
Current Applications of Single-Cell RNA Sequencing in Glioblastoma: A Scoping Review

 ,
,  , , , , , , , and
, , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
3. Review
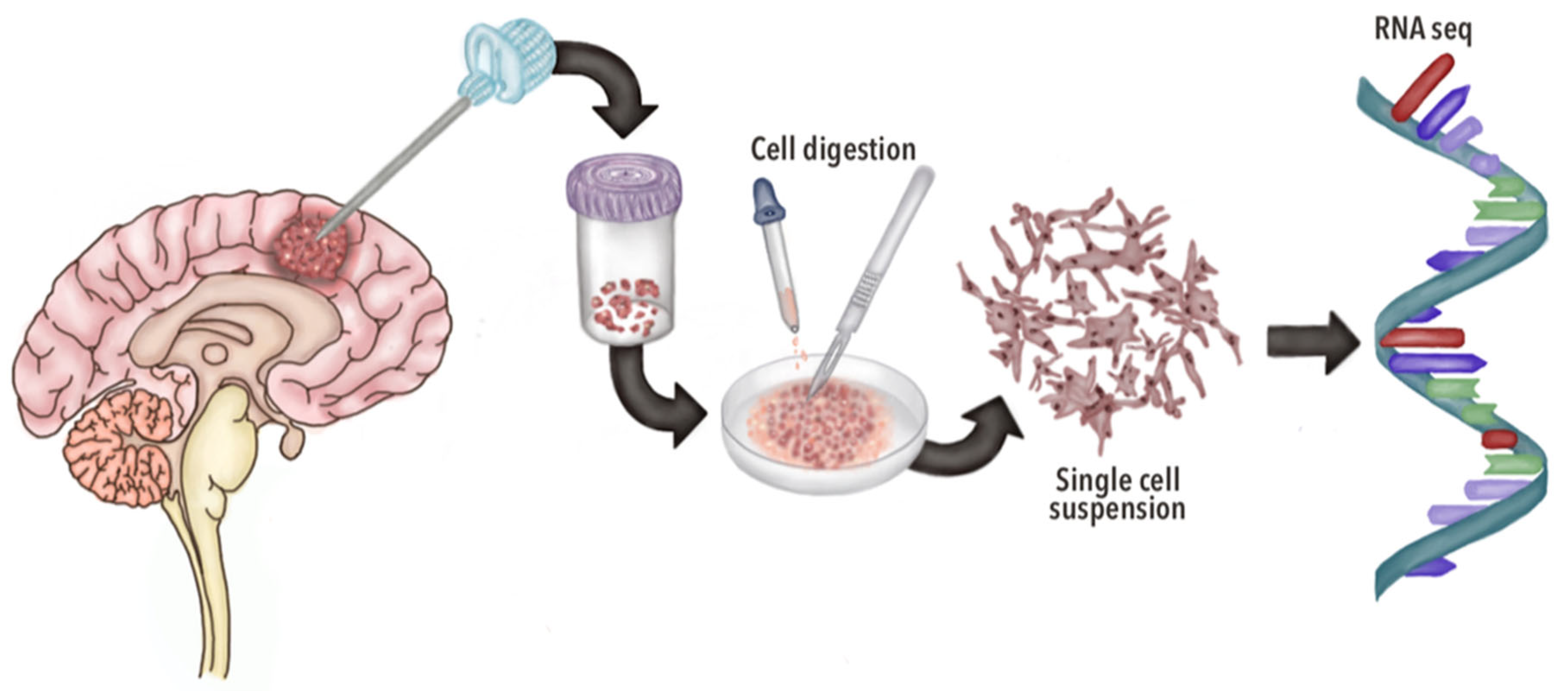
3.1. General Aspects of scRNA-seq
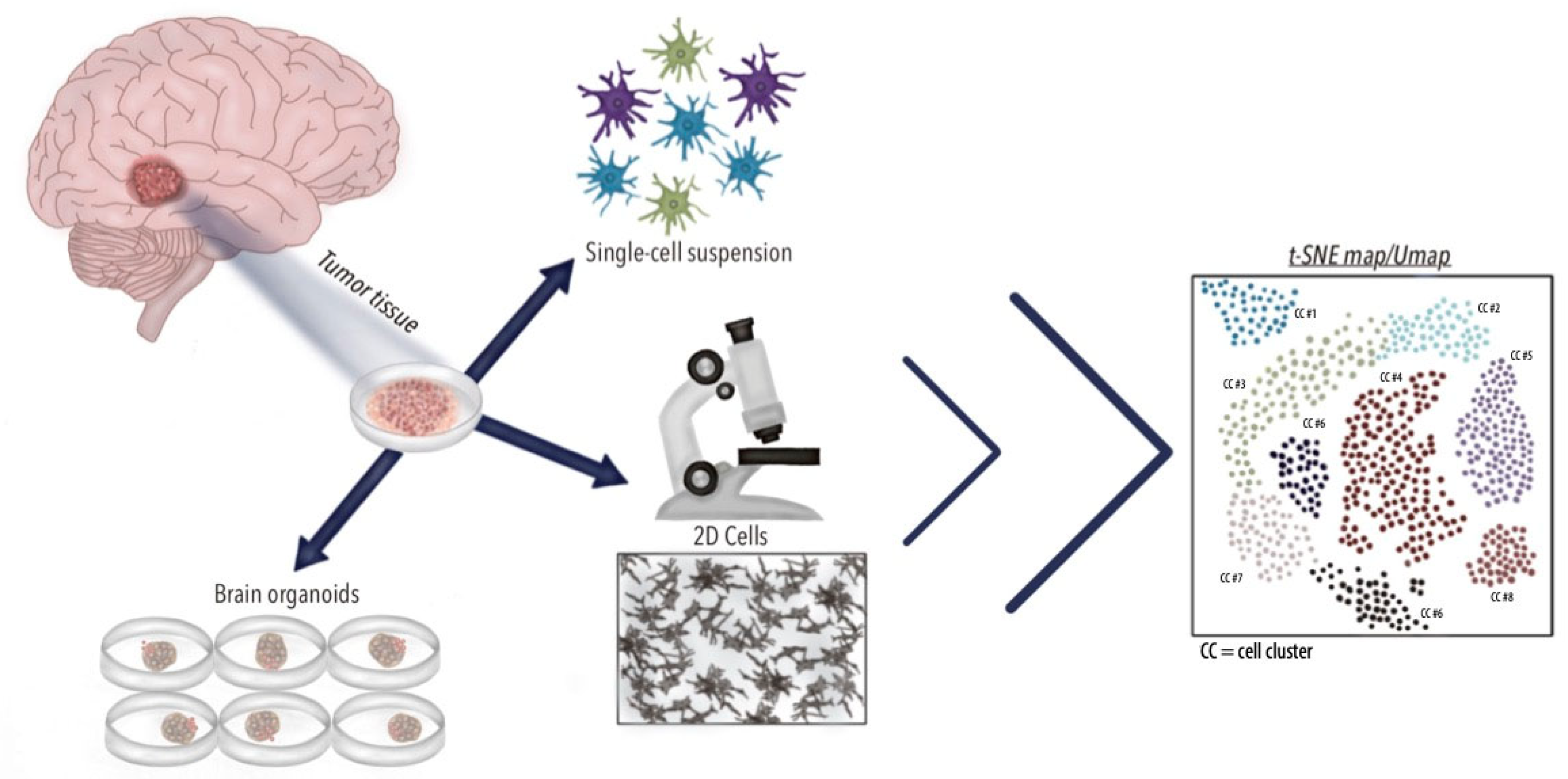
3.2. In Vitro and In Vivo Applications of scRNA-seq in GBM
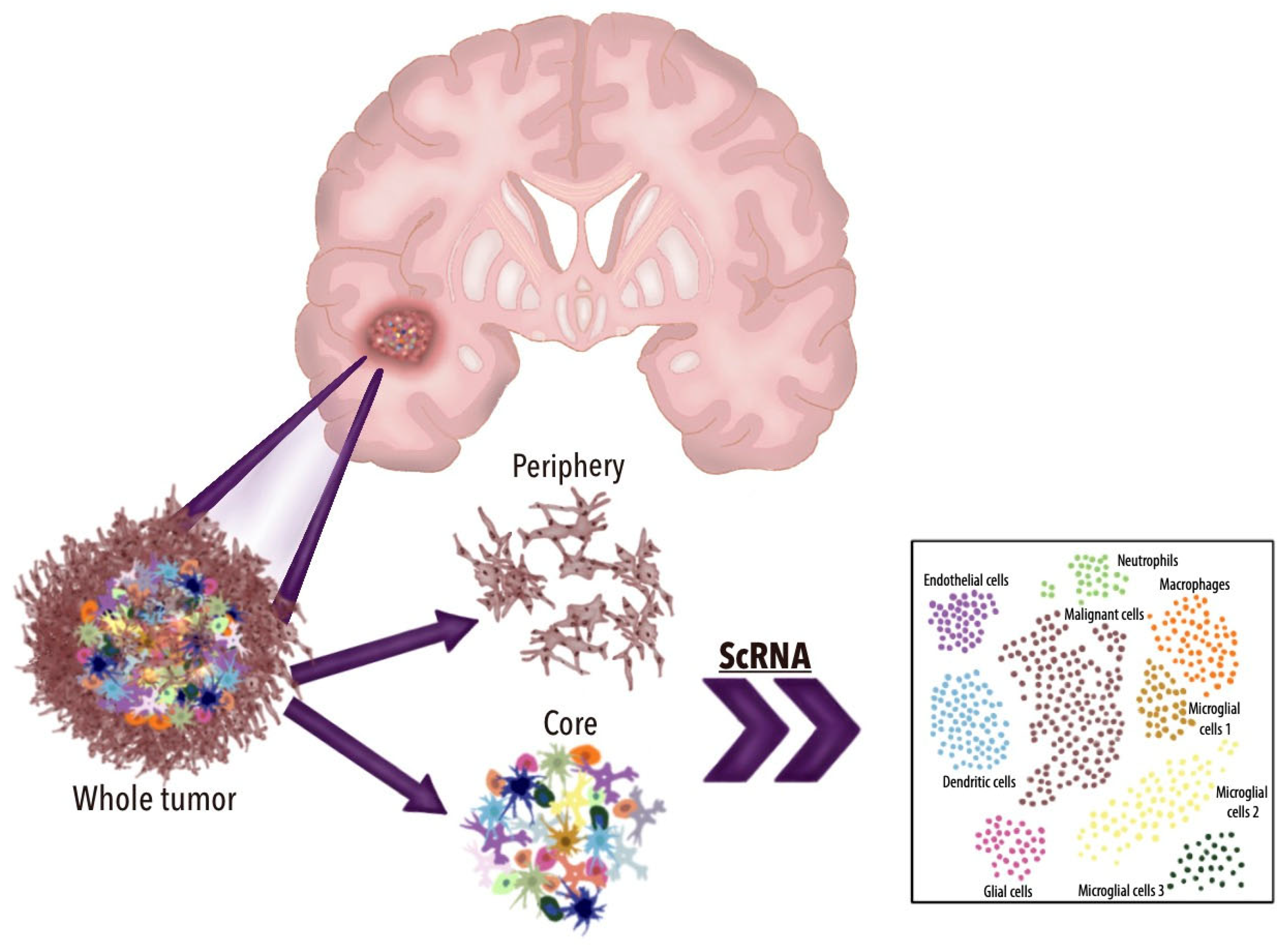
3.3. TME Dynamics in GBM and scRNA-seq Applications
3.4. Potential Diagnostic Applications of scRNA-seq in GBM
3.5. Prognostic Applications of scRNA-seq in GBM
3.6. Therapeutic Applications of scRNA-seq in GBM
3.6.1. Precision Medicine
3.6.2. Chemotherapy
3.6.3. Immunotherapy
3.6.4. Radiotherapy
3.6.5. Mechanisms of Resistance
3.6.6. Technical Limitations and Challenges of scRNA-seq in GBM Research
3.7. Future Perspectives
4. Limitations
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Disclosures
Abbreviations
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Price, M.; Neff, C.; Nagarajan, N.; Kruchko, C.; Waite, K.A.; Cioffi, G.; Cordeiro, B.B.; Willmarth, N.; Penas-Prado, M.; Gilbert, M.R.; et al. CBTRUS Statistical Report: American Brain Tumor Association & NCI Neuro-Oncology Branch Adolescent and Young Adult Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016–2020. Neuro Oncol. 2024, 26, iii1–iii53. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Lopez, P.D.; Corrales-Garcia, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef]
- Johnson, K.C.; Anderson, K.J.; Courtois, E.T.; Gujar, A.D.; Barthel, F.P.; Varn, F.S.; Luo, D.; Seignon, M.; Yi, E.; Kim, H.; et al. Single-cell multimodal glioma analyses identify epigenetic regulators of cellular plasticity and environmental stress response. Nat. Genet. 2021, 53, 1456–1468. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, D.; Peng, M.; Tang, L.; Ouyang, J.; Xiong, F.; Guo, C.; Tang, Y.; Zhou, Y.; Liao, Q.; et al. Single-cell RNA sequencing in cancer research. J. Exp. Clin. Cancer Res. 2021, 40, 81. [Google Scholar] [CrossRef]
- Wu, H.; Guo, C.; Wang, C.; Xu, J.; Zheng, S.; Duan, J.; Li, Y.; Bai, H.; Xu, Q.; Ning, F.; et al. Single-cell RNA sequencing reveals tumor heterogeneity, microenvironment, and drug-resistance mechanisms of recurrent glioblastoma. Cancer Sci. 2023, 114, 2609–2621. [Google Scholar] [CrossRef]
- Jain, S.; Rick, J.W.; Joshi, R.S.; Beniwal, A.; Spatz, J.; Gill, S.; Chang, A.C.; Choudhary, N.; Nguyen, A.T.; Sudhir, S.; et al. Single-cell RNA sequencing and spatial transcriptomics reveal cancer-associated fibroblasts in glioblastoma with protumoral effects. J. Clin. Investig. 2023, 133, e147087. [Google Scholar] [CrossRef]
- Zheng, P.; Ren, D.; Cong, Y.; Xiaoxue, Z.; Zhang, Y. Single-Cell Sequencing Reveals Necroptosis-Related Prognostic Genes of Glioblastoma. Oxid. Med. Cell Longev. 2023, 2023, 2926655. [Google Scholar] [CrossRef]
- LeBlanc, V.G.; Trinh, D.L.; Aslanpour, S.; Hughes, M.; Livingstone, D.; Jin, D.; Ahn, B.Y.; Blough, M.D.; Cairncross, J.G.; Chan, J.A.; et al. Single-cell landscapes of primary glioblastomas and matched explants and cell lines show variable retention of inter- and intratumor heterogeneity. Cancer Cell 2022, 40, 379–392.e9. [Google Scholar] [CrossRef]
- Yeo, A.T.; Rawal, S.; Delcuze, B.; Christofides, A.; Atayde, A.; Strauss, L.; Balaj, L.; Rogers, V.A.; Uhlmann, E.J.; Varma, H.; et al. Single-cell RNA sequencing reveals evolution of immune landscape during glioblastoma progression. Nat. Immunol. 2022, 23, 971–984. [Google Scholar] [CrossRef] [PubMed]
- Yesudhas, D.; Dharshini, S.A.P.; Taguchi, Y.H.; Gromiha, M.M. Tumor Heterogeneity and Molecular Characteristics of Glioblastoma Revealed by Single-Cell RNA-Seq Data Analysis. Genes 2022, 13, 428. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Zhang, Y.; Li, G.; Li, Y.; Xie, H.; Chen, X. New insights for precision treatment of glioblastoma from analysis of single-cell lncRNA expression. J. Cancer Res. Clin. Oncol. 2021, 147, 1881–1895. [Google Scholar] [CrossRef]
- Chen, A.X.; Gartrell, R.D.; Zhao, J.; Upadhyayula, P.S.; Zhao, W.; Yuan, J.; Minns, H.E.; Dovas, A.; Bruce, J.N.; Lasorella, A.; et al. Single-cell characterization of macrophages in glioblastoma reveals MARCO as a mesenchymal pro-tumor marker. Genome Med. 2021, 13, 88. [Google Scholar] [CrossRef]
- Xie, Y.; He, L.; Lugano, R.; Zhang, Y.; Cao, H.; He, Q.; Chao, M.; Liu, B.; Cao, Q.; Wang, J.; et al. Key molecular alterations in endothelial cells in human glioblastoma uncovered through single-cell RNA sequencing. JCI Insight 2021, 6, e150861. [Google Scholar] [CrossRef]
- Mathewson, N.D.; Ashenberg, O.; Tirosh, I.; Gritsch, S.; Perez, E.M.; Marx, S.; Jerby-Arnon, L.; Chanoch-Myers, R.; Hara, T.; Richman, A.R.; et al. Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell 2021, 184, 1281–1298.e26. [Google Scholar] [CrossRef]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406. [Google Scholar] [CrossRef]
- Liu, H.; Yang, Q.; Xiong, Y.; Xiong, Z.; Li, X. Improved Prognostic Prediction of Glioblastoma using a PAS Detected from Single-cell RNA-seq. J. Cancer 2020, 11, 3751–3761. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef] [PubMed]
- Little, S.E.; Popov, S.; Jury, A.; Bax, D.A.; Doey, L.; Al-Sarraj, S.; Jurgensmeier, J.M.; Jones, C. Receptor tyrosine kinase genes amplified in glioblastoma exhibit a mutual exclusivity in variable proportions reflective of individual tumor heterogeneity. Cancer Res. 2012, 72, 1614–1620. [Google Scholar] [CrossRef]
- Lai, W.; Li, D.; Kuang, J.; Deng, L.; Lu, Q. Integrated analysis of single-cell RNA-seq dataset and bulk RNA-seq dataset constructs a prognostic model for predicting survival in human glioblastoma. Brain Behav. 2022, 12, e2575. [Google Scholar] [CrossRef]
- Yu, K.; Hu, Y.; Wu, F.; Guo, Q.; Qian, Z.; Hu, W.; Chen, J.; Wang, K.; Fan, X.; Wu, X.; et al. Surveying brain tumor heterogeneity by single-cell RNA-sequencing of multi-sector biopsies. Natl. Sci. Rev. 2020, 7, 1306–1318. [Google Scholar] [CrossRef]
- Lemee, J.M.; Clavreul, A.; Menei, P. Intratumoral heterogeneity in glioblastoma: Don’t forget the peritumoral brain zone. Neuro Oncol. 2015, 17, 1322–1332. [Google Scholar] [CrossRef]
- Lee, J.K.; Wang, J.; Sa, J.K.; Ladewig, E.; Lee, H.O.; Lee, I.H.; Kang, H.J.; Rosenbloom, D.S.; Camara, P.G.; Liu, Z.; et al. Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat. Genet. 2017, 49, 594–599. [Google Scholar] [CrossRef]
- Pine, A.R.; Cirigliano, S.M.; Nicholson, J.G.; Hu, Y.; Linkous, A.; Miyaguchi, K.; Edwards, L.; Singhania, R.; Schwartz, T.H.; Ramakrishna, R.; et al. Tumor Microenvironment Is Critical for the Maintenance of Cellular States Found in Primary Glioblastomas. Cancer Discov. 2020, 10, 964–979. [Google Scholar] [CrossRef]
- Sullivan, J.P.; Nahed, B.V.; Madden, M.W.; Oliveira, S.M.; Springer, S.; Bhere, D.; Chi, A.S.; Wakimoto, H.; Rothenberg, S.M.; Sequist, L.V.; et al. Brain tumor cells in circulation are enriched for mesenchymal gene expression. Cancer Discov. 2014, 4, 1299–1309. [Google Scholar] [CrossRef]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lonnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.E.; Westermann, A.J.; Gorski, S.A.; Vogel, J. Single-cell RNA-seq: Advances and future challenges. Nucleic Acids Res. 2014, 42, 8845–8860. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wu, W.; Zhang, J.; Zhao, Z.; Li, L.; Zhu, M.; Wu, M.; Wu, F.; Zhou, F.; Du, Y.; et al. Natural Coevolution of Tumor and Immunoenvironment in Glioblastoma. Cancer Discov. 2022, 12, 2820–2837. [Google Scholar] [CrossRef]
- Chaicharoenaudomrung, N.; Kunhorm, P.; Noisa, P. Three-dimensional cell culture systems as an in vitro platform for cancer and stem cell modeling. World J. Stem Cells 2019, 11, 1065–1083. [Google Scholar] [CrossRef]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef]
- Pombo Antunes, A.R.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef]
- Sharma, P.; Aaroe, A.; Liang, J.; Puduvalli, V.K. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neurooncol. Adv. 2023, 5, vdad009. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef]
- Eisenbarth, D.; Wang, Y.A. Glioblastoma heterogeneity at single cell resolution. Oncogene 2023, 42, 2155–2165. [Google Scholar] [CrossRef]
- Abdelfattah, N.; Kumar, P.; Wang, C.; Leu, J.S.; Flynn, W.F.; Gao, R.; Baskin, D.S.; Pichumani, K.; Ijare, O.B.; Wood, S.L.; et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat. Commun. 2022, 13, 767. [Google Scholar] [CrossRef] [PubMed]
- De Kleer, I.; Willems, F.; Lambrecht, B.; Goriely, S. Ontogeny of myeloid cells. Front. Immunol. 2014, 5, 423. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Lo, C.W.S.; Ma, W.; Chu, A.T.W.; Tong, A.H.Y.; Chung, B.H.Y. Revealing the role of SPP1+ macrophages in glioma prognosis and therapeutic targeting by investigating tumor-associated macrophage landscape in grade 2 and 3 gliomas. Cell Biosci. 2024, 14, 37. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.C.; Gutierrez-Vazquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 729–740. [Google Scholar] [CrossRef]
- Mi, Y.; Guo, N.; Luan, J.; Cheng, J.; Hu, Z.; Jiang, P.; Jin, W.; Gao, X. The Emerging Role of Myeloid-Derived Suppressor Cells in the Glioma Immune Suppressive Microenvironment. Front. Immunol. 2020, 11, 737. [Google Scholar] [CrossRef]
- Sawant, D.V.; Yano, H.; Chikina, M.; Zhang, Q.; Liao, M.; Liu, C.; Callahan, D.J.; Sun, Z.; Sun, T.; Tabib, T.; et al. Adaptive plasticity of IL-10+ and IL-35+ T(reg) cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 2019, 20, 724–735. [Google Scholar] [CrossRef]
- Ravi, V.M.; Neidert, N.; Will, P.; Joseph, K.; Maier, J.P.; Kuckelhaus, J.; Vollmer, L.; Goeldner, J.M.; Behringer, S.P.; Scherer, F.; et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat. Commun. 2022, 13, 925. [Google Scholar] [CrossRef]
- Han, S.; Feng, S.; Ren, M.; Ma, E.; Wang, X.; Xu, L.; Xu, M. Glioma cell-derived placental growth factor induces regulatory B cells. Int. J. Biochem. Cell Biol. 2014, 57, 63–68. [Google Scholar] [CrossRef]
- Slyper, M.; Porter, C.B.M.; Ashenberg, O.; Waldman, J.; Drokhlyansky, E.; Wakiro, I.; Smillie, C.; Smith-Rosario, G.; Wu, J.; Dionne, D.; et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med. 2020, 26, 792–802. [Google Scholar] [CrossRef]
- Kinker, G.S.; Greenwald, A.C.; Tal, R.; Orlova, Z.; Cuoco, M.S.; McFarland, J.M.; Warren, A.; Rodman, C.; Roth, J.A.; Bender, S.A.; et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat. Genet. 2020, 52, 1208–1218. [Google Scholar] [CrossRef]
- Kulkarni, A.; Anderson, A.G.; Merullo, D.P.; Konopka, G. Beyond bulk: A review of single cell transcriptomics methodologies and applications. Curr. Opin. Biotechnol. 2019, 58, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Hernandez Martinez, A.; Madurga, R.; Garcia-Romero, N.; Ayuso-Sacido, A. Unravelling glioblastoma heterogeneity by means of single-cell RNA sequencing. Cancer Lett. 2022, 527, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Filbin, M.G.; Tirosh, I.; Hovestadt, V.; Shaw, M.L.; Escalante, L.E.; Mathewson, N.D.; Neftel, C.; Frank, N.; Pelton, K.; Hebert, C.M.; et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 2018, 360, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef]
- Annovazzi, L.; Mellai, M.; Caldera, V.; Valente, G.; Schiffer, D. SOX2 expression and amplification in gliomas and glioma cell lines. Cancer Genom. Proteom. 2011, 8, 139–147. [Google Scholar]
- Yuan, J.; Levitin, H.M.; Frattini, V.; Bush, E.C.; Boyett, D.M.; Samanamud, J.; Ceccarelli, M.; Dovas, A.; Zanazzi, G.; Canoll, P.; et al. Single-cell transcriptome analysis of lineage diversity in high-grade glioma. Genome Med. 2018, 10, 57. [Google Scholar] [CrossRef]
- Kanamori, M.; Morishita, Y.; Shimoda, Y.; Yamamori, E.; Sato, S.; Osada, Y.; Osawa, S.I.; Shibahara, I.; Saito, R.; Sonoda, Y.; et al. Distant recurrence in the cerebellar dentate nucleus through the dentato-rubro-thalamo-cortical pathway in supratentorial glioma cases. Acta Neurochir. 2024, 166, 83. [Google Scholar] [CrossRef]
- Seiz, M.; Nolte, I.; Pechlivanis, I.; Freyschlag, C.F.; Schmieder, K.; Vajkoczy, P.; Tuettenberg, J. Far-distant metastases along the CSF pathway of glioblastoma multiforme during continuous low-dose chemotherapy with temozolomide and celecoxib. Neurosurg. Rev. 2010, 33, 375–381; discussion 381. [Google Scholar] [CrossRef]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef]
- Kim, Y.; Varn, F.S.; Park, S.H.; Yoon, B.W.; Park, H.R.; Lee, C.; Verhaak, R.G.W.; Paek, S.H. Perspective of mesenchymal transformation in glioblastoma. Acta Neuropathol. Commun. 2021, 9, 50. [Google Scholar] [CrossRef]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jung, J.; Babikir, H.; Shamardani, K.; Jain, S.; Feng, X.; Gupta, N.; Rosi, S.; Chang, S.; Raleigh, D.; et al. A single-cell atlas of glioblastoma evolution under therapy reveals cell-intrinsic and cell-extrinsic therapeutic targets. Nat. Cancer 2022, 3, 1534–1552. [Google Scholar] [CrossRef] [PubMed]
- Garofano, L.; Migliozzi, S.; Oh, Y.T.; D’Angelo, F.; Najac, R.D.; Ko, A.; Frangaj, B.; Caruso, F.P.; Yu, K.; Yuan, J.; et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat. Cancer 2021, 2, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Do, H.T.T.; Lee, C.H.; Cho, J. Chemokines and their Receptors: Multifaceted Roles in Cancer Progression and Potential Value as Cancer Prognostic Markers. Cancers 2020, 12, 287. [Google Scholar] [CrossRef]
- Guo, P.; Hu, B.; Gu, W.; Xu, L.; Wang, D.; Huang, H.J.; Cavenee, W.K.; Cheng, S.Y. Platelet-derived growth factor-B enhances glioma angiogenesis by stimulating vascular endothelial growth factor expression in tumor endothelia and by promoting pericyte recruitment. Am. J. Pathol. 2003, 162, 1083–1093. [Google Scholar] [CrossRef]
- Fischer, I.; Gagner, J.P.; Law, M.; Newcomb, E.W.; Zagzag, D. Angiogenesis in gliomas: Biology and molecular pathophysiology. Brain Pathol. 2005, 15, 297–310. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, M.; Gan, H.; Wang, H.; Lee, J.H.; Fang, D.; Kitange, G.J.; He, L.; Hu, Z.; Parney, I.F.; et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun. 2018, 9, 2949. [Google Scholar] [CrossRef]
- Hide, T.; Komohara, Y. Oligodendrocyte Progenitor Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1234, 107–122. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, Q.; Bai, J.; Zhao, Z.; Zhang, J. Transcriptome analysis revealed CENPF associated with glioma prognosis. Math. Biosci. Eng. 2021, 18, 2077–2096. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Mendez, G.; Ozpinar, A.; Raskin, J.; Gultekin, S.H.; Ross, D.A. Case comparison and literature review of glioblastoma: A tale of two tumors. Surg. Neurol. Int. 2014, 5, 121. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Romero, N.; Palacin-Aliana, I.; Madurga, R.; Carrion-Navarro, J.; Esteban-Rubio, S.; Jimenez, B.; Collazo, A.; Perez-Rodriguez, F.; Ortiz de Mendivil, A.; Fernandez-Carballal, C.; et al. Bevacizumab dose adjustment to improve clinical outcomes of glioblastoma. BMC Med. 2020, 18, 142. [Google Scholar] [CrossRef]
- Miao, D.; Margolis, C.A.; Vokes, N.I.; Liu, D.; Taylor-Weiner, A.; Wankowicz, S.M.; Adeegbe, D.; Keliher, D.; Schilling, B.; Tracy, A.; et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat. Genet. 2018, 50, 1271–1281. [Google Scholar] [CrossRef]
- Shembrey, C.; Huntington, N.D.; Hollande, F. Impact of Tumor and Immunological Heterogeneity on the Anti-Cancer Immune Response. Cancers 2019, 11, 1217. [Google Scholar] [CrossRef]
- Flemming, A. Tumour heterogeneity determines immune response. Nat. Rev. Immunol. 2019, 19, 662–663. [Google Scholar] [CrossRef]
- Petrecca, K.; Guiot, M.C.; Panet-Raymond, V.; Souhami, L. Failure pattern following complete resection plus radiotherapy and temozolomide is at the resection margin in patients with glioblastoma. J. Neurooncol. 2013, 111, 19–23. [Google Scholar] [CrossRef]
- Teng, J.; da Hora, C.C.; Kantar, R.S.; Nakano, I.; Wakimoto, H.; Batchelor, T.T.; Chiocca, E.A.; Badr, C.E.; Tannous, B.A. Dissecting inherent intratumor heterogeneity in patient-derived glioblastoma culture models. Neuro Oncol. 2017, 19, 820–832. [Google Scholar] [CrossRef]
- Ordonez-Rubiano, E.G.; Combita, A.; Baldoncini, M.; Payan-Gomez, C.; Gomez-Amarillo, D.F.; Hakim, F.; Camargo, J.; Zorro-Sepulveda, V.; Luzzi, S.; Zorro, O.; et al. Cellular Senescence in Diffuse Gliomas: From Physiopathology to Possible Treatments. World Neurosurg. 2024, 191, 138–148. [Google Scholar] [CrossRef]
- Wang, Q.; Tan, Y.; Fang, C.; Zhou, J.; Wang, Y.; Zhao, K.; Jin, W.; Wu, Y.; Liu, X.; Liu, X.; et al. Single-cell RNA-seq reveals RAD51AP1 as a potent mediator of EGFRvIII in human glioblastomas. Aging 2019, 11, 7707–7722. [Google Scholar] [CrossRef] [PubMed]
- Saurty-Seerunghen, M.S.; Bellenger, L.; El-Habr, E.A.; Delaunay, V.; Garnier, D.; Chneiweiss, H.; Antoniewski, C.; Morvan-Dubois, G.; Junier, M.P. Capture at the single cell level of metabolic modules distinguishing aggressive and indolent glioblastoma cells. Acta Neuropathol. Commun. 2019, 7, 155. [Google Scholar] [CrossRef] [PubMed]
- Di, Z.; Yang, B.; Li, M.; Wu, Y.; Ji, H. Batch effects correction in scRNA-seq based on biological-noise decoupling autoencoder and central-cross loss. Comput. Biol. Chem. 2024, 113, 108261. [Google Scholar] [CrossRef] [PubMed]
- Monnier, L.; Cournede, P.H. A novel batch-effect correction method for scRNA-seq data based on Adversarial Information Factorization. PLoS Comput. Biol. 2024, 20, e1011880. [Google Scholar] [CrossRef]
- Oh, J.; Chang, C.; Long, Q. Accounting for technical noise in Bayesian graphical models of single-cell RNA-sequencing data. Biostatistics 2022, 24, 161–176. [Google Scholar] [CrossRef]
- Hastings, J.; Lee, D.; O’Connell, M.J. Batch-effect correction in single-cell RNA sequencing data using JIVE. Bioinform. Adv. 2024, 4, vbae134. [Google Scholar] [CrossRef]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin. Transl. Med. 2022, 12, e694. [Google Scholar] [CrossRef]
- Yekula, A.; Tracz, J.; Rincon-Torroella, J.; Azad, T.; Bettegowda, C. Single-Cell RNA Sequencing of Cerebrospinal Fluid as an Advanced Form of Liquid Biopsy for Neurological Disorders. Brain Sci. 2022, 12, 812. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Author and Year | Cells Identified | Most Important Genes Identified | Conclusions |
|---|---|---|---|
| Wu et al. [7] 2023 | 10,144 cells from primary GBM and 19,982 from recurrent GBM lesions. Tumor cells. Endothelial cells. Immune cells. | TOP2A, MKI67, UBE2C, CENPF, PBK, VEGF. | Identified three separate cell types, from GBM lesions, from which 22 clusters were retrieved. Identified malignant cells in single-cell analysis using copy number variations. Identified high expression of proliferation-related genes in cell clusters. Detected VEGFA overexpression in almost all clusters. |
| Jain et al. [8] 2023 | Serial trypsinization of 4385 GBM cells. Cancer-associated fibroblasts, epithelial cells, endothelial cells, and pericytes, immune cells. | ACTA2. COL1A1. TNC, S100A4. PDPN. PDGFRB. | Identified cancer-associated fibroblasts in GBM samples, and identified proximity to mesenchymal glioblastoma stem cells, endothelial cells, and M2 macrophages. |
| Zheng et al. [9] 2023 | Fibroblasts. Chondrocytes Astrocytes. T_cells. Tissue_stem_cells. Monocyte. | 571 genes related to necroptosis. ADORA2A. KDR. LAG3. EEF1B2. NDUFB2. RPL13. PTEN. EGFR. TTN. | A risk model was constructed using a Cox regression model with least absolute shrinkage and selection operator analysis, which included ten necroptosis-related genes. |
| LeBlanc et al. [10] 2022 | >8000 single-cell genome, and >75,000 single-cell transcriptome profiles from 10 primary tumors and 2 recurrent tumors. | NRCAM. NCAM2. SHISA9 ACTA2. PDGFRB. VWF. MOG. MAG. ACTA2. PDGFRB. | Patient-derived explants (PDEs) can serve as a more accurate model for studying the complex heterogeneity of GBMs. |
| Yeo et al. [11] 2022 | de novo mouse-made cells: 27,633 CD45- and 36,304 CD45+ cells. Dendritic cells (i.e., conventional or plasmacytoid), macrophages, T cells and natural killer cells, microglia, neutrophils, B cells, and mast cells. Distinct populations of EGFR+ cancer cells. | Upregulated pathways INFα/β/γ, cell migration, angiogenesis, oligodendrocyte differentiation, myelination and cell adhesion, and overexpression. Csfr3, Ccr1, Cxcr2, and Cxcr4 highly expressed in PMN-MDSCs. | Demonstrated relevant changes in the innate immune cell composition of the GBM microenvironment, with accumulation of myeloid-derived suppressor cells that promote immunosuppression. |
| Yesudhas et al. [12] 2022 | 3389 cells from four primary GBMs. | 94 differentially expressed genes (DEGs) between tumor and periphery cells. CX3CR1, GAPDH, FN1, PDGFRA, HTRA1, ANXA2 THBS1, GFAP, PTN, TNC, VIM. | Insights into the heterogeneity of GBM and identifies novel disease-specific biomarkers, presenting potential avenues for the development of targeted therapies in GBM management. |
| Meng et al. [13] 2021 | 3589 cells from 4 cases. | DLL3. NEFL. NKX2-2. GABRA1. SOX2. SYT1. OLIG2. SLC12A5. FGFR3. ILR4. PDGFA. TRADD. EGFR. RELB. AKT2. CHI3L1(YKL40). NES. MET. | Reveals critical insights into intratumoral heterogeneity. This approach holds promise for improving the oncological management and outcomes of GBMs. |
| Chen et al. [14] 2021 | 17,132 cells from 50 cases. CD14 macrophages, CD3 T cells. SOX2 neuroglial cells. | 499 genes in total. CSF1. CSF2. HGF. MCP-1. SDF-1. MFGE8. PDC001. PW039-705. PW035-710All. PJ052. PJ053. | MARCO macrophages found in GBMs correlate with worse prognosis. MARCO expression changes with anti-PD1 therapy. This indicates its potential as a biomarker for treatment response in GBM. |
| Xie et al. [15] 2021 | Endothelial cells. Macrophages. Microglia. Neutrophils. T cells. B cells. Neuroglial cells. Vascular mural cells. | KLF2. TIMP3. SLC2A1. SLCO1A2. ABCG2. ABCB1. SLCO1A2. NET1. ATP10A. MYO1B. SPARC. ITGA5. PGF. NOTCH4. CD93. FABP1A. GNG11. SELE. VACM1. IL1B. | BBB transporters, including SLC2A1, ABCG2, ABCB1, SLCO1A2, and ATP10A, were elevated in endothelial cells, which impacts drug penetration and efficacy in brain tissue. |
| Mathewson et al. [16] 2021 | 8252 cells from 31 cases. T cells: CD8 T cells—CD4 conventional T cells—CD4 regulatory T cells—cycling T cells. | PRF1. GZMB. GZMA, GZMH. CLEC2D. NKG7. GNLY. KLRD1. FGFBP2. FCGR3A. S1PR5. KLRC1. KLRC3. KLRB1. KLRC2. | CLEC2D–CD161 pathway inhibition can enhance anti-tumor immune microenvironmental. NK-like receptor expression in GBM-infiltrating T cells implies that targeting these receptors could strengthen T-cell-based therapies. |
| Couturier et al. [17] 2020 | 53,586 glioblastoma cells. Glioblastoma stem cells. | TOP2A. FOXM1. USP1. APOD, OLIG2. SOX11. S100A10. HLA-4. APOE. HSPA1B. | Discovered a conserved trilineage hierarchy in glioblastoma centered around glial progenitor-like cells. |
| Liu et al. [18] 2020 | 3589 cells from 154 GBM patients in the TCGAGBM dataset and 155 GBM patients in the GSE16011 dataset. | FERMT1. COL22A1. LOXL1. PCDHB3. TCAF2. HOXB2. HOXD11, PTPRN. TSHZ2. | Prognostic model that incorporated factors such as radiotherapy status, and age to predict survival probabilities, suggesting that these genes could serve as potential prognostic biomarkers. |
| Neftel et al. [19] 2019 | 7930 cells from 28 cases. Macrophages. Oligodendrocytes. T cells. Astrocytes | 5730 genes in total. HILPDA. DDIT3. ENO2 and LDHA. MGH125. MGH102. EGFR. PDGFRA. CDK4. | High-level amplifications of EGFR, PDGFRA, and CDK4 influence cellular states within the GBM microenvironment. PDGFRA and CDK4 amplifications correlate with the expansion of NPC and OPC, respectively. |
| Darmanis et al. [20] 2017 | 3589 cells from 4 cases. Tumor cells. Vascular cells. Oligodendrocytes. OPCs. Neurons. Astrocytes. | MBP. OPALIN. GPR17. L1CAM. ALDH1L1. WIF1. NTSR2. PECAM-1. NFIB. SOX9. Higher expression of hypoxia and adhesion-related genes in the tumor core. | Identified infiltrating neoplastic cells in peripheral regions of the core lesions, representing intratumor heterogeneity. Identified consistent gene signature between patients. Identified myeloid cell populations in the tumor core and surrounding peritumoral space. |
| Patel et al. [21] 2013 | 430 cells from 5 cases. NPC. Neurons. Mesenchymal cells. | EGFR. PDGFRA. PDFGA. FGFR1. FGF1. NOTCH2. JAG1. | Identified intratumor heterogeneity by identifying different GBM subtypes within the tumors. High tumor heterogeneity was associated with poor prognosis. |
| Müller et al. [22] 2017 | 672 cells identified. Tumor-associated macrophages (TAMs) from 5 GBMs. | Upregulated genes in blood-derived TAMs include those of immunosuppressive cytokines (specific genes not mentioned). | Blood-derived TAMs infiltrate pretreatment GBMs and exhibit immunosuppressive characteristics, presenting a barrier to immunotherapy. |
| Little et al. [23] 2012 | 41,997 cells were counted across 190 distinct loci. | EGFR (upregulated), PDGFRA (upregulated). | Intratumoral heterogeneity in glioblastoma complicates treatment strategies, as different cell populations with distinct gene amplifications may contribute variably to disease progression and response to therapies. |
| Lai et al. [24] 2022 | 2305 cancer cells from tumor cores. | LITAF (Downregulated), MTHFD2 (Upregulated), NRXN3 (Upregulated), OSMR (Upregulated), RUFY2. | Novel prognostic model for predicting survival in GBM patients by integrating scRNA-seq and bulk RNA-seq datasets. |
| Yu et al. [25] 2020 | 6148 cells identified (from 7928 single-cell transcriptomes). | EGFR (Upregulated) cells, PTPRZ1 (Upregulated), SOX2 (Upregulated), MKI67 (Marker for proliferation), HYDIN, FOXJ1. | scRNA-seq can uncover distinct cellular states and gene expression profiles that are critical for understanding tumor progression and therapeutic resistance in GBM. Emphasis made on the importance of multi-sector biopsies to capture the heterogeneity of gliomas effectively. |
| Lemée et al. [26] 2015 | Not specified. | Genes related to stem cell phenotype: CD133, Sox2, nestin, musashi 1 (upregulated). Invasion-related genes: Galectin-1, Rac1, Rac3, RhoA GTPases, p27, avb3 integrin (upregulated). Cell adhesion-related genes: CDH20, PCDH19 (upregulated). Migration-related genes: SNAI2, NANOG, USP6, DISC1 (upregulated). Immune response: TLR4 (upregulated). Angiogenesis: HEG1, VEGFR2 (upregulated). | Emphasis made on the importance of understanding the peritumoral brain zone (PBZ) in GBM, highlighting that it contains tumor and stromal cells that promote growth and invasion. |
| Lee et al. [27] 2017 | 305 single cells from 7 samples of 3 patients. | EGFR, PIK3CA. | Different single cells exhibited various EGFR alterations, indicating late events in tumor evolution. The presence of transcriptional heterogeneity suggests that 5-ALA (-) tumors can still harbor aggressive tumor markers despite being perceived as being less aggressive. |
| Pine et al. [28] 2020 | 62,885 cells identified. Neural progenitor-like cells (NPC-like), Oligodendrocyte progenitor-like cells (OPC-like), Astrocyte-like cells (AC-like), Mesenchymal-like cells (MES-like). | SOX4 (upregulated), BCAN (upregulated and associated with invasiveness), DLL3 (upregulated), KPNA2 (upregulated and promotes metabolic reprogramming). | Compared scRNA-seq across four patient-derived glioblastoma stem cell models, including glioma spheres, brain organoids, glioblastoma cerebral organoids, and patient-derived xenografts. Successfully recapitulated cellular states commonly found in primary tumors. |
| Sullivan et al. [29] 2014 | Not specified. | SERPINE1, TGFB1, TGFBR2, and VIM (all upregulated). ASCL1, GFAP, NCAM1, and SOX9 (all downregulated), TWIST1, and NF-kB. EGFR amplification. | Circulating tumor cells exhibit higher mesenchymal and lower neural differentiation, contributing to invasiveness and possibly rare metastases. |
| Jacob et al. [30] 2020 | scRNA-seq data from organoids derived from 53 patient cases and established 70 glioblastoma organoid (GBO) samples. | EGFR (including variant III—EGFRvIII), SOX2, and NESTIN. | Organoids retained transcriptomic signatures, cell-type diversity, and molecular properties of parental tumors. |
| Potential Diagnostic |
|---|
|
| Therapeutic |
|
| Prognostic role of scRNA-seq in GBM |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ordóñez-Rubiano, E.G.; Rincón-Arias, N.; Shelton, W.J.; Salazar, A.F.; Sierra, M.A.; Bertani, R.; Gómez-Amarillo, D.F.; Hakim, F.; Baldoncini, M.; Payán-Gómez, C.; et al. Current Applications of Single-Cell RNA Sequencing in Glioblastoma: A Scoping Review. Brain Sci. 2025, 15, 309. https://doi.org/10.3390/brainsci15030309
Ordóñez-Rubiano EG, Rincón-Arias N, Shelton WJ, Salazar AF, Sierra MA, Bertani R, Gómez-Amarillo DF, Hakim F, Baldoncini M, Payán-Gómez C, et al. Current Applications of Single-Cell RNA Sequencing in Glioblastoma: A Scoping Review. Brain Sciences. 2025; 15(3):309. https://doi.org/10.3390/brainsci15030309
Chicago/Turabian StyleOrdóñez-Rubiano, Edgar G., Nicolás Rincón-Arias, William J. Shelton, Andres F. Salazar, María Alejandra Sierra, Raphael Bertani, Diego F. Gómez-Amarillo, Fernando Hakim, Matías Baldoncini, César Payán-Gómez, and et al. 2025. "Current Applications of Single-Cell RNA Sequencing in Glioblastoma: A Scoping Review" Brain Sciences 15, no. 3: 309. https://doi.org/10.3390/brainsci15030309
APA StyleOrdóñez-Rubiano, E. G., Rincón-Arias, N., Shelton, W. J., Salazar, A. F., Sierra, M. A., Bertani, R., Gómez-Amarillo, D. F., Hakim, F., Baldoncini, M., Payán-Gómez, C., Cómbita, A. L., Ordonez-Rubiano, S. C., & Parra-Medina, R. (2025). Current Applications of Single-Cell RNA Sequencing in Glioblastoma: A Scoping Review. Brain Sciences, 15(3), 309. https://doi.org/10.3390/brainsci15030309