
Murine Non-Transgenic Models of Alzheimer’s Disease Pathology: Focus on Risk Factors

 , ,
, ,  and
and
Abstract
1. Introduction
2. Methods
3. Results
3.1. Evolution of AD Definition
3.2. Pathological Changes in AD
3.2.1. Aβ and Its Pathological Forms
3.2.2. Phosphorylated Tau and NFTs
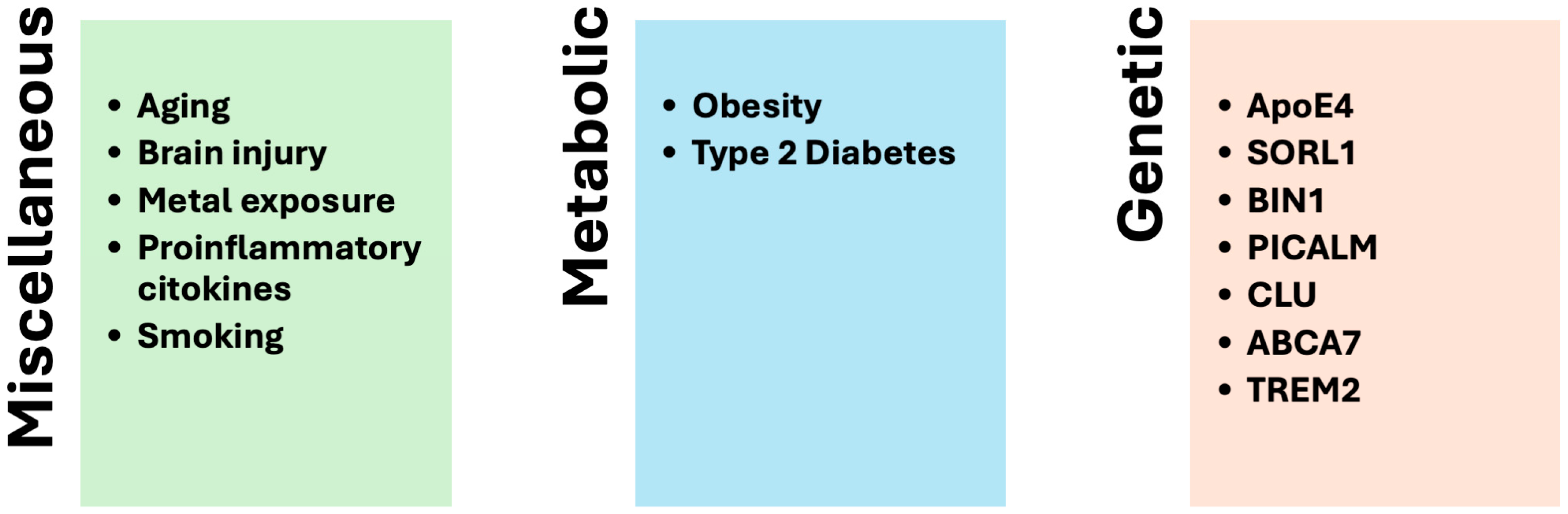
3.3. Factors Related to SAD
3.3.1. Aging
3.3.2. Metal Exposure
3.3.3. Brain Injury
3.3.4. Proinflammatory Cytokines
3.3.5. Type 2 Diabetes Mellitus
3.3.6. Obesity
3.3.7. Smoking
3.3.8. Diet
3.3.9. Alcohol Consumption
3.3.10. Genetics
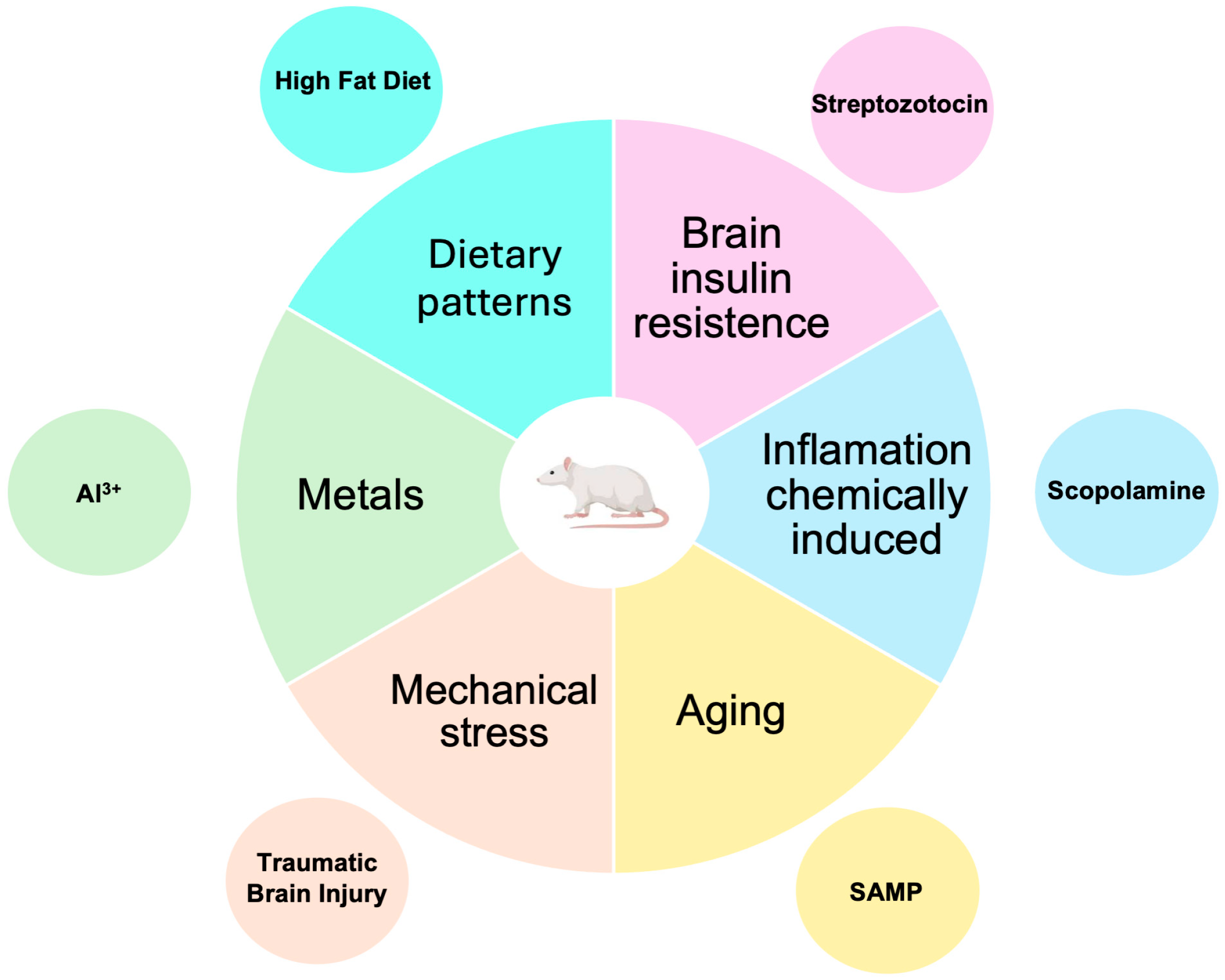
3.4. Non-Transgenic Models
3.4.1. Models Induced by the Administration of Streptozotocin (STZ)

{kind=link}
{kind=link}
{kind=link}
| Model | Animal | Aβ | P-Tau | Neuro-Inflammation | Other Changes | Time | Selected References |
|---|---|---|---|---|---|---|---|
| Stretptozotocyn | Mouse Rat | ✔ | ✔ | ✔ | Oxidative and nitrative stress, glucose hypometabolism cerebral state of insulin resistance, progressive cholinergic deficiency, increased exploratory activity, impaired learning and spatial memory. | 3 weeks | [81] |
| Scopolamine | Mouse Rat | ✔ | ✔ | ✔ | Neuronal apoptosis, changes in the expression of choline acetyltransferase (ChAT), acetylcholinesterase (AChE), and acetylcholine (ACh) receptors. Changes in memory evaluated by MWM, passive avoidance. | 1 to 6 weeks | [84] |
| Aging | Mouse | ✔ | ✔ | ✔ | Oxidative stress, cognitive deficit, especially in spatial memory and learning tasks. | 7–8 months | [85] |
| Mechanical stress | Mouse Rat | ✔ | ✔ | ✔ | Cognitive dysfunction, including impairments in memory and object recognition, along with behavioral disturbances such as anxiety and symptoms resembling depression. | 1 month | [86] |
| Aluminum | Rat | ✔ | ✔ | Lipoperoxidation increased malondialdehyde levels, lower superoxide dismutase activity, and catalase activity. | 4–6 weeks | [87] | |
| Dietary patterns High-fat diet | Rat Mice | ✔ | ✔ | ✔ | Dysregulations in autophagy, apoptosis. Oxidative stress and decreases in long-term potentiation. | 8–30 weeks | [88] |
3.4.2. Models Induced by the Administration of Scopolamine
3.4.3. Models Induced by Aging
3.4.4. Models Induced by Mechanical Stress
3.4.5. Models Induced by Metals
3.4.6. Dietary Patterns
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α7 nAChR | α7-nicotinic acetylcholine receptor |
| ACh | acetylcholine |
| AChE | acetylcholinesterase |
| AD | Alzheimer’s disease |
| ChAT | cholineacetyltransferase |
| ATP | adenosine triphosphate |
| Al3+ | aluminum |
| AD | Alzheimer’s disease |
| APP | amyloid β precursor protein |
| Aβ | amyloid-β |
| fAβ | amyloid-β fibril |
| oAβ | amyloid-β oligomer |
| ALS | amyotrophic lateral sclerosis |
| BACE1 | β secretase |
| PrPc | cellular prion protein |
| CNS | central nervous system |
| CTE | chronic traumatic encephalopathy |
| Cu2+ | copper |
| d-gal | d-galactose |
| FAD | familiar form of AD |
| RAGE | glycation product receptor |
| GSK3β | glycogen synthase kinase 3β |
| H2O2 | hydrogen peroxide |
| HFD | high-fat diet |
| ·OH | hydroxyl radical |
| P-Tau | hyperphosphorylated tau |
| HIF-1α | hypoxia-inducible factor 1-alpha |
| IGF | insulin-like growth factor |
| icv | intracerebroventricular |
| ip | intraperitoneal |
| Fe2+ | iron |
| MDA | malondialdehyde |
| MCI | mild cognitive impairment |
| Mn2+ | manganese |
| MWM | Morris water maze |
| MPO | myeloperoxidase |
| NMDA | N-methyl-d-aspartate |
| NFTs | neurofibrillary tangles |
| NO | nitric oxide |
| PD | Parkinson’s disease |
| PSEN1 | Presenilin 1 |
| PSEN2 | Presenilin 2 |
| P-tau | hyperphosphorylated tau |
| ROS | reactive oxygen species |
| GSH | reduced glutathione |
| SAM | senescence-accelerated mouse |
| SAD | sporadic form of AD |
| STZ | streptozotocin |
| TBI | traumatic brain injury |
| WD | Wilson’s disease |
| Zn2+ | zinc |
References
- Gao, S.; Burney, H.N.; Callahan, C.M.; Purnell, C.E.; Hendrie, H.C. Incidence of Dementia and Alzheimer Disease Over Time: A Meta-Analysis. J. Am. Geriatr. Soc. 2019, 67, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular Basis of Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A. What causes Alzheimer’s disease? Folia. Neuropathol. 2013, 51, 169–188. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.; van der Lee, S.J.; Naj, A.C.; Bellenguez, C.; Jun, G.; Logsdon, B.A.; Williams, J. Genetic mechanisms of Alzheimer’s disease. Nat. Neurosci. 2020, 23, 304–316. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Zhang, Y. Animal models of Alzheimer’s disease: Applications, evaluation, and perspectives. Zool. Res. 2022, 43, 1026–1040. [Google Scholar] [CrossRef]
- Van Dam, D.; De Deyn, P.P. Non human primate models for Alzheimer’s disease-related research and drug discovery. Expert. Opin. Drug Discov. 2016, 12, 187–200. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Transgenic mouse models of Alzheimer’s disease: Progress and challenges. J. Alzheimer’s Dis. 2017, 57, 341–363. [Google Scholar]
- Zhong, M.Z.; Peng, T.; Duarte, M.L.; Wang, M.; Cai, D. Updates on mouse models of Alzheimer’s disease. Mol. Neurodegener. 2024, 19, 23. [Google Scholar] [CrossRef]
- Wenk, G.L. Animal models of Alzheimer’s disease: Are they valid and useful? Acta Neurobiol. Exp. 1990, 50, 219–223. [Google Scholar]
- Ameen-Ali, K.E.; Wharton, S.B.; Simpson, J.E.; Heath, P.R.; Sharp, P.; Berwick, J. Review: Neuropathology and behavioural features of transgenic murine models of Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2017, 43, 553–570. [Google Scholar] [CrossRef]
- Sarasa, M.; Pesini, P. Natural non-trasgenic animal models for research in Alzheimer’s disease. Curr. Alzheimer Res. 2009, 6, 171–178. [Google Scholar]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Perl, D.P. Neuropathology of Alzheimer’s disease. Mt. Sinai J. Med. 2010, 77, 32–42. [Google Scholar] [PubMed]
- Alzheimer, A. Alzheimer’s disease (AD) is an irreversible, progressive neurodegenerative disorder that occurs gradually and results in memory loss, unusual behavior, personality changes, and a decline in thinking abilities. AD is named after Uber eine eijenartige Erkrankung der Hirnride. Allg. Z. Psychiatr. 1907, 64, 146–148. [Google Scholar]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Re-search Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar]
- Gilbert, B.J. The role of amyloid β in the pathogenesis of Alzheimer’s disease. J. Clin. Pathol. 2013, 66, 362–366. [Google Scholar] [CrossRef]
- Dickson, D.W. The pathogenesis of senile plaques. J. Neuropathol. Exp. Neurol. 1997, 56, 321–339. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 19, 5541–5554. [Google Scholar]
- Tiana, G.; Simona, F.; Broglia, R.A.; Colombo, G. Thermodynamics of beta-amyloid fibril formation. J. Chem. Phys. 2004, 120, 8307–8317. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Rowan, M.J.; Selkoe, D.J. Amyloid-beta oligomers: Their production, toxicity and therapeutic inhibition. Biochem. Soc. Trans. 2002, 30, 552–557. [Google Scholar] [CrossRef]
- Morris, J.C.; Storandt, M.; McKeel, D.W., Jr.; Rubin, E.H.; Price, J.L.; Grant, E.A.; Berg, L. Cerebral amyloid deposition and diffuse plaques in “normal” aging: Evidence for presymptomatic and very mild Alzheimer’s disease. Neurology 1996, 46, 707–719. [Google Scholar] [PubMed]
- Hayden, E.Y.; Teplow, D.B. Amyloid β-protein oligomers and Alzheimer’s disease. Alzheimers Res. Ther. 2013, 5, 60. [Google Scholar]
- Gyure, K.A.; Durham, R.; Stewart, W.F.; Smialek, J.E.; Troncoso, J.C. Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch. Pathol. Lab. Med. 2001, 125, 489–492. [Google Scholar] [PubMed]
- Walsh, D.M.; Klyubin, I.; Shankar, G.M.; Townsend, M.; Fadeeva, J.V.; Betts, V.; Podlisny, M.B.; Cleary, J.P.; Ashe, K.H.; Rowan, M.J.; et al. The role of cell-derived oligomers of Abeta in Alzheimer’s disease and avenues for therapeutic intervention. Biochem. Soc. Trans. 2005, 33, 1087–1090. [Google Scholar]
- Yan, S.S.; Chen, D.; Yan, S.; Guo, L.; Du, H.; Chen, J.X. RAGE is a key cellular target for Abeta-induced perturbation in Alzheimer’s disease. Front. Biosci. 2012, 4, 240–250. [Google Scholar]
- Koerich, S.; Parreira, G.M.; de Almeida, D.L.; Vieira, R.P.; de Oliveira, A.C.P. Receptors for Advanced Glycation End Products (RAGE): Promising Targets Aiming at the Treatment of Neurodegenerative Conditions. Curr. Neuropharmacol. 2023, 21, 219–234. [Google Scholar]
- Wang, Z.C.; Zhao, J.; Li, S. Dysregulation of synaptic and extrasynaptic N-methyl-D-aspartate receptors induced by amyloid-β. Neurosci. Bull. 2013, 29, 752–760. [Google Scholar]
- Ni, R.; Marutle, A.; Nordberg, A. Modulation of α7 nicotinic acetylcholine receptor and fibrillar amyloid-β interactions in Alzheimer’s disease brain. J. Alzheimers Dis. 2013, 33, 841–851. [Google Scholar]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 87–95. [Google Scholar]
- Opazo, C.; Huang, X.; Cherny, R.A.; Moir, R.D.; Roher, A.E.; White, A.R.; Cappai, R.; Masters, C.L.; Tanzi, R.E.; Inestrosa, N.C.; et al. Metalloenzyme-like activity of Alzheimer’s disease beta-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H(2)O(2). J. Biol. Chem. 2002, 277, 40302–40308. [Google Scholar]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed]
- Mietelska-Porowska, A.; Wasik, U.; Goras, M.; Filipek, A.; Niewiadomska, G. Tau protein modifications and interactions: Their role in function and dysfunction. Int. J. Mol. Sci. 2014, 15, 4671–4713. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.T.; Patel, T.; Kang, S.G.; Yarahmady, A.; Srinivasan, M.; Julien, O.; Heras, J.; Mok, S.A. Disease-Associated Mutations in Tau Encode for Changes in Aggregate Structure Conformation. ACS Chem. Neurosci. 2023, 14, 4282–4297. [Google Scholar] [CrossRef] [PubMed]
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated Tau in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2022, 23, 12841. [Google Scholar] [CrossRef]
- Sinsky, J.; Pichlerova, K.; Hanes, J. Tau Protein Interaction Partners and Their Roles in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2021, 22, 9207. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R. Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta 2005, 1739, 240–250. [Google Scholar]
- Ramkumar, A.; Jong, B.Y.; Ori-McKenney, K.M. ReMAPping the microtubule landscape: How phosphorylation dictates the activities of microtubule-associated proteins. Dev. Dyn. 2018, 247, 138–155. [Google Scholar]
- Reddy, P.H.; Reddy, T.P. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr. Alzheimer Res. 2011, 8, 393–409. [Google Scholar]
- Tolnay, M.; Probst, A. The neuropathological spectrum of neurodegenerative tauopathies. IUBMB Life 2003, 55, 299–305. [Google Scholar]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152. [Google Scholar]
- Zetterberg, H.; Mattsson, N. Understanding the cause of sporadic Alzheimer’s disease. Expert. Rev. Neurother. 2014, 14, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Guglielmotto, M.; Aragno, M.; Autelli, R.; Giliberto, L.; Novo, E.; Colombatto, S.; Danni, O.; Parola, M.; Smith, M.A.; Perry, G.; et al. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: Role of oxidative stress and HIF1alpha. J. Neurochem. 2009, 108, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Atwood, C.S.; Hartshorn, M.A.; Multhaup, G.; Goldstein, L.E.; Scarpa, R.C.; Cuajungco, M.P.; Gray, D.N.; Lim, J.; Moir, R.D.; et al. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 1999, 38, 7609–7616. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R.; Oliver, C.N. Metal-catalyzed oxidation of proteins. Physiological consequences. J. Biol. Chem. 1991, 266, 2005–2008. [Google Scholar] [CrossRef]
- Islam, F.; Shohag, S.; Akhter, S.; Islam, M.R.; Sultana, S.; Mitra, S.; Chandran, D.; Khandaker, M.U.; Ashraf, G.M.; Idris, A.M.; et al. Exposure of metal toxicity in Alzheimer’s disease: An extensive review. Front. Pharmacol. 2022, 13, 903099. [Google Scholar] [CrossRef]
- Kawahara, M.; Kato-Negishi, M.; Tanaka, K.I. Dietary Trace Elements and the Pathogenesis of Neurodegenerative Diseases. Nutrients 2023, 15, 2067. [Google Scholar] [CrossRef]
- Ramos-Cejudo, J.; Wisniewski, T.; Marmar, C.; Zetterberg, H.; Blennow, K.; de Leon, M.J.; Fossati, S. Traumatic Brain Injury and Alzheimer’s Disease: The Cerebrovascular Link. EBioMedicine 2018, 28, 21–30. [Google Scholar] [CrossRef]
- McKee, A.C.; Stern, R.A.; Nowinski, C.J.; Stein, T.D.; Alvarez, V.E.; Daneshvar, D.H.; Lee, H.S.; Wojtowicz, S.M.; Hall, G.; Baugh, C.M.; et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013, 136 Pt 1, 43–64. [Google Scholar] [CrossRef]
- Washington, P.M.; Morffy, N.; Parsadanian, M.; Zapple, D.N.; Burns, M.P. Experimental traumatic brain injury induces rapid aggregation and oligomerization of amyloid-beta in an Alzheimer’s disease mouse model. J. Neurotrauma 2014, 31, 125–134. [Google Scholar] [CrossRef]
- McKee, A.C.; Cairns, N.J.; Dickson, D.W.; Folkerth, R.D.; Keene, C.D.; Litvan, I.; Perl, D.P.; Stein, T.D.; Vonsattel, J.P.; Stewart, W.; et al. TBI/CTE group. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016, 131, 75–86. [Google Scholar] [CrossRef]
- Zotova, E.; Nicoll, J.A.; Kalaria, R.; Holmes, C.; Boche, D. Inflammation in Alzheimer’s disease: Relevance to pathogenesis and therapy. Alzheimers Res. Ther. 2010, 2, 1. [Google Scholar] [PubMed]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [PubMed]
- Weisman, D.; Hakimian, E.; Ho, G.J. Interleukins, inflammation, and mechanisms of Alzheimer’s disease. Vitam. Horm. 2006, 74, 505–530. [Google Scholar] [PubMed]
- Rubio-Perez, J.M.; Morillas-Ruiz, J.M. A review: Inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J. 2012, 2012, 756357. [Google Scholar]
- Mrak, R.E.; Griffin, W.S. Potential inflammatory biomarkers in Alzheimer’s disease. J. Alzheimers Dis. 2005, 8, 369–375. [Google Scholar]
- Banks, W.A.; Farr, S.A.; Morley, J.E. Entry of blood-borne cytokines into the central nervous system: Effects on cognitive processes. Neuroimmunomodulation 2003, 10, 319–327. [Google Scholar]
- Shaftel, S.S.; Griffin, W.S.; O’Banion, M.K. The role of interleukin-1 in neuroinflammation and Alzheimer disease: An evolving perspective. J. Neuroinflammation 2008, 5, 7. [Google Scholar] [CrossRef]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937–1942. [Google Scholar]
- Neumann, K.F.; Rojo, L.; Navarrete, L.P.; Farías, G.; Reyes, P.; Maccioni, R.B. Insulin resistance and Alzheimer’s disease: Molecular links & clinical implications. Curr. Alzheimer Res. 2008, 5, 438–447. [Google Scholar]
- Son, S.M.; Song, H.; Byun, J.; Park, K.S.; Jang, H.C.; Park, Y.J.; Mook-Jung, I. Altered APP processing in insulin-resistant conditions is mediated by autophagosome accumulation via the inhibition of mammalian target of rapamycin pathway. Diabetes 2012, 61, 3126–3138. [Google Scholar]
- Bjorkhem, I.; Meaney, S. Brain cholesterol: Long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 806–815. [Google Scholar] [PubMed]
- Dietschy, J.M.; Turley, S.D. Cholesterol metabolism in the brain. Curr. Opin. Lipidol. 2001, 12, 105–112. [Google Scholar] [PubMed]
- Simons, M.; Keller, P.; Dichgans, J.; Schulz, J.B. Cholesterol and Alzheimer’s disease: Is there a link? Neurology 2001, 57, 1089–1093. [Google Scholar] [PubMed]
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011, 10, 819–828. [Google Scholar]
- Durazzo, T.C.; Mattsson, N.; Weiner, M.W. Smoking and increased Alzheimer’s disease risk: A review of potential mechanisms. Alzheimers Dement. 2014, 10, S122–S145. [Google Scholar]
- Rueff-Barroso, C.R.; Trajano, E.T.; Alves, J.N.; Paiva, R.O.; Lanzetti, M.; Pires, K.M.; Bezerra, F.S.; Pinho, R.A.; Valenca, S.S.; Porto, L.C. Organ-related cigarette smoke-induced oxidative stress is strain-dependent. Med. Sci. Monit. 2010, 16, BR218–BR226. [Google Scholar]
- Ho, Y.S.; Yang, X.; Yeung, S.C.; Chiu, K.; Lau, C.F.; Tsang, A.W.; Mak, J.C.; Chang, R.C. Cigarette smoking accelerated brain aging and induced pre-Alzheimer-like neuropathology in rats. PLoS ONE 2012, 7, e36752. [Google Scholar]
- Khanna, A.; Guo, M.; Mehra, M.; Royal, W., 3rd. Inflammation and oxidative stress induced by cigarette smoke in Lewis rat brains. J. Neuroimmunol. 2013, 254, 69–75. [Google Scholar]
- Calder, P.C.; Bosco, N.; Bourdet-Sicard, R.; Capuron, L.; Delzenne, N.; Doré, J.; Franceschi, C.; Lehtinen, M.J.; Recker, T.; Salvioli, S.; et al. Health relevance of the modification of low grade inflammation in ageing (inflammageing) and the role of nutrition. Ageing Res. Rev. 2017, 40, 95–119. [Google Scholar]
- Miquel, S.; Champ, C.; Day, J.; Aarts, E.; Bahr, B.A.; Bakker, M.; Bánáti, D.; Calabrese, V.; Cederholm, T.; Cryan, J.; et al. Poor cognitive ageing: Vulnerabilities, mechanisms and the impact of nutritional interventions. Ageing Res. Rev. 2018, 42, 40–55. [Google Scholar]
- McGrattan, A.M.; McGuinness, B.; McKinley, M.C.; Kee, F.; Passmore, P.; Woodside, J.V.; McEvoy, C.T. Diet and Inflammation in Cognitive Ageing and Alzheimer’s Disease. Curr. Nutr. Rep. 2019, 8, 53–65. [Google Scholar] [PubMed]
- Peng, B.; Yang, Q.B.; Joshi, R.; Liu, Y.; Akbar, M.; Song, B.J.; Zhou, S.; Wang, X. Role of Alcohol Drinking in Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 2316. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Risk Reduction of Cognitive Decline and Dementia: WHO Guidelines; World Health Organization: Geneva, Switzerland, 2019; Available online: https://www.who.int/publications/i/item/9789241550543 (accessed on 20 January 2024).
- Kuusisto, J.; Koivisto, K.; Kervinen, K.; Mykkänen, L.; Helkala, E.L.; Vanhanen, M.; Hänninen, T.; Pyörälä, K.; Kesäniemi, Y.A.; Riekkinen, P.; et al. Association of apolipoprotein E phenotypes with late onset Alzheimer’s disease: Population based study. BMJ 1994, 309, 636–638. [Google Scholar] [PubMed]
- Barabash, A.; Marcos, A.; Ancín, I.; Vázquez-Alvarez, B.; de Ugarte, C.; Gil, P.; Fernández, C.; Encinas, M.; López-Ibor, J.J.; Cabranes, J.A. APOE, ACT and CHRNA7 genes in the conversion from amnestic mild cognitive impairment to Alzheimer’s disease. Neurobiol. Aging 2009, 30, 1254–1264. [Google Scholar] [CrossRef]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar]
- Kamat, P.K. Streptozotocin induced Alzheimer’s disease like changes and the underlying neural degeneration and regeneration mechanism. Neural Regen. Res. 2015, 10, 1050–1052. [Google Scholar]
- Salkovic-Petrisic, M.; Knezovic, A.; Hoyer, S.; Riederer, P. What have we learned from the streptozotocin-induced animal model of sporadic Alzheimer’s disease, about the therapeutic strategies in Alzheimer’s research. J. Neural Transm. 2013, 120, 233–252. [Google Scholar]
- Rai, S.; Kamat, P.K.; Nath, C.; Shukla, R. Glial activation and post-synaptic neurotoxicity: The key events in Streptozotocin (ICV) induced memory impairment in rats. Pharmacol. Biochem. Behav. 2014, 117, 104–117. [Google Scholar]
- Labak, M.; Foniok, T.; Kirk, D.; Rushforth, D.; Tomanek, B.; Jasiński, A.; Grieb, P. Metabolic changes in rat brain following intracerebroventricular injections of streptozotocin: A model of sporadic Alzheimer’s disease. Acta Neurochir. Suppl. 2010, 106, 177–181. [Google Scholar]
- Chen, Y.; Liang, Z.; Blanchard, J.; Dai, C.L.; Sun, S.; Lee, M.H.; Grundke-Iqbal, I.; Iqbal, K.; Liu, F.; Gong, C.X. A non-transgenic mouse model (icv-STZ mouse) of Alzheimer’s disease: Similarities to and differences from the transgenic model (3xTg-AD mouse). Mol. Neurobiol. 2013, 47, 711–725. [Google Scholar]
- Chen, Y.; Liang, Z.; Tian, Z.; Blanchard, J.; Dai, C.L.; Chalbot, S.; Iqbal, K.; Liu, F.; Gong, C.X. Intracerebroventricular streptozotocin exacerbates Alzheimer-like changes of 3xTg-AD mice. Mol. Neurobiol. 2014, 49, 547–562. [Google Scholar]
- Grieb, P. Intracerebroventricular Streptozotocin Injections as a Model of Alzheimer’s Disease: In Search of a Relevant Mechanism. Mol. Neurobiol. 2016, 53, 1741–1752. [Google Scholar] [CrossRef]
- Tang, K.S. The cellular and molecular processes associated with scopolamine-induced memory deficit: A model of Alzheimer’s biomarkers. Life Sci. 2019, 233, 116695. [Google Scholar]
- Morley, J.E.; Armbrecht, H.J.; Farr, S.A.; Kumar, V.B. The senescence accelerated mouse (SAMP8) as a model for oxidative stress and Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 650–656. [Google Scholar]
- Levy Nogueira, M.; Epelbaum, S.; Steyaert, J.M.; Dubois, B.; Schwartz, L. Mechanical stress models of Alzheimer’s disease pathology. Alzheimers Dement. 2016, 12, 324–333. [Google Scholar]
- Song, J. Animal Model of Aluminum-Induced Alzheimer’s Disease. Adv. Exp. Med. Biol. 2018, 1091, 113–127. [Google Scholar]
- Valentin-Escalera, J.; Leclerc, M.; Calon, F. High-Fat Diets in Animal Models of Alzheimer’s Disease: How Can Eating Too Much Fat Increase Alzheimer’s Disease Risk? J. Alzheimers. Dis. 2024, 97, 977–1005. [Google Scholar]
- Bagheri, M.; Joghataei, M.T.; Mohseni, S.; Roghani, M. Genistein ameliorates learning and memory deficits in amyloid β(1–40) rat model of Alzheimer’s disease. Neurobiol. Learn Mem. 2011, 95, 270–276. [Google Scholar]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef]
- Safar, M.M.; Arab, H.H.; Rizk, S.M.; El-Maraghy, S.A. Bone Marrow-Derived Endothelial Progenitor Cells Protect Against Scopolamine-Induced Alzheimer-Like Pathological Aberrations. Mol. Neurobiol. 2016, 53, 1403–1418. [Google Scholar] [CrossRef]
- Llorens-Martín, M.; Jurado, J.; Hernández, F.; Avila, J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar]
- Lu, C.; Wang, Y.; Xu, T.; Li, Q.; Wang, D.; Zhang, L.; Fan, B.; Wang, F.; Liu, X. Genistein Ameliorates Scopolamine-Induced Amnesia in Mice Through the Regulation of the Cholinergic Neurotransmission, Antioxidant System and the ERK/CREB/BDNF Signaling. Front. Pharmacol. 2018, 9, 1153. [Google Scholar]
- Sun, D.; Unnithan, R.R.; French, C. Scopolamine Impairs Spatial Information Recorded With “Miniscope” Calcium Imaging in Hippocampal Place Cells. Front. Neurosci. 2021, 15, 640350. [Google Scholar]
- Shen, H.; Zheng, Y.; Chen, R.; Huang, X.; Shi, G. Neuroprotective effects of quercetin 3-O-sophoroside from Hibiscus rosa-sinensis Linn. on scopolamine-induced amnesia in mice. J. Funct. Foods 2021, 76, 104291. [Google Scholar] [CrossRef]
- Ghasemi, S.; Moradzadeh, M.; Hosseini, M.; Beheshti, F.; Sadeghnia, H.R. Beneficial effects of Urtica dioica on scopolamine-induced memory impairment in rats: Protection against acetylcholinesterase activity and neuronal oxidative damage. Drug Chem. Toxicol. 2019, 42, 167–175. [Google Scholar]
- Kameyama, T.; Nabeshima, T.; Kozawa, T. Step-down-type passive avoidance- and escape-learning method. Suitability for experimental amnesia models. J. Pharmacol. Methods 1986, 16, 39–52. [Google Scholar]
- Utkan, T.; Gocmez, S.S.; Ozer, C.; Gacar, N.; Aricioglu, F. Selective and nonselective neuronal NOS inhibitors impair cognitive function in the three panel runway and passive avoidance tasks in rats. Pharmacol. Biochem. Behav. 2012, 101, 515–519. [Google Scholar] [PubMed]
- Buege, J.A.; Aust, S.D. Microsomal lipid peroxidation. Methods Enzym. 1978, 52, 302–310. [Google Scholar]
- Sanchez-Varo, R.; Mejias-Ortega, M.; Fernandez-Valenzuela, J.J.; Nu√±ez-Diaz, C.; Caceres-Palomo, L.; Vegas-Gomez, L.; Sanchez-Mejias, E.; Trujillo-Estrada, L.; Garcia-Leon, J.A.; Moreno-Gonzalez, I.; et al. Transgenic Mouse Models of Alzheimer’s Disease: An Integrative Analysis. Int. J. Mol. Sci. 2022, 23, 5404. [Google Scholar] [CrossRef]
- Morley, J.E.; Farr, S.A.; Kumar, V.B.; Armbrecht, H.J. The SAMP8 mouse: A model to develop therapeutic interventions for Alzheimer’s disease. Curr. Pharm. Des. 2012, 18, 1123–1130. [Google Scholar]
- Kurokawa, T.; Ozaki, N.; Ishibashi, S. Difference between senescence-accelerated prone and resistant mice in response to insulin in the heart. Mech. Ageing Dev. 1998, 102, 25–32. [Google Scholar] [CrossRef]
- Pallas, M.; Camins, A.; Smith, M.A.; Perry, G.; Lee, H.G.; Casadesus, G. From aging to Alzheimer’s disease: Unveiling “the switch” with the senescence-accelerated mouse model (SAMP8). J. Alzheimers Dis. 2008, 15, 615–624. [Google Scholar] [CrossRef]
- Takeda, T. Senescence-accelerated mouse (SAM) with special references to neurodegeneration models, SAMP8 and SAMP10 mice. Neurochem. Res. 2009, 34, 639–659. [Google Scholar] [CrossRef]
- Cheng, X.R.; Zhou, W.X.; Zhang, Y.X. The behavioral, pathological and therapeutic features of the senescence-accelerated mouse prone 8 strain as an Alzheimer’s disease animal model. Ageing Res. Rev. 2014, 13, 13–37. [Google Scholar] [CrossRef]
- Rebrin, I.; Zicker, S.; Wedekind, K.J.; Paetau-Robinson, I.; Packer, L.; Sohal, R.S. Effect of antioxidant-enriched diets on glutathione redox status in tissue homogenates and mitochondria of the senescence-accelerated mouse. Free Radic. Biol. Med. 2005, 39, 549–557. [Google Scholar] [CrossRef]
- Prieto, R.; Gutiérrez-González, R.; Pascual, J.M.; Roda, J.M.; Cerdán, S.; Matias-Guiu, J.; Barcia, J.A. Modelos experimentales de traumatismo craneoencefálico. Experimental models of traumatic brain injury. Neurocirugia 2009, 20, 225–244. [Google Scholar] [CrossRef]
- Ojo, J.O.; Mouzon, B.; Greenberg, M.B.; Bachmeier, C.; Mullan, M.; Crawford, F. Repetitive mild traumatic brain injury augments tau pathology and glial activation in aged hTau mice. J. Neuropathol. Exp. Neurol. 2013, 72, 137–151. [Google Scholar] [CrossRef]
- Breunig, J.J.; Guillot-Sestier, M.V.; Town, T. Brain injury, neuroinflammation and Alzheimer’s disease. Front. Aging Neurosci. 2013, 5, 26. [Google Scholar] [CrossRef]
- Payno, M.; Villanueva, C.; Matías-Guiu, J. Modelos experimentales de la enfermedad de Alzheimer. Experimental models in Alzheimer’s disease. Neurologia 2009, 24, 255–262. [Google Scholar]
- Levy Nogueira, M.; Hamraz, M.; Abolhassani, M.; Bigan, E.; Lafitte, O.; Steyaert, J.M.; Dubois, B.; Schwartz, L. Mechanical stress increases brain amyloid β, tau, and α-synuclein concentrations in wild-type mice. Alzheimers Dement. 2018, 14, 444–453. [Google Scholar] [CrossRef]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Huang, X.; Atwood, C.S.; Moir, R.D.; Hartshorn, M.A.; Vonsattel, J.P.; Tanzi, R.E.; Bush, A.I. Zinc-induced Alzheimer’s Abeta1-40 aggregation is mediated by conformational factors. J. Biol. Chem. 1997, 272, 26464–26470. [Google Scholar] [CrossRef]
- Campbell, A. The potential role of aluminium in Alzheimer’s disease. Nephrol. Dial. Transplant. 2002, 17 (Suppl. S2), 17–20. [Google Scholar] [CrossRef]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Wang, H.; Boldogh, I.; Rao, K.S.; Mitra, S.; Hegde, M.L. New perspectives on oxidized genome damage and repair inhibition by pro-oxidant metals in neurological diseases. Biomolecules 2014, 4, 678–703. [Google Scholar] [CrossRef]
- Kawahara, M.; Kato-Negishi, M. Link between Aluminum and the Pathogenesis of Alzheimer’s Disease: The Integration of the Aluminum and Amyloid Cascade Hypotheses. Int. J. Alzheimers Dis. 2011, 2011, 276393. [Google Scholar] [CrossRef]
- Lukiw, W.J.; LeBlanc, H.J.; Carver, L.A.; McLachlan, D.R.; Bazan, N.G. Run-on gene transcription in human neocortical nuclei. Inhibition by nanomolar aluminum and implications for neurodegenerative disease. J. Mol. Neurosci. 1998, 11, 67–78. [Google Scholar] [CrossRef]
- Scott, C.W.; Fieles, A.; Sygowski, L.A.; Caputo, C.B. Aggregation of tau protein by aluminum. Brain Res. 1993, 628, 77–84. [Google Scholar] [CrossRef]
- Walton, J.R.; Wang, M.X. APP expression, distribution and accumulation are altered by aluminum in a rodent model for Alzheimer’s disease. J. Inorg. Biochem. 2009, 103, 1548–1554. [Google Scholar] [CrossRef]
- Bitra, V.R.; Rapaka, D.; Mathala, N.; Akula, A. Effect of wheat grass powder on aluminum induced Alzheimer’s disease in Wistar rats. Asian Pac. J. Trop. Med. 2014, 7, S278–S281. [Google Scholar] [CrossRef]
- Bazzari, F.H.; Abdallah, D.M.; El-Abhar, H.S. Chenodeoxycholic Acid Ameliorates AlCl3-Induced Alzheimer’s Disease Neurotoxicity and Cognitive Deterioration via Enhanced Insulin Signaling in Rats. Molecules 2019, 24, 1992. [Google Scholar] [CrossRef]
- Liaquat, L.; Ahmad, S.; Sadir, S.; Batool, Z.; Khaliq, S.; Tabassum, S.; Emad, S.; Madiha, S.; Shahzad, S.; Haider, S. Development of AD like symptoms following co-administration of AlCl3 and d-gal in rats: A neurochemical, biochemical and behavioural study. Pak. J. Pharm. Sci. 2017, 30 (Suppl. S2), 647–653. [Google Scholar]
- Haider, S.; Liaquat, L.; Ahmad, S.; Batool, Z.; Siddiqui, R.A.; Tabassum, S.; Shahzad, S.; Rafiq, S.; Naz, N. Naringenin protects AlCl3/d-galactose induced neurotoxicity in rat model of AD via attenuation of acetylcholinesterase levels and inhibition of oxidative stress. PLoS ONE 2020, 15, e0227631. [Google Scholar]
- Livingston, G.; Sommerlad, A.; Orgeta, V.; Costafreda, S.G.; Huntley, J.; Ames, D.; Ballard, C.; Banerjee, S.; Burns, A.; Cohen-Mansfield, J.; et al. Dementia prevention, intervention, and care. Lancet 2017, 390, 2673–2734. [Google Scholar]
- Samadi, M.; Moradi, S.; Moradinazar, M.; Mostafai, R.; Pasdar, Y. Dietary pattern in relation to the risk of Alzheimer’s disease: A systematic review. Neurol. Sci. 2019, 40, 2031–2043. [Google Scholar] [CrossRef]
- Hill, E.; Clifton, P.; Goodwill, A.M.; Dennerstein, L.; Campbell, S.; Szoeke, C. Dietary patterns and β-amyloid deposition in aging Australian women. Alzheimers Dement. 2018, 4, 535–541. [Google Scholar]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar]
- Nuzzo, D.; Picone, P.; Baldassano, S.; Caruana, L.; Messina, E.; Gammazza, A.; Cappello, F.; Mulè, F.; Carlo, M. Insulin resistance as common molecular denominator linking obesity to Alzheimer’s disease. Curr. Alzheimer Res. 2015, 12, 723–735. [Google Scholar]
- Baranowski, B.J.; Bott, K.N.; MacPherson, R.E.K. Evaluation of neuropathological effects of a high-fat high-sucrose diet in middle-aged male C57BL6/J mice. Physiol. Rep. 2018, 6, e13729. [Google Scholar] [CrossRef]
- Busquets, O.; Ettcheto, M.; Pallàs, M.; Beas-Zarate, C.; Verdaguer, E.; Auladell, C.; Folch, J.; Camins, A. Long-term exposition to a high fat diet favors the appearance of β-amyloid depositions in the brain of C57BL/6J mice. A potential model of sporadic Alzheimer’s disease. Mech. Ageing Dev. 2017, 162, 38–45. [Google Scholar]
- Niu, L.; Han, D.W.; Xu, R.L.; Han, B.; Zhou, X.; Wu, H.W.; Li, S.H.; Qu, C.X.; Liu, M. A high-sugar high-fat diet induced metabolic syndrome shows some symptoms of Alzheimer’s disease in rats. J. Nutr. Heal. Aging 2016, 20, 509–513. [Google Scholar]
- Salas, I.H.; Weerasekera, A.; Ahmed, T.; Callaerts-Vegh, Z.; Himmelreich, U.; D’Hooge, R.; Balschun, D.; Saido, T.C.; De Strooper, B.; Dotti, C.G. High fat diet treatment impairs hippocampal long-term potentiation without alterations of the core neuropathological features of Alzheimer disease. Neurobiol. Dis. 2018, 13, 82–96. [Google Scholar]
- Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Mitschelen, M.; Koller, A.; Szalai, G.; Sonntag, W.E.; Ungvari, Z.; Csiszar, A. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: Effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1212–1226. [Google Scholar] [PubMed]
- Chang, W.; Huang, D.; Lo, Y.M.; Tee, Q.; Kuo, P.; Wu, J.S.; Huang, W.; Shen, S. Protective effect of caffeic acid against Alzheimer’s disease pathogenesis via modulating cerebral insulin signaling, β-amyloid accumulation, and synaptic plasticity in hyperinsulinemic rats. J. Agric. Food Chem. 2019, 67, 7684–7693. [Google Scholar] [CrossRef] [PubMed]
- Nakandakari, S.C.B.R.; Muñoz, V.R.; Kuga, G.K.; Gaspar, R.C.; Sant’Ana, M.R.; Pavan, I.C.B.; da Silva, L.G.S.; Morelli, A.P.; Simabuco, F.M.; da Silva, A.S.R.; et al. Short-term high-fat diet modulates several inflammatory, ER stress, and apoptosis markers in the hippocampus of young mice. Brain Behav. Immun. 2019, 79, 284–293. [Google Scholar]
- Saraceno, C.; Musardo, S.; Marcello, E.; Pelucchi, S.; Di Luca, M. Modeling Alzheimer’s disease: From past to future. Front. Pharmacol. 2013, 4, 77. [Google Scholar]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef]
- Tag, S.H.; Kim, B.; Bae, J.; Chang, K.A.; Im, H.I. Neuropathological and behavioral features of an APP/PS1/MAPT (6xTg) transgenic model of Alzheimer’s disease. Mol. Brain 2022, 15, 51. [Google Scholar] [CrossRef]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef]
- Trejo-Lopez, J.A.; Yachnis, A.T.; Prokop, S. Neuropathology of Alzheimer’s Disease. Neurotherapeutics 2022, 19, 173–185. [Google Scholar] [CrossRef]
- Moya-Alvarado, G.; Gershoni-Emek, N.; Perlson, E.; Bronfman, F.C. Neurodegeneration and Alzheimer’s disease (AD). What Can Proteomics Tell Us About the Alzheimer’s Brain? Mol. Cell Proteom. 2016, 15, 409–425. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Rodríguez, M.; Vega López, J.M.; Martínez-Rosas, M.; Nicolás-Vázquez, M.I.; Mera Jiménez, E. Murine Non-Transgenic Models of Alzheimer’s Disease Pathology: Focus on Risk Factors. Brain Sci. 2025, 15, 322. https://doi.org/10.3390/brainsci15030322
Hernández-Rodríguez M, Vega López JM, Martínez-Rosas M, Nicolás-Vázquez MI, Mera Jiménez E. Murine Non-Transgenic Models of Alzheimer’s Disease Pathology: Focus on Risk Factors. Brain Sciences. 2025; 15(3):322. https://doi.org/10.3390/brainsci15030322
Chicago/Turabian StyleHernández-Rodríguez, Maricarmen, Juan Manuel Vega López, Martín Martínez-Rosas, María Inés Nicolás-Vázquez, and Elvia Mera Jiménez. 2025. "Murine Non-Transgenic Models of Alzheimer’s Disease Pathology: Focus on Risk Factors" Brain Sciences 15, no. 3: 322. https://doi.org/10.3390/brainsci15030322
APA StyleHernández-Rodríguez, M., Vega López, J. M., Martínez-Rosas, M., Nicolás-Vázquez, M. I., & Mera Jiménez, E. (2025). Murine Non-Transgenic Models of Alzheimer’s Disease Pathology: Focus on Risk Factors. Brain Sciences, 15(3), 322. https://doi.org/10.3390/brainsci15030322