. Adenosine enhances glial glutamate efflux via A2a adenosine receptors. Life Sci. 2001, 68, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Nishizaki, T.; Nagai, K.; Nomura, T.; Tada, H.; Kanno, T.; Tozaki, H.; Li, X.X.; Kondoh, T.; Kodama, N.; Takahashi, E.; et al. A new neuromodulatory pathway with a glial contribution mediated via A(2a) adenosine receptors. Glia 2002, 39, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-P.; Wu, K.-C.; Lin, C.-Y.; Chern, Y. Emerging roles of dysregulated adenosine homeostasis in brain disorders with a specific focus on neurodegenerative diseases. J. Biomed. Sci. 2021, 28, 70. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol. Aging 1997, 18, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Fukumitsu, N.; Ishii, K.; Kimura, Y.; Oda, K.; Hashimoto, M.; Suzuki, M.; Ishiwata, K. Adenosine A1 receptors using 8-dicyclopropylmethyl-1-[11C]methyl-3-propylxanthine PET in Alzheimer’s disease. Ann. Nucl. Med. 2008, 22, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Illes, P.; Ulrich, H.; Chen, J.F.; Tang, Y. Purinergic receptors in cognitive disturbances. Neurobiol. Dis. 2023, 185, 106229. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Battistello, E.; Casetta, I.; Gragnaniello, D.; Poloni, T.E.; Medici, V.; Cirrincione, A.; Varani, K.; Vincenzi, F.; Borea, P.A.; et al. Upregulation of Cortical A2A Adenosine Receptors Is Reflected in Platelets of Patients with Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 80, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Arendash, G.W.; Schleif, W.; Rezai-Zadeh, K.; Jackson, E.K.; Zacharia, L.C.; Cracchiolo, J.R.; Shippy, D.; Tan, J. Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain beta-amyloid production. Neuroscience 2006, 142, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Currais, A.; Kato, K.; Canuet, L.; Ishii, R.; Tanaka, T.; Takeda, M.; Soriano, S. Caffeine modulates tau phosphorylation and affects Akt signaling in postmitotic neurons. J. Mol. Neurosci. 2011, 43, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Kellett, K.A.; Hooper, N.M. The Role of Tissue Non-specific Alkaline Phosphatase (TNAP) in Neurodegenerative Diseases: Alzheimer’s Disease in the Focus. Subcell. Biochem. 2015, 76, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Sebastián-Serrano, Á.; de Diego-García, L.; Martínez-Frailes, C.; Ávila, J.; Zimmermann, H.; Millán, J.L.; Miras-Portugal, M.T.; Díaz-Hernández, M. Tissue-nonspecific Alkaline Phosphatase Regulates Purinergic Transmission in the Central Nervous System During Development and Disease. Comput. Struct. Biotechnol. J. 2014, 13, 95–100. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ellis, J.A.; Jackman, M.R.; Luzio, J.P. The post-synthetic sorting of endogenous membrane proteins examined by the simultaneous purification of apical and basolateral plasma membrane fractions from Caco-2 cells. Biochem. J. 1992, 283 Pt 2, 553–560. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Augusto, E.; Matos, M.; Sévigny, J.; El-Tayeb, A.; Bynoe, M.S.; Müller, C.E.; Cunha, R.A.; Chen, J.F. Ecto-5′-nucleotidase (CD73)-mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions. J. Neurosci. 2013, 33, 11390–11399. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gonçalves, F.Q.; Lopes, J.P.; Silva, H.B.; Lemos, C.; Silva, A.C.; Gonçalves, N.; Tomé, Â.R.; Ferreira, S.G.; Canas, P.M.; Rial, D.; et al. Synaptic and memory dysfunction in a β-amyloid model of early Alzheimer’s disease depends on increased formation of ATP-derived extracellular adenosine. Neurobiol. Dis. 2019, 132, 104570. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.A. How does adenosine control neuronal dysfunction and neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Zebisch, M.; Sträter, N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abbracchio, M.P.; Burnstock, G.; Boeynaems, J.M.; Barnard, E.A.; Boyer, J.L.; Kennedy, C.; Knight, G.E.; Fumagalli, M.; Gachet, C.; Jacobson, K.A.; et al. International Union of Pharmacology LVIII: Update on the P2Y G protein-coupled nucleotide receptors: From molecular mechanisms and pathophysiology to therapy. Pharmacol. Rev. 2006, 58, 281–341. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Franke, H.; Verkhratsky, A.; Burnstock, G.; Illes, P. Pathophysiology of astroglial purinergic signalling. Purinergic Signal. 2012, 8, 629–657. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Silva-Vilches, C.; Ring, S.; Mahnke, K. ATP and Its Metabolite Adenosine as Regulators of Dendritic Cell Activity. Front. Immunol. 2018, 9, 2581. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dubyak, G.R. P2X7 receptor regulation of non-classical secretion from immune effector cells. Cell. Microbiol. 2012, 14, 1697–1706. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mitra, S.; Sarkar, A. Microparticulate P2X7 and GSDM-D mediated regulation of functional IL-1β release. Purinergic Signal. 2019, 15, 119–123. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ronning, K.E.; Déchelle-Marquet, P.A.; Che, Y.; Guillonneau, X.; Sennlaub, F.; Delarasse, C. The P2X7 Receptor, a Multifaceted Receptor in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 11747. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Khakh, B.S.; North, R.A. Neuromodulation by extracellular ATP and P2X receptors in the CNS. Neuron 2012, 76, 51–69. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Marchenkova, A.; Vilotti, S.; Ntamati, N.; van den Maagdenberg, A.M.; Nistri, A. Inefficient constitutive inhibition of P2X3 receptors by brain natriuretic peptide system contributes to sensitization of trigeminal sensory neurons in a genetic mouse model of familial hemiplegic migraine. Mol. Pain 2016, 12, 1744806916646110. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Illes, P.; Rubini, P.; Ulrich, H.; Zhao, Y.; Tang, Y. Regulation of Microglial Functions by Purinergic Mechanisms in the Healthy and Diseased CNS. Cells 2020, 9, 1108. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Territo, P.R.; Zarrinmayeh, H. P2X7 Receptors in Neurodegeneration: Potential Therapeutic Applications From Basic to Clinical Approaches. Front. Cell Neurosci. 2021, 15, 617036. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Illes, P. P2X7 Receptors Amplify CNS Damage in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 5996. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hirayama, Y.; Anzai, N.; Kinouchi, H.; Koizumi, S. P2X7 Receptors in Astrocytes: A Switch for Ischemic Tolerance. Molecules 2022, 27, 3655. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Di Lauro, C.; Bianchi, C.; Sebastián-Serrano, Á.; Soria-Tobar, L.; Alvarez-Castelao, B.; Nicke, A.; Díaz-Hernández, M. P2X7 receptor blockade reduces tau induced toxicity, therapeutic implications in tauopathies. Prog. Neurobiol. 2022, 208, 102173. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Han, J.; Resing, D.; Liu, H.; Yue, X.; Miller, R.L.; Schoch, K.M.; Miller, T.M.; Perlmutter, J.S.; Egan, T.M.; et al. Synthesis and in vitro characterization of a P2X7 radioligand [123I]TZ6019 and its response to neuroinflammation in a mouse model of Alzheimer disease. Eur. J. Pharmacol. 2018, 820, 8–17. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Francistiová, L.; Bianchi, C.; Di Lauro, C.; Sebastián-Serrano, Á.; de Diego-García, L.; Kobolák, J.; Dinnyés, A.; Díaz-Hernández, M. The Role of P2X7 Receptor in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 94. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, S.J. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 2008, 372, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Wilkaniec, A.; Gąssowska, M.; Czapski, G.A.; Cieślik, M.; Sulkowski, G.; Adamczyk, A. P2X7 receptor-pannexin 1 interaction mediates extracellular alpha-synuclein-induced ATP release in neuroblastoma SH-SY5Y cells. Purinergic Signal. 2017, 13, 347–361. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jiang, T.; Hoekstra, J.; Heng, X.; Kang, W.; Ding, J.; Liu, J.; Chen, S.; Zhang, J. P2X7 receptor is critical in α-synuclein—Mediated microglial NADPH oxidase activation. Neurobiol. Aging 2015, 36, 2304–2318. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic signalling. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S172–S181. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Woods, L.T.; Ajit, D.; Camden, J.M.; Erb, L.; Weisman, G.A. Purinergic receptors as potential therapeutic targets in Alzheimer’s disease. Neuropharmacology 2016, 104, 169–179. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Woods, L.T.; Forti, K.M.; Shanbhag, V.C.; Camden, J.M.; Weisman, G.A. P2Y receptors for extracellular nucleotides: Contributions to cancer progression and therapeutic implications. Biochem. Pharmacol. 2021, 187, 114406. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ajit, D.; Woods, L.T.; Camden, J.M.; Thebeau, C.N.; El-Sayed, F.G.; Greeson, G.W.; Erb, L.; Petris, M.J.; Miller, D.C.; Sun, G.Y.; et al. Loss of P2Y2 nucleotide receptors enhances early pathology in the TgCRND8 mouse model of Alzheimer’s disease. Mol. Neurobiol. 2014, 49, 1031–1042. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bours, M.J.; Dagnelie, P.C.; Giuliani, A.L.; Wesselius, A.; Di Virgilio, F. P2 receptors and extracellular ATP: A novel homeostatic pathway in inflammation. Front. Biosci.(Schol. Ed.) 2011, 3, 1443–1456. [Google Scholar] [CrossRef] [PubMed]
- Weisman, G.A.; Ajit, D.; Garrad, R.; Peterson, T.S.; Woods, L.T.; Thebeau, C.; Camden, J.M.; Erb, L. Neuroprotective roles of the P2Y(2) receptor. Purinergic Signal. 2012, 8, 559–578. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Delekate, A.; Füchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Lobo, P.; Reid, M.J.; Jimenez-Sanchez, M.; Verkhratsky, A.; Perez-Nievas, B.G.; Noble, W. Astrocyte adaptation in Alzheimer’s disease: A focus on astrocytic P2X7R. Essays Biochem. 2023, 67, 119–130. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- de Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bours, M.J.; Swennen, E.L.; Di Virgilio, F.; Cronstein, B.N.; Dagnelie, P.C. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 2006, 112, 358–404. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, P.; Ye, Y. Filamentous recombinant human Tau activates primary astrocytes via an integrin receptor complex. Nat. Commun. 2021, 12, 95. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, C.C.; Hu, J.; Zhao, N.; Wang, J.; Wang, N.; Cirrito, J.R.; Kanekiyo, T.; Holtzman, D.M.; Bu, G. Astrocytic LRP1 Mediates Brain Aβ Clearance and Impacts Amyloid Deposition. J. Neurosci. 2017, 37, 4023–4031. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Martin, E.; Amar, M.; Dalle, C.; Youssef, I.; Boucher, C.; Le Duigou, C.; Brückner, M.; Prigent, A.; Sazdovitch, V.; Halle, A.; et al. New role of P2X7 receptor in an Alzheimer’s disease mouse model. Mol. Psychiatry 2019, 24, 108–125. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, J.; He, W.; Zhang, J. A richer and more diverse future for microglia phenotypes. Heliyon 2023, 9, e14713. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, J.; Li, X.; Jiang, X.; Yang, M.; Yang, R.; Burnstock, G.; Xiang, Z.; Yuan, H. Microvesicles shed from microglia activated by the P2X7-p38 pathway are involved in neuropathic pain induced by spinal nerve ligation in rats. Purinergic Signal. 2017, 13, 13–26. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Honda, S.; Sasaki, Y.; Ohsawa, K.; Imai, Y.; Nakamura, Y.; Inoue, K.; Kohsaka, S. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J. Neurosci. 2001, 21, 1975–1982. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Di Virgilio, F.; Vultaggio-Poma, V.; Falzoni, S.; Giuliani, A.L. Extracellular ATP: A powerful inflammatory mediator in the central nervous system. Neuropharmacology 2023, 224, 109333. [Google Scholar] [CrossRef] [PubMed]
- Sanz, J.M.; Chiozzi, P.; Ferrari, D.; Colaianna, M.; Idzko, M.; Falzoni, S.; Fellin, R.; Trabace, L.; Di Virgilio, F. Activation of microglia by amyloid {beta} requires P2X7 receptor expression. J. Immunol. 2009, 182, 4378–4385. [Google Scholar] [CrossRef] [PubMed]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Jansen, A.H.P.; Kooijman, L.; Bossers, K.; Hol, E.M. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 2014, 35, 2746–2760. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Madry, C.; Kyrargyri, V.; Arancibia-Cárcamo, I.L.; Jolivet, R.; Kohsaka, S.; Bryan, R.M.; Attwell, D. Microglial Ramification, Surveillance, and Interleukin-1β Release Are Regulated by the Two-Pore Domain K+ Channel THIK-1. Neuron 2018, 97, 299–312.e6. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Drinkall, S.; Lawrence, C.B.; Ossola, B.; Russell, S.; Bender, C.; Brice, N.B.; Dawson, L.A.; Harte, M.; Brough, D. The two pore potassium channel THIK-1 regulates NLRP3 inflammasome activation. Glia 2022, 70, 1301–1316. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rifat, A.; Ossola, B.; Bürli, R.W.; Dawson, L.A.; Brice, N.L.; Rowland, A.; Lizio, M.; Xu, X.; Page, K.; Fidzinski, P.; et al. Differential contribution of THIK-1 K+ channels and P2X7 receptors to ATP-mediated neuroinflammation by human microglia. J. Neuroinflamm. 2024, 21, 58. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jeppesen, D.K.; Zhang, Q.; Franklin, J.L.; Coffey, R.J. Extracellular vesicles and nanoparticles: Emerging complexities. Trends Cell Biol. 2023, 33, 667–681. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schnatz, A.; Müller, C.; Brahmer, A.; Krämer-Albers, E.M. Extracellular Vesicles in neural cell interaction and CNS homeostasis. FASEB Bioadv. 2021, 3, 577–592. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gomes, A.R.; Sangani, N.B.; Fernandes, T.G.; Diogo, M.M.; Curfs, L.M.G.; Reutelingsperger, C.P. Extracellular Vesicles in CNS Developmental Disorders. Int. J. Mol. Sci. 2020, 21, 9428. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hill, A.F. Extracellular Vesicles and Neurodegenerative Diseases. J. Neurosci. 2019, 39, 9269–9273. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ridder, K.; Keller, S.; Dams, M.; Rupp, A.K.; Schlaudraff, J.; Del Turco, D.; Starmann, J.; Macas, J.; Karpova, D.; Devraj, K.; et al. Extracellular vesicle-mediated transfer of genetic information between the hematopoietic system and the brain in response to inflammation. PLoS Biol. 2014, 12, e1001874. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrügger, U.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404, Erratum in J. Extracell. Vesicles 2024, 13, e12451. https://doi.org/10.1002/jev2.12451. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carotti, V.; Rigalli, J.P.; van Asbeck-van der Wijst, J.; Hoenderop, J.G.J. Interplay between purinergic signalling and extracellular vesicles in health and disease. Biochem. Pharmacol. 2022, 203, 115192. [Google Scholar] [CrossRef] [PubMed]
- Verweij, F.J.; Balaj, L.; Boulanger, C.M.; Carter, D.R.F.; Compeer, E.B.; D’Angelo, G.; El Andaloussi, S.; Goetz, J.G.; Gross, J.C.; Hyenne, V.; et al. The power of imaging to understand extracellular vesicle biology in vivo. Nat. Methods 2021, 18, 1013–1026. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gomes, P.; Tzouanou, F.; Skolariki, K.; Vamvaka-Iakovou, A.; Noguera-Ortiz, C.; Tsirtsaki, K.; Waites, C.L.; Vlamos, P.; Sousa, N.; Costa-Silva, B.; et al. Extracellular vesicles and Alzheimer’s disease in the novel era of Precision Medicine: Implications for disease progression, diagnosis and treatment. Exp. Neurol. 2022, 358, 114183. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liang, T.; Wu, Z.; Li, J.; Wu, S.; Shi, W.; Wang, L. The emerging double-edged sword role of exosomes in Alzheimer’s disease. Front. Aging Neurosci. 2023, 15, 1209115. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Weng, S.; Lai, Q.L.; Wang, J.; Zhuang, L.; Cheng, L.; Mo, Y.; Liu, L.; Zhao, Z.; Zhang, Y.; Qiao, S. The Role of Exosomes as Mediators of Neuroinflammation in the Pathogenesis and Treatment of Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 899944. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Phinney, D.G.; Di Giuseppe, M.; Njah, J.; Sala, E.; Shiva, S.; St Croix, C.M.; Stolz, D.B.; Watkins, S.C.; Di, Y.P.; Leikauf, G.D.; et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat. Commun. 2015, 6, 8472. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gupta, A.; Pulliam, L. Exosomes as mediators of neuroinflammation. J. Neuroinflamm. 2014, 11, 68. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Berriel Pinho, V.H.; Daher, J.P.L.; Kanaan, S.; Medeiros, T. Extracellular vesicles in Alzheimer’s disease. Arq. Neuropsiquiatr. 2024, 82, s00441779296. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ludwig, N.; Azambuja, J.H.; Rao, A.; Gillespie, D.G.; Jackson, E.K.; Whiteside, T.L. Adenosine receptors regulate exosome production. Purinergic Signal. 2020, 16, 231–240. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kou, M.; Huang, L.; Yang, J.; Chiang, Z.; Chen, S.; Liu, J.; Guo, L.; Zhang, X.; Zhou, X.; Xu, X.; et al. Mesenchymal stem cell-derived extracellular vesicles for immunomodulation and regeneration: A next generation therapeutic tool? Cell Death Dis. 2022, 13, 580. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Smyth, L.A.; Ratnasothy, K.; Tsang, J.Y.; Boardman, D.; Warley, A.; Lechler, R.; Lombardi, G. CD73 expression on extracellular vesicles derived from CD4+ CD25+ Foxp3+ T cells contributes to their regulatory function. Eur. J. Immunol. 2013, 43, 2430–2440. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.; Rissiek, A.; Winzer, R.; Puig, B.; Rissiek, B.; Haag, F.; Mittrücker, H.W.; Magnus, T.; Tolosa, E. Generation and Function of Non-cell-bound CD73 in Inflammation. Front. Immunol. 2019, 10, 1729. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Voloboueva, L.A.; Duan, M.; Ouyang, Y.; Emery, J.F.; Stoy, C.; Giffard, R.G. Overexpression of mitochondrial Hsp70/Hsp75 protects astrocytes against ischemic injury in vitro. J. Cereb. Blood Flow Metab. 2008, 28, 1009–1016. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guo, S.; Kim, W.J.; Lok, J.; Lee, S.R.; Besancon, E.; Luo, B.H.; Stins, M.F.; Wang, X.; Dedhar, S.; Lo, E.H. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 7582–7587. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dave, K.M.; Zhao, W.; Hoover, C.; D’Souza, A.; SManickam, D. Extracellular Vesicles Derived from a Human Brain Endothelial Cell Line Increase Cellular ATP Levels. AAPS PharmSciTech 2021, 22, 18, Erratum in AAPS PharmSciTech 2023, 24, 63. https://doi.org/10.1208/s12249-023-02526-7. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Di Iorio, P.; Ciccarelli, R. Adenine-Based Purines and Related Metabolizing Enzymes: Evidence for Their Impact on Tumor Extracellular Vesicle Activities. Cells 2021, 10, 188. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ceruti, S.; Colombo, L.; Magni, G.; Viganò, F.; Boccazzi, M.; Deli, M.A.; Sperlágh, B.; Abbracchio, M.P.; Kittel, A. Oxygen-glucose deprivation increases the enzymatic activity and the microvesicle-mediated release of ectonucleotidases in the cells composing the blood-brain barrier. Neurochem. Int. 2011, 59, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Yerneni, S.S.; Menshikova, E.V.; Gillespie, D.G.; Jackson, E.K.; Whiteside, T.L. Simultaneous Inhibition of Glycolysis and Oxidative Phosphorylation Triggers a Multi-Fold Increase in Secretion of Exosomes: Possible Role of 2′3′-cAMP. Sci. Rep. 2020, 10, 6948, Erratum in Sci. Rep. 2020, 10, 14027. https://doi.org/10.1038/s41598-020-70340-3. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ludwig, N.; Whiteside, T.L.; Reichert, T.E. Challenges in Exosome Isolation and Analysis in Health and Disease. Int. J. Mol. Sci. 2019, 20, 4684. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ludwig, N.; Yerneni, S.S.; Razzo, B.M.; Whiteside, T.L. Exosomes from HNSCC promote angiogenesis through reprogramming of endothelial cells. Mol. Cancer Res. 2018, 16, 1798–1808. [Google Scholar] [CrossRef]
- Chen, T.; Guo, J.; Yang, M.; Zhu, X.; Cao, X. Chemokine-containing exosomes are released from heat-stressed tumor cells via lipid raft-dependent pathway and act as efficient tumor vaccine. J. Immunol. 2011, 186, 2219–2228. [Google Scholar] [CrossRef]
- Keklikoglou, I.; Cianciaruso, C.; Güç, E.; Squadrito, M.L.; Spring, L.M.; Tazzyman, S.; Lambein, L.; Poissonnier, A.; Ferraro, G.B.; Baer, C.; et al. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat. Cell Biol. 2019, 21, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Garcia, N.A.; Ontoria-Oviedo, I.; González-King, H.; Diez-Juan, A.; Sepúlveda, P. Glucose starvation in cardiomyocytes enhances exosome secretion and promotes angiogenesis in endothelial cells. PLoS ONE 2015, 10, e0138849. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.E.; Scholz-Romero, K.; Sweeney, E.; Peiris, H.; Kobayashi, M.; Duncombe, G.; Mitchell, M.D.; Salomon, C. The effect of glucose on the release and bioactivity of exosomes from first trimester trophoblast cells. J. Clin. Endocrinol. Metab. 2015, 100, E1280–E1288. [Google Scholar] [CrossRef]
- Lamichhane, T.N.; Leung, C.A.; Douti, L.Y.; Jay, S.M. Ethanol induces enhanced vascularization bioactivity of endothelial cell-derived extracellular vesicles via regulation of microRNAs and long non-coding RNAs. Sci. Rep. 2017, 7, 13794. [Google Scholar] [CrossRef] [PubMed]
- Turola, E.; Furlan, R.; Bianco, F.; Matteoli, M.; Verderio, C. Microglial microvesicle secretion and intercellular signaling. Front. Physiol. 2012, 3, 149. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Qu, Y.; Ramachandra, L.; Mohr, S.; Franchi, L.; Harding, C.V.; Nunez, G.; Dubyak, G.R. P2X7 receptor-stimulated secretion of MHC class II-containing exosomes requires the ASC/NLRP3 inflammasome but is independent of caspase-1. J. Immunol. 2009, 182, 5052–5062. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, T.; Ma, S.; Lv, J.; Wang, X.; Afewerky, H.K.; Li, H.; Lu, Y. The emerging role of exosomes in Alzheimer’s disease. Ageing Res. Rev. 2021, 68, 101321. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Mustapic, M.; Kapogiannis, D.; Eitan, E.; Lobach, I.V.; Goetzl, L.; Schwartz, J.B.; Miller, B.L. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer’s disease. FASEB J. 2016, 30, 3853–3859. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Perez-Gonzalez, R.; Gauthier, S.A.; Kumar, A.; Levy, E. The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J. Biol. Chem. 2012, 287, 43108–43115. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lombardi, M.; Gabrielli, M.; Adinolfi, E.; Verderio, C. Role of ATP in Extracellular Vesicle Biogenesis and Dynamics. Front. Pharmacol. 2021, 12, 654023. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Huang, M.; Zheng, M.; Dai, C.; Song, Q.; Zhang, Q.; Li, Q.; Gu, X.; Chen, H.; Jiang, G.; et al. ADSCs-derived extracellular vesicles alleviate neuronal damage, promote neurogenesis and rescue memory loss in mice with Alzheimer’s disease. J. Control Release. 2020, 327, 688–702. [Google Scholar] [CrossRef] [PubMed]
- Katsuda, T.; Tsuchiya, R.; Kosaka, N.; Yoshioka, Y.; Takagaki, K.; Oki, K.; Takeshita, F.; Sakai, Y.; Kuroda, M.; Ochiya, T. Human adipose tissue-derived mesenchymal stem cells secrete functional neprilysin-bound exosomes. Sci. Rep. 2013, 3, 1197. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lee, M.; Ban, J.J.; Yang, S.; Im, W.; Kim, M. The exosome of adipose-derived stem cells reduces β-amyloid pathology and apoptosis of neuronal cells derived from the transgenic mouse model of Alzheimer’s disease. Brain Res. 2018, 1691, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Turovsky, E.A.; Golovicheva, V.V.; Varlamova, E.G.; Danilina, T.I.; Goryunov, K.V.; Shevtsova, Y.A.; Pevzner, I.B.; Zorova, L.D.; Babenko, V.A.; Evtushenko, E.A.; et al. Mesenchymal stromal cell-derived extracellular vesicles afford neuroprotection by modulating PI3K/AKT pathway and calcium oscillations. Int. J. Biol. Sci. 2022, 18, 5345–5368. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Discher, D.E.; Péault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. Npj. Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kaniowska, D.; Wenk, K.; Rademacher, P.; Weiss, R.; Fabian, C.; Schulz, I.; Guthardt, M.; Lange, F.; Greiser, S.; Schmidt, M.; et al. Extracellular Vesicles of Mesenchymal Stromal Cells Can be Taken Up by Microglial Cells and Partially Prevent the Stimulation Induced by β-amyloid. Stem Cell Rev. Rep. 2022, 18, 1113–1126. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cone, A.S.; Yuan, X.; Sun, L.; Duke, L.C.; Vreones, M.P.; Carrier, A.N.; Kenyon, S.M.; Carver, S.R.; Benthem, S.D.; Stimmell, A.C.; et al. Mesenchymal stem cell-derived extracellular vesicles ameliorate Alzheimer’s disease-like phenotypes in a preclinical mouse model. Theranostics 2021, 11, 8129–8142. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, Y.A.; Lu, C.H.; Ke, C.C.; Chiu, S.J.; Jeng, F.S.; Chang, C.W.; Yang, B.H.; Liu, R.S. Mesenchymal Stem Cell-Derived Exosomes Ameliorate Alzheimer’s Disease Pathology and Improve Cognitive Deficits. Biomedicines 2021, 9, 594. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bernstein, H.G.; Hölzl, G.; Dobrowolny, H.; Hildebrandt, J.; Trübner, K.; Krohn, M.; Bogerts, B.; Pahnke, J. Vascular and extravascular distribution of the ATP-binding cassette transporters ABCB1 and ABCC1 in aged human brain and pituitary. Mech. Ageing Dev. 2014, 141–142, 12–21. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cecchin, R.; Troyer, Z.; Witwer, K.; Morris, K.V. Extracellular vesicles: The next generation in gene therapy delivery. Mol. Ther. 2023, 31, 1225–1230. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
| Purinergic Receptor | Localization in the CNS | Receptor Activation Effect | Reference |
|---|---|---|---|
| A1R | Presynaptic neurons | Inhibition of neurotransmitter release | [76] |
| Postsynaptic neurons | Neuron depolarization | [76] | |
| Oligodendrocytes | Premature differentiation Promotion of myelination | [77,78] | |
| Astrocytes | Promotion of proliferation | [77] | |
| A2R | Presynaptic neurons | Increase in neurotransmitter release | [76] |
| Postsynaptic neurons | Increase in cellular excitability | [76] | |
| Astrocytes | Increase in glutamate release 1 | [79,80] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewandowska, J.; Majewski, J.; Roszek, K. Extracellular Vesicles and Purinergic Signaling in Alzheimer’s Disease—Joining Forces for Novel Therapeutic Approach. Brain Sci. 2025, 15, 570. https://doi.org/10.3390/brainsci15060570
Lewandowska J, Majewski J, Roszek K. Extracellular Vesicles and Purinergic Signaling in Alzheimer’s Disease—Joining Forces for Novel Therapeutic Approach. Brain Sciences. 2025; 15(6):570. https://doi.org/10.3390/brainsci15060570
Chicago/Turabian StyleLewandowska, Julita, Jakub Majewski, and Katarzyna Roszek. 2025. "Extracellular Vesicles and Purinergic Signaling in Alzheimer’s Disease—Joining Forces for Novel Therapeutic Approach" Brain Sciences 15, no. 6: 570. https://doi.org/10.3390/brainsci15060570
APA StyleLewandowska, J., Majewski, J., & Roszek, K. (2025). Extracellular Vesicles and Purinergic Signaling in Alzheimer’s Disease—Joining Forces for Novel Therapeutic Approach. Brain Sciences, 15(6), 570. https://doi.org/10.3390/brainsci15060570