
Modeling ALS with Patient-Derived iPSCs: Recent Advances and Future Potentials


Abstract
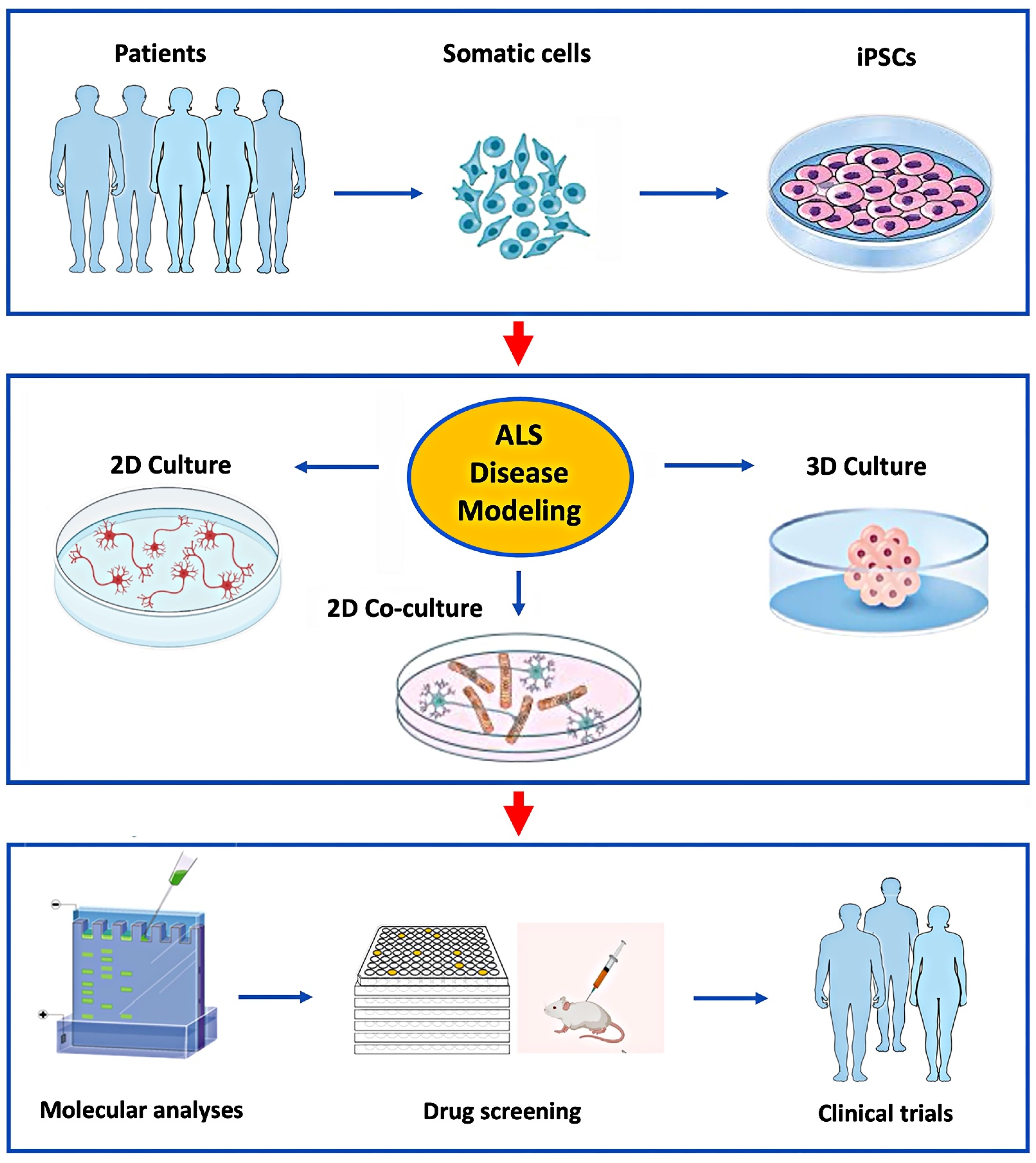
1. Introduction
2. Human iPSC-Derived Motor Neuron Culturing
3. Co-Culturing hiPSC-Derived Motor Neurons with Neuroglia
3.1. Astrocyte Co-Cultures
3.2. Microglia Co-Cultures
4. HiPSC-Derived Organoids
4.1. Brain Organoids
4.2. Spinal Cord Organoids
4.3. Neuromuscular Organoids
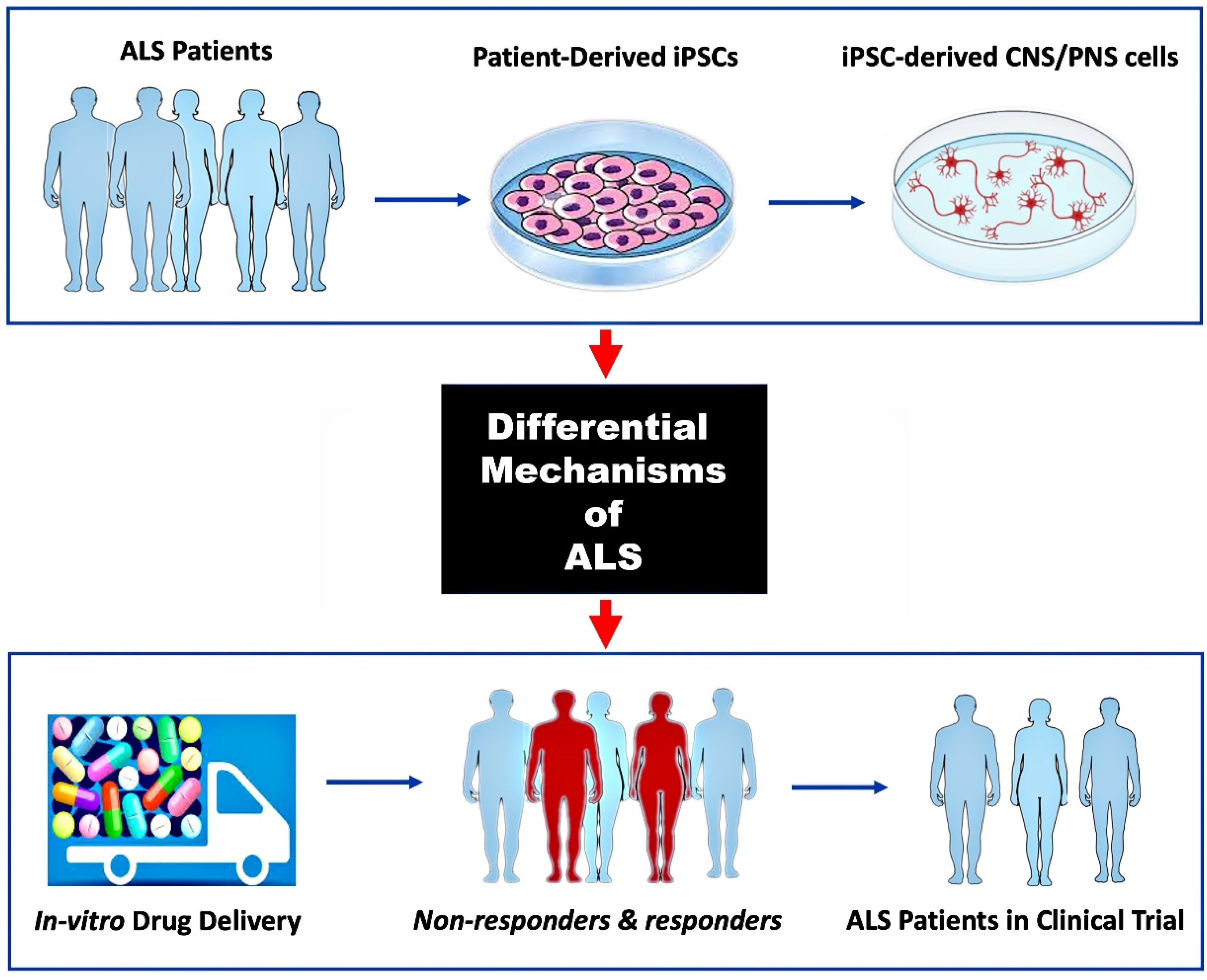
5. Discussion
6. Conclusions and Future Direction
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D | Two dimensional |
| 3D | Three dimensional |
| ALI-COs | Air–liquid interface–cerebral organoids |
| ALS | Amyotrophic lateral sclerosis |
| ALS-FTD | Amyotrophic lateral sclerosis with frontotemporal dementia |
| CNS | Central nervous system |
| C RISPR-Cas9 | Clustered regularly interspersed short palindromic repeats |
| DPR | Dipeptide protein repeat |
| ESCs | Embryonic stem cells |
| fALS | Familial amyotrophic lateral sclerosis |
| FUS | Fused in sarcoma |
| FTD | Frontotemporal dementia |
| h iPSCs | Human induced pluripotent stem cells |
| HRE | Hexanucleotide repeat expansion |
| iPSCs | Induced pluripotent stem cells |
| LMC | Lateral motor columns |
| MMC | Median motor columns |
| MNs | Motor neurons |
| NMOs | Neuromuscular organoids |
| NPCs | Neuronal progenitor cells |
| PNS | Peripheral nervous system |
| phMNs | Phrenic motor neurons |
| RAN | Repeat-associated non-AUG |
| ROPI | Ropinirole hydrochloride |
| sALS | Sporadic amyotrophic lateral sclerosis |
References
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Lomen-Hoerth, C.; Anderson, T.; Miller, B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 2002, 59, 1077–1079. [Google Scholar] [CrossRef] [PubMed]
- Vasta, R.; Chia, R.; Traynor, B.J.; Chiò, A. Unraveling the complex interplay between genes, environment, and climate in ALS. EBioMedicine 2022, 75, 103795. [Google Scholar] [CrossRef] [PubMed]
- Pham, J.; Keon, M.; Brennan, S.; Saksena, N. Connecting RNA-Modifying Similarities of TDP-43, FUS, and SOD1 with MicroRNA Dysregulation Amidst A Renewed Network Perspective of Amyotrophic Lateral Sclerosis Proteinopathy. Int. J. Mol. Sci. 2020, 21, 3464. [Google Scholar] [CrossRef]
- Burrell, J.R.; Kiernan, M.C.; Vucic, S.; Hodges, J.R. Motor Neuron dysfunction in frontotemporal dementia. Brain 2011, 134, 2582–2594. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Liu, G.; David, B.T.; Trawczynski, M.; Fessler, R.G. Advances in Pluripotent Stem Cells: History, Mechanisms, Technologies, and Applications. Stem Cell Rev. Rep. 2020, 16, 3–32. [Google Scholar] [CrossRef]
- González, F.; Boué, S.; Belmonte, J.C.I. Methods for making induced pluripotent stem cells: Reprogramming à la carte. Nat. Rev. Genet. 2011, 12, 231–242. [Google Scholar] [CrossRef]
- Ragagnin, A.M.G.; Shadfar, S.; Vidal, M.; Jamali, S.; Atkin, J.D. Motor Neuron Susceptibility in ALS/FTD. Front. Neurosci. 2019, 13, 532. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Maragakis, N.J. Mini-Review: Induced Pluripotent Stem Cells and the Search for New Cell-Specific ALS Thera-peutic Targets. Neurosci. Lett. 2021. [CrossRef]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Hadano, S.; Aoki, M.; Saya, H.; Sobue, G.; et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, T.; Zerbo, R.A.; Balbi, M.; Torazza, C.; Frumento, G.; Fedele, E.; Bonanno, G.; Milanese, M. Nearly 30 Years of Animal Models to Study Amyotrophic Lateral Sclerosis: A Historical Overview and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 12236. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.M.C.; Greensmith, L.; Malaspina, A.; Fratta, P.; Hanna, M.G.; Schiavo, G.; Isaacs, A.M.; Orrell, R.W.; Cunningham, T.J.; Arozena, A.A. Opinion: More Mouse Models and More Translation Needed for ALS. Mol. Neurodegener. 2023, 14, 30. [Google Scholar] [CrossRef] [PubMed]
- Centeno, E.G.Z.; Cimarosti, H.; Bithell, A. 2D versus 3D Human Induced Pluripotent Stem Cell-Derived Cultures for Neurodegenerative Disease Modelling. Mol. Neurodegener. 2018, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Huo, Z.; Chen, Y.; Zhao, Z.; Meng, F.; Wang, X.; Liu, S.; Zhang, H.; Zhou, F.; Liu, J.; et al. Induced Pluripotent Stem Cells and Their Applications in Amyotrophic Lateral Sclerosis. Cells 2023, 12, 971. [Google Scholar] [CrossRef]
- Giacomelli, E.; Vahsen, B.F.; Calder, E.L.; Xu, Y.; Scaber, J.; Gray, E.; Dafinca, R.; Talbot, K.; Studer, L. Human stem cell models of neurodegeneration: From basic science of amyotrophic lateral sclerosis to clinical translation. Cell Stem Cell 2022, 29, 11–35. [Google Scholar] [CrossRef]
- Guo, W.; Fumagalli, L.; Prior, R.; Van Den Bosch, L. Current Advances and Limitations in Modeling ALS/FTD in a Dish Using Induced Pluripotent Stem Cells. Front. Neurosci. 2017, 11, 671. [Google Scholar] [CrossRef]
- Van Damme, P.; Robberecht, W.; Van Den Bosch, L. Modelling amyotrophic lateral sclerosis: Progress and possibilities. Dis. Model. Mech. 2017, 10, 537–549. [Google Scholar] [CrossRef]
- Okano, H.; Morimoto, S.; Kato, C.; Nakahara, J.; Takahashi, S. Induced Pluripotent Stem Cells-Based Disease Modeling, Drug Screening, Clinical Trials, and Reverse Translational Research for Amyotrophic Lateral Sclerosis. J. Neurochem. 2023, 167, 603–614. [Google Scholar] [CrossRef]
- Liu, R.; Meng, X.; Yu, X.; Wang, G.; Dong, Z.; Zhou, Z.; Qi, M.; Yu, X.; Ji, T.; Wang, F. From 2D to 3D Co-Culture Systems: A Review of Co-Culture Models to Study the Neural Cells Interaction. Int. J. Mol. Sci. 2022, 23, 13116. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R. A Yeast Model for Understanding ALS: Fast, Cheap, and Easy to Control. PLoS Biol. 2011, 9, e1001053. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Farr, G.W.; Hall, D.H.; Li, F.; Furtak, K.; Dreier, L.; Horwich, A.L. An ALS-Linked Mutant SOD1 Produces a Locomotor Defect Associated with Aggregation and Synaptic Dysfunction When Expressed in Neurons of Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000350. [Google Scholar] [CrossRef] [PubMed]
- Casci, I.; Pandey, U.B. A fruitful endeavor: Modeling ALS in the fruit fly. Brain Res. 2015, 1607, 47–74. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, T.; Lyon, A.N.; Pineda, R.H.; Wang, C.; Janssen, P.M.L.; Canan, B.D.; Burghes, A.H.M.; Beattie, C.E. A genetic model of amyotrophic lateral sclerosis in zebrafish displays phenotypic hallmarks of motoneuron disease. Dis. Model. Mech. 2010, 3, 652–662. [Google Scholar] [CrossRef]
- Stephenson, J.; Amor, S. Modelling amyotrophic lateral sclerosis in mice. Drug Discov. Today Dis. Model. 2017, 25-26, 35–44. [Google Scholar] [CrossRef]
- Morimoto, S.; Takahashi, S.; Ito, D.; Daté, Y.; Okada, K.; Kato, C.; Nakamura, S.; Ozawa, F.; Chyi, C.M.; Nishiyama, A.; et al. Phase 1/2a clinical trial in ALS with ropinirole, a drug candidate identified by iPSC drug discovery. Cell Stem Cell 2023, 30, 766–780.e9. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef]
- Workman, M.J.; Lim, R.G.; Wu, J.; Frank, A.; Ornelas, L.; Panther, L.; Galvez, E.; Perez, D.; Meepe, I.; Lei, S.; et al. Large-scale differentiation of iPSC-derived motor neurons from ALS and control subjects. Neuron 2023, 111, 1191–1204.e5. [Google Scholar] [CrossRef]
- Ogaki, K.; Li, Y.; Atsuta, N.; Tomiyama, H.; Funayama, M.; Watanabe, H.; Nakamura, R.; Yoshino, H.; Yato, S.; Tamura, A.; et al. Analysis of C9orf72 repeat expansion in 563 Japanese patients with amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 2527.e11–2527.e16. [Google Scholar] [CrossRef]
- Wei, Q.; Zhou, Q.; Chen, Y.; Ou, R.; Cao, B.; Xu, Y.; Yang, J.; Shang, H.-F. Analysis of SOD1 mutations in a Chinese population with amyotrophic lateral sclerosis: A case-control study and literature review. Sci. Rep. 2017, 7, srep44606. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.-W.; Chen, H.; Liu, H.; Lu, J.; Qian, K.; Huang, C.-L.; Zhong, X.; Fan, F.; Zhang, S.-C. Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat. Commun. 2015, 6, 6626. [Google Scholar] [CrossRef] [PubMed]
- Velasco, S.; Ibrahim, M.M.; Kakumanu, A.; Garipler, G.; Aydin, B.; Al-Sayegh, M.A.; Hirsekorn, A.; Abdul-Rahman, F.; Satija, R.; Ohler, U.; et al. A Multi-Step Transcriptional and Chromatin State Cascade Underlies Motor Neuron Programming from Embryonic Stem Cells. Cell Stem Cell 2017, 20, 205–217.e8. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-K.; Pfaff, S.L. Synchronization of Neurogenesis and Motor Neuron Specification by Direct Coupling of bHLH and Homeodomain Transcription Factors. Neuron 2003, 38, 731–745. [Google Scholar] [CrossRef]
- Davis-Anderson, K.; Micheva-Viteva, S.; Solomon, E.; Hovde, B.; Cirigliano, E.; Harris, J.; Twary, S.; Iyer, R. CRISPR/Cas9 Directed Reprogramming of iPSC for Accelerated Motor Neuron Differentiation Leads to Dysregulation of Neuronal Fate Patterning and Function. Int. J. Mol. Sci. 2023, 24, 16161. [Google Scholar] [CrossRef]
- Liu, M.L.; Zang, T.; Zhang, C.L. Direct Lineage Reprogramming Reveals Disease-Specific Phenotypes of Motor Neurons from Human ALS Patients. Cell Rep. 2016, 14, 115–128. [Google Scholar] [CrossRef]
- Thiry, L.; Sirois, J.; Durcan, T.M.; Stifani, S. Generation of Human IPSC-Derived Phrenic-like Motor Neurons to Model Respiratory Motor Neuron Degeneration in ALS. Commun. Biol. 2024. [Google Scholar] [CrossRef]
- Stoklund Dittlau, K.; Van Den Bosch, L. Why Should We Care about Astrocytes in a Motor Neuron Disease? Front. Mol. Med. 2023, 3, 1047540. [Google Scholar] [CrossRef]
- Kunze, A.; Lengacher, S.; Dirren, E.; Aebischer, P.; Magistretti, P.J.; Renaud, P. Astrocyte-Neuron Co-Culture on Microchips Based on the Model of SOD Mutation to Mimic ALS. Integr. Biol. 2013, 5, 964–975. [Google Scholar] [CrossRef]
- Di Giorgio, F.P.; Carrasco, M.A.; Siao, M.C.; Maniatis, T.; Eggan, K. Non-Cell Autonomous Effect of Glia on Motor Neurons in an Embryonic Stem Cell-Based ALS Model. Nat. Neurosci. 2007, 10, 608–614. [Google Scholar] [CrossRef]
- Marchetto, M.C.N.; Muotri, A.R.; Mu, Y.; Smith, A.M.; Cezar, G.G.; Gage, F.H. Non-Cell-Autonomous Effect of Human SOD1G37R Astrocytes on Motor Neurons Derived from Human Embryonic Stem Cells. Cell Stem Cell 2008, 3, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from Familial and Sporadic ALS Patients Are Toxic to Motor Neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Luchena, C.; Zuazo-Ibarra, J.; Valero, J.; Matute, C.; Alberdi, E.; Capetillo-Zarate, E. A Neuron, Microglia, and Astrocyte Triple Co-culture Model to Study Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 844534. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, S.E.; Medeiros, M.; Porfirio, J.; Tavares, W.; Pessôa, L.; Grinberg, L.; Leite, R.E.; Ferretti-Rebustini, R.E.L.; Suemoto, C.K.; Filho, W.J.; et al. Similar Microglial Cell Densities across Brain Structures and Mammalian Species: Implications for Brain Tissue Function. J. Neurosci. 2020, 40, 4622–4643. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Vahsen, B.F.; Gray, E.; Candalija, A.; Cramb, K.M.L.; Scaber, J.; Dafinca, R.; Katsikoudi, A.; Xu, Y.; Farrimond, L.; Wade-Martins, R.; et al. Human iPSC co-culture model to investigate the interaction between microglia and motor neurons. Sci. Rep. 2022, 12, 12606. [Google Scholar] [CrossRef]
- Geirsdottir, L.; David, E.; Keren-Shaul, H.; Weiner, A.; Bohlen, S.C.; Neuber, J.; Balic, A.; Giladi, A.; Sheban, F.; Dutertre, C.-A.; et al. Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program. Cell 2019, 179, 1609–1622.e16. [Google Scholar] [CrossRef]
- Fattorelli, N.; Martinez-Muriana, A.; Wolfs, L.; Geric, I.; De Strooper, B.; Mancuso, R. Stem-cell-derived human microglia transplanted into mouse brain to study human disease. Nat. Protoc. 2021, 16, 1013–1033. [Google Scholar] [CrossRef]
- Kirchhoff, F.; Dringen, R.; Giaume, C. Pathways of neuron-astrocyte interactions and their possible role in neuroprotection. Eur. Arch. Psychiatry Clin. Neurosci. 2001, 251, 159–169. [Google Scholar] [CrossRef]
- Stoklund Dittlau, K.; Terrie, L.; Baatsen, P.; Kerstens, A.; De Swert, L.; Janky, R.; Corthout, N.; Masrori, P.; Van Damme, P.; Hyttel, P.; et al. FUS-ALS HiPSC-Derived Astrocytes Impair Human Motor Units through Both Gain-of-Toxicity and Loss-of-Support Mechanisms. Mol. Neurodegener. 2023, 18, 5. [Google Scholar] [CrossRef]
- McCauley, M.E.; Baloh, R.H. Inflammation in ALS/FTD Pathogenesis. Acta Neuropathol. 2019, 137, 715–730. [Google Scholar] [CrossRef]
- Gonzalez-Fernandez, C.; González, P.; Rodríguez, F. New Insights into Wnt Signaling Alterations in Amyotrophic Lateral Sclerosis: A Potential Therapeutic Target? Neural Regen. Res. 2020, 15, 1580–1589. [Google Scholar] [CrossRef]
- Gagliardi, D.; Costamagna, G.; Taiana, M.; Andreoli, L.; Biella, F.; Bersani, M.; Bresolin, N.; Comi, G.P.; Corti, S. Insights into Disease Mechanisms and Potential Therapeutics for C9orf72-Related Amyotrophic Lateral Sclerosis/Frontotemporal Dementia. Ageing Res. Rev. 2020, 64, 101172. [Google Scholar] [CrossRef]
- Schmitz, A.; Pinheiro Marques, J.; Oertig, I.; Maharjan, N.; Saxena, S. Emerging Perspectives on Dipeptide Repeat Proteins in C9ORF72 ALS/FTD. Front. Cell. Neurosci. 2021, 15, 637548. [Google Scholar] [CrossRef]
- Marchi, P.M.; Marrone, L.; Brasseur, L.; Coens, A.; Webster, C.P.; Bousset, L.; Destro, M.; Smith, E.F.; Walther, C.G.; Alfred, V.; et al. C9ORF72-Derived Poly-GA DPRs Undergo Endocytic Uptake in IAstrocytes and Spread to Motor Neurons. Life Sci. Alliance 2022, 5, e202101276. [Google Scholar] [CrossRef]
- Nihei, Y.; Mori, K.; Werner, G.; Arzberger, T.; Zhou, Q.; Khosravi, B.; Japtok, J.; Hermann, A.; Sommacal, A.; Weber, M.; et al. Poly-Glycine–Alanine Exacerbates C9orf72 Repeat Expansion-Mediated DNA Damage via Sequestration of Phosphorylated ATM and Loss of Nuclear HnRNPA. Acta Neuropathol. 2020, 139, 99–118. [Google Scholar] [CrossRef]
- Li, C.Y.; Yang, T.M.; Ou, R.W.; Wei, Q.Q.; Shang, H.F. Genome-Wide Genetic Links between Amyotrophic Lateral Sclerosis and Autoimmune Diseases. BMC Med. 2021, 19, 27. [Google Scholar] [CrossRef]
- Burberry, A.; Suzuki, N.; Wang, J.Y.; Moccia, R.; Mordes, D.A.; Stewart, M.H.; Suzuki-Uematsu, S.; Ghosh, S.; Singh, A.; Merkle, F.T.; et al. Loss-of-Function Mutations in the C9ORF72 Mouse Ortholog Cause Fatal Autoimmune Disease. Sci. Transl. Med. 2016, 8, 347ra93. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W.; Mochly-Rosen, D. Fragmented Mitochondria Released from Microglia Trigger A1 Astrocytic Response and Propagate Inflammatory Neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Shi, Y.; Lin, S.; Staats, K.A.; Li, Y.; Chang, W.H.; Hung, S.T.; Hendricks, E.; Linares, G.R.; Wang, Y.; Son, E.Y.; et al. Haploinsufficiency Leads to Neurodegeneration in C9ORF72 ALS/FTD Human Induced Motor Neurons. Nat. Med. 2018, 24, 313–325. [Google Scholar] [CrossRef]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.-H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef]
- Lopez-Lengowski, K.; Kathuria, A.; Gerlovin, K.; Karmacharya, R. Co-Culturing Microglia and Cortical Neurons Differentiated from Human Induced Pluripotent Stem Cells. J. Vis. Exp. 2021, e62480. [Google Scholar] [CrossRef]
- Haenseler, W.; Sansom, S.N.; Buchrieser, J.; Newey, S.E.; Moore, C.S.; Nicholls, F.J.; Chintawar, S.; Schnell, C.; Antel, J.P.; Allen, N.D.; et al. A Highly Efficient Human Pluripotent Stem Cell Microglia Model Displays a Neuronal-Co-culture-Specific Expression Profile and Inflammatory Response. Stem Cell Rep. 2017, 8, 1727–1742. [Google Scholar] [CrossRef]
- Vahsen, B.F.; Nalluru, S.; Morgan, G.R.; Farrimond, L.; Carroll, E.; Xu, Y.; Cramb, K.M.L.; Amein, B.; Scaber, J.; Katsikoudi, A.; et al. C9orf72-ALS Human IPSC Microglia Are pro-Inflammatory and Toxic to Co-Cultured Motor Neurons via MMP. Nat. Commun. 2023, 14, 5898. [Google Scholar] [CrossRef]
- Spiller, K.J.; Khan, T.; Dominique, M.A.; Restrepo, C.R.; Cotton-Samuel, D.; Levitan, M.; Jafar-Nejad, P.; Zhang, B.; Soriano, A.; Rigo, F.; et al. Reduction of matrix metalloproteinase 9 (MMP-9) protects motor neurons from TDP-43-triggered death in rNLS8 mice. Neurobiol. Dis. 2019, 124, 133–140. [Google Scholar] [CrossRef]
- Kaplan, A.; Spiller, K.J.; Towne, C.; Kanning, K.C.; Choe, G.T.; Geber, A.; Akay, T.; Aebischer, P.; Henderson, C.E. Neuronal Matrix Metalloproteinase-9 Is a Determinant of Selective Neurodegeneration. Neuron 2014, 81, 333–348. [Google Scholar] [CrossRef]
- Goshi, N.; Morgan, R.K.; Lein, P.J.; Seker, E. A Primary Neural Cell Culture Model to Study Neuron, Astrocyte, and Microglia Interactions in Neuroinflammation. J. Neuroinflamm. 2020, 17, 155. [Google Scholar] [CrossRef]
- Suarez-Martinez, E.; Suazo-Sanchez, I.; Celis-Romero, M.; Carnero, A. 3D and Organoid Culture in Research: Physiology, Hereditary Genetic Diseases and Cancer. Cell Biosci. 2022, 12, 39. [Google Scholar] [CrossRef]
- Dawoody Nejad, L.; Julian, L.M. Stem Cell-Derived Organoid Models for SARS-CoV-2 and Its Molecular Interaction with Host Cells. Mol. Biol. Rep. 2023, 12, 10627–10635. [Google Scholar] [CrossRef]
- Agboola, O.S.; Hu, X.; Shan, Z.; Wu, Y.; Lei, L. Brain Organoid: A 3D Technology for Investigating Cellular Composition and Interactions in Human Neurological Development and Disease Models in Vitro. Stem Cell Res. Ther. 2021, 12, 430. [Google Scholar] [CrossRef]
- Huch, M.; Koo, B.K. Modeling Mouse and Human Development Using Organoid Cultures. Development 2015, 142, 3113–3125. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral Organoids Model Human Brain Development and Microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Giandomenico, S.L.; Mierau, S.B.; Gibbons, G.M.; Wenger, L.M.D.; Masullo, L.; Sit, T.; Sutcliffe, M.; Boulanger, J.; Tripodi, M.; Derivery, E.; et al. Cerebral Organoids at the Air–Liquid Interface Generate Diverse Nerve Tracts with Functional Output. Nat. Neurosci. 2019, 22, 669–679. [Google Scholar] [CrossRef]
- Eichmüller, O.L.; Knoblich, J.A. Human Cerebral Organoids - a New Tool for Clinical Neurology Research. Nat. Rev. Neurol. 2022, 18, 661–680. [Google Scholar] [CrossRef]
- Szebényi, K.; Wenger, L.M.D.; Sun, Y.; Dunn, A.W.E.; Limegrover, C.A.; Gibbons, G.M.; Conci, E.; Paulsen, O.; Mierau, S.B.; Balmus, G.; et al. Human ALS/FTD brain organoid slice cultures display distinct early astrocyte and targetable neuronal pathology. Nat. Neurosci. 2021, 24, 1542–1554. [Google Scholar] [CrossRef]
- Smedley, G.D.; Walker, K.E.; Yuan, S.H. The Role of PERK in Understanding Development of Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 8146. [Google Scholar] [CrossRef]
- Guo, R.; Chen, Y.; Zhang, J.; Zhou, Z.; Feng, B.; Du, X.; Liu, X.; Ma, J.; Cui, H. Neural Differentiation and Spinal Cord Organoid Generation from Induced Pluripotent Stem Cells (IPSCs) for ALS Modelling and Inflammatory Screening. Mol. Neurobiol. 2023, 61, 4732–4749. [Google Scholar] [CrossRef]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “Dying-Back” Phenomenon of Motor Neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef]
- Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C. Skeletal Muscle in Als: An Unappreciated Therapeutic Opportunity? Cells 2021, 10, 525. [Google Scholar] [CrossRef]
- Gao, C.; Shi, Q.; Pan, X.; Chen, J.; Zhang, Y.; Lang, J.; Wen, S.; Liu, X.; Cheng, T.L.; Lei, K. Neuromuscular Organoids Model Spinal Neuromuscular Pathologies in C9orf72 Amyotrophic Lateral Sclerosis. Cell Rep. 2024, 43, 113892. [Google Scholar] [CrossRef]
- Massih, B.; Veh, A.; Schenke, M.; Mungwa, S.; Seeger, B.; Selvaraj, B.T.; Chandran, S.; Reinhardt, P.; Sterneckert, J.; Hermann, A.; et al. A 3D Cell Culture System for Bioengineering Human Neuromuscular Junctions to Model ALS. Front. Cell Dev. Biol. 2023, 11, 996952. [Google Scholar] [CrossRef]
- Murray, L.M.; Talbot, K.; Gillingwater, T.H. Review: Neuromuscular Synaptic Vulnerability in Motor Neurone Disease: Amyotrophic Lateral Sclerosis and Spinal Muscular Atrophy. Neuropathol. Appl. Neurobiol. 2010, 36, 133–156. [Google Scholar] [CrossRef]
- Xu, C.; Tabebordbar, M.; Iovino, S.; Ciarlo, C.; Liu, J.; Castiglioni, A.; Price, E.; Liu, M.; Barton, E.R.; Kahn, C.R.; et al. XA Zebrafish Embryo Culture System Defines Factors That Promote Vertebrate Myogenesis across Species. Cell 2013, 155, 909–921. [Google Scholar] [CrossRef]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced Pluripotent Stem Cells Generated from Patients with ALS Can Be Differentiated into Motor Neurons. Science 2008, 321, 1218–1221. [Google Scholar] [CrossRef]
- Bakooshli, M.A.; Lippmann, E.S.; Mulcahy, B.; Iyer, N.; Nguyen, C.T.; Tung, K.; Stewart, B.A.; Van Den Dorpel, H.; Fuehrmann, T.; Shoichet, M.; et al. A 3d Culture Model of Innervated Human Skeletal Muscle Enables Studies of the Adult Neuromuscular Junction. elife 2019, 8, e44530. [Google Scholar] [CrossRef]
- Lin, C.Y.; Yoshida, M.; Li, L.T.; Ikenaka, A.; Oshima, S.; Nakagawa, K.; Sakurai, H.; Matsui, E.; Nakahata, T.; Saito, M.K. IPSC-Derived Functional Human Neuromuscular Junctions Model the Pathophysiology of Neuromuscular Diseases. J. Clin. Investig. Insight 2019, 4, e124299. [Google Scholar] [CrossRef]
- Dittlau, K.S.; Krasnow, E.N.; Fumagalli, L.; Vandoorne, T.; Terrie, L.; Baatsen, P.; Giacomazzi, G.; Sampaolesi, M.; Thorrez, L.; Van Damme, P.; et al. Modeling ALS—Human Neuromuscular Junctions in a Dish. Amyotroph. Lateral Scler. Front. Degener. 2019, 11, 2304989. [Google Scholar]
- Faustino Martins, J.M.; Fischer, C.; Urzi, A.; Vidal, R.; Kunz, S.; Ruffault, P.L.; Kabuss, L.; Hube, I.; Gazzerro, E.; Birchmeier, C.; et al. Self-Organizing 3D Human Trunk Neuromuscular Organoids. Cell Stem Cell 2020, 26, 172–186.e6. [Google Scholar] [CrossRef]
- Andersen, J.; Revah, O.; Miura, Y.; Thom, N.; Amin, N.D.; Kelley, K.W.; Singh, M.; Chen, X.; Thete, M.V.; Walczak, E.M.; et al. Generation of Functional Human 3D Cortico-Motor Assembloids. Cell 2020, 183, 1913–1929.e26. [Google Scholar] [CrossRef]
- Santhanam, N.; Kumanchik, L.; Guo, X.; Sommerhage, F.; Cai, Y.; Jackson, M.; Martin, C.; Saad, G.; McAleer, C.W.; Wang, Y.; et al. Stem cell derived phenotypic human neuromuscular junction model for dose response evaluation of therapeutics. Biomaterials 2018, 166, 64–78. [Google Scholar] [CrossRef]
- Osaki, T.; Uzel, S.G.M.; Kamm, R.D. Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci. Adv. 2018, 4, eaat5847. [Google Scholar] [CrossRef]
- Pereira, J.D.; DuBreuil, D.M.; Devlin, A.C.; Held, A.; Sapir, Y.; Berezovski, E.; Hawrot, J.; Dorfman, K.; Chander, V.; Wainger, B.J. Human Sensorimotor Organoids Derived from Healthy and Amyotrophic Lateral Sclerosis Stem Cells Form Neuromuscular Junctions. Nat. Commun. 2021, 12, 4744. [Google Scholar] [CrossRef]
- McCampbell, A.; Cole, T.; Wegener, A.J.; Tomassy, G.S.; Setnicka, A.; Farley, B.J.; Schoch, K.M.; Hoye, M.L.; Shabsovich, M.; Sun, L.; et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J. Clin. Investig. 2018, 128, 3558–3567. [Google Scholar] [CrossRef]
- Pașca, A.M.; Park, J.-Y.; Shin, H.-W.; Qi, Q.; Revah, O.; Krasnoff, R.; O’hara, R.; Willsey, A.J.; Palmer, T.D.; Pașca, S.P. Human 3D cellular model of hypoxic brain injury of prematurity. Nat. Med. 2019, 25, 784–791. [Google Scholar] [CrossRef]
- Lobsiger, C.S.; Cleveland, D.W. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat. Neurosci. 2007, 10, 1355–1360. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and Edaravone: A Tale of Two Amyotrophic Lateral Sclerosis Drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef]
- Bonaventura, G.; Iemmolo, R.; Attaguile, G.A.; La Cognata, V.; Pistone, B.S.; Raudino, G.; D’agata, V.; Cantarella, G.; Barcellona, M.L.; Cavallaro, S. iPSCs: A Preclinical Drug Research Tool for Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 4596. [Google Scholar] [CrossRef]
- Beghini, D.G.; Kasai-Brunswick, T.H.; Henriques-Pons, A. Induced Pluripotent Stem Cells in Drug Discovery and Neurodegenerative Disease Modelling. Int. J. Mol. Sci. 2024, 25, 2392. [Google Scholar] [CrossRef]
- Pasteuning-Vuhman, S.; de Jongh, R.; Timmers, A.; Pasterkamp, R.J. Towards Advanced IPSC-Based Drug Development for Neurodegenerative Disease. Trends Mol. Med. 2021, 27, 263–279. [Google Scholar] [CrossRef]
- Doss, M.X.; Sachinidis, A. Current Challenges of IPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef]
- Monteduro, A.G.; Rizzato, S.; Caragnano, G.; Trapani, A.; Giannelli, G.; Maruccio, G. Organs-on-Chips Technologies—A Guide from Disease Models to Opportunities for Drug Development. Biosens. Bioelectron. 2023, 231, 115271. [Google Scholar] [CrossRef] [PubMed]
- Busek, M.; Aizenshtadt, A.; Amirola-Martinez, M.; Delon, L.; Krauss, S. Academic User View: Organ-on-a-Chip Technology. Biosensors 2022, 12, 126. [Google Scholar] [CrossRef] [PubMed]
- Morello, G.; Salomone, S.; D’agata, V.; Conforti, F.L.; Cavallaro, S. From Multi-Omics Approaches to Precision Medicine in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2020, 14, 577755. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Cell Type | Objective/Study | Key Findings |
|---|---|---|
| iPSC-derived MNs | sALS iPSC-derived MNs [11] |
|
| hiPSC-derived MNs [27] |
| |
| ISL1 and LHX3 transgenes were delivered by CRISPR/Cas9 techniques in the human iPSC genome [33] |
| |
| hiPSC-derived phMN-enriched cultures [35] | Calibrated activation of RA and SHH signaling in hiPSC-derived NPCs facilitates a cervical identity of dorsal NPCs to produce phMN-like neurons [35]. | |
| Co-culturing hiPSC-derived MNs with Neuroglia | Co-cultured FUS-ALS iPSC-derived astrocytes and MNs in a microfluidic device with skeletal myocytes [48] | FUS astrocytes are deleterious to MN neurite outgrowth, network integration, and neuromuscular junction (NMJ) formation and functionality [48]. |
| iPSC-derived human astrocytes co-cultured with Hb9-GFP mouse MNs [53] |
| |
| Co-cultured hiPSC-derived microglia–MN [61] |
| |
| HiPSC-derived Organoids | C9 ALI-COs developed from ALS/FTD iPSCs [72] | ALI-COs develop a consistent microarchitecture and mature cortical circuit-forming disease-relevant phenotypes. Although lacking microglia and vasculatures, C9 ALI-COs show abnormalities specific to neurons and astrocytes [72]. |
| C9-knockdown hiPSCs differentiated into MNs, astrocytes, and SCOs [74] | SCOs created with lower levels of the C9orf72 protein show characteristic cellular compositions similar to those in the spinal cord and considerably increased inflammatory markers [74]. | |
| hiPSC-derived NMOs with the C9orf72 HRE mutation [77] | DPRs and RNA foci are seen in neurons and astrocytes. Treatment with GSK2606414 results in a doubling of muscle contraction, decreased autophagy, and DPR aggregation [77]. | |
| Functional NMJs generated from five iPSC lines [89] | NMJs in organoid cultures show abnormal contraction and immunocytochemistry, with their early loss being a crucial component of ALS models [89]. | |
| SOD1 hiPSC-derived MNs co-cultured with myoblast-derived human skeletal muscle in a 3D hydrogel-based model [78] | MNs with SOD1 mutations initially display normal morphology (e.g., postsynaptic folding) but later exhibit a pathogenic phenotype [78]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dawoody Nejad, L.; Pioro, E.P. Modeling ALS with Patient-Derived iPSCs: Recent Advances and Future Potentials. Brain Sci. 2025, 15, 134. https://doi.org/10.3390/brainsci15020134
Dawoody Nejad L, Pioro EP. Modeling ALS with Patient-Derived iPSCs: Recent Advances and Future Potentials. Brain Sciences. 2025; 15(2):134. https://doi.org/10.3390/brainsci15020134
Chicago/Turabian StyleDawoody Nejad, Ladan, and Erik P. Pioro. 2025. "Modeling ALS with Patient-Derived iPSCs: Recent Advances and Future Potentials" Brain Sciences 15, no. 2: 134. https://doi.org/10.3390/brainsci15020134
APA StyleDawoody Nejad, L., & Pioro, E. P. (2025). Modeling ALS with Patient-Derived iPSCs: Recent Advances and Future Potentials. Brain Sciences, 15(2), 134. https://doi.org/10.3390/brainsci15020134