
On the Cut-Off Value of the Anteroposterior Diameter of the Midbrain Atrophy in Spinocerebellar Ataxia Type 2 Patients

 , , , ,
, , , ,  ,
,
Abstract
1. Introduction
2. Preliminary
3. Materials and Methods
3.1. Patients and Methods
3.1.1. Screening of the Control Group and Patients
3.1.2. Conducting the Cranial MRI Study
3.1.3. Manual Measurement of Specific Anatomical Structures
3.2. Statistical Analysis
4. Results
4.1. Demographics
4.2. Derivation of SCA2 Atrophy Risk Score
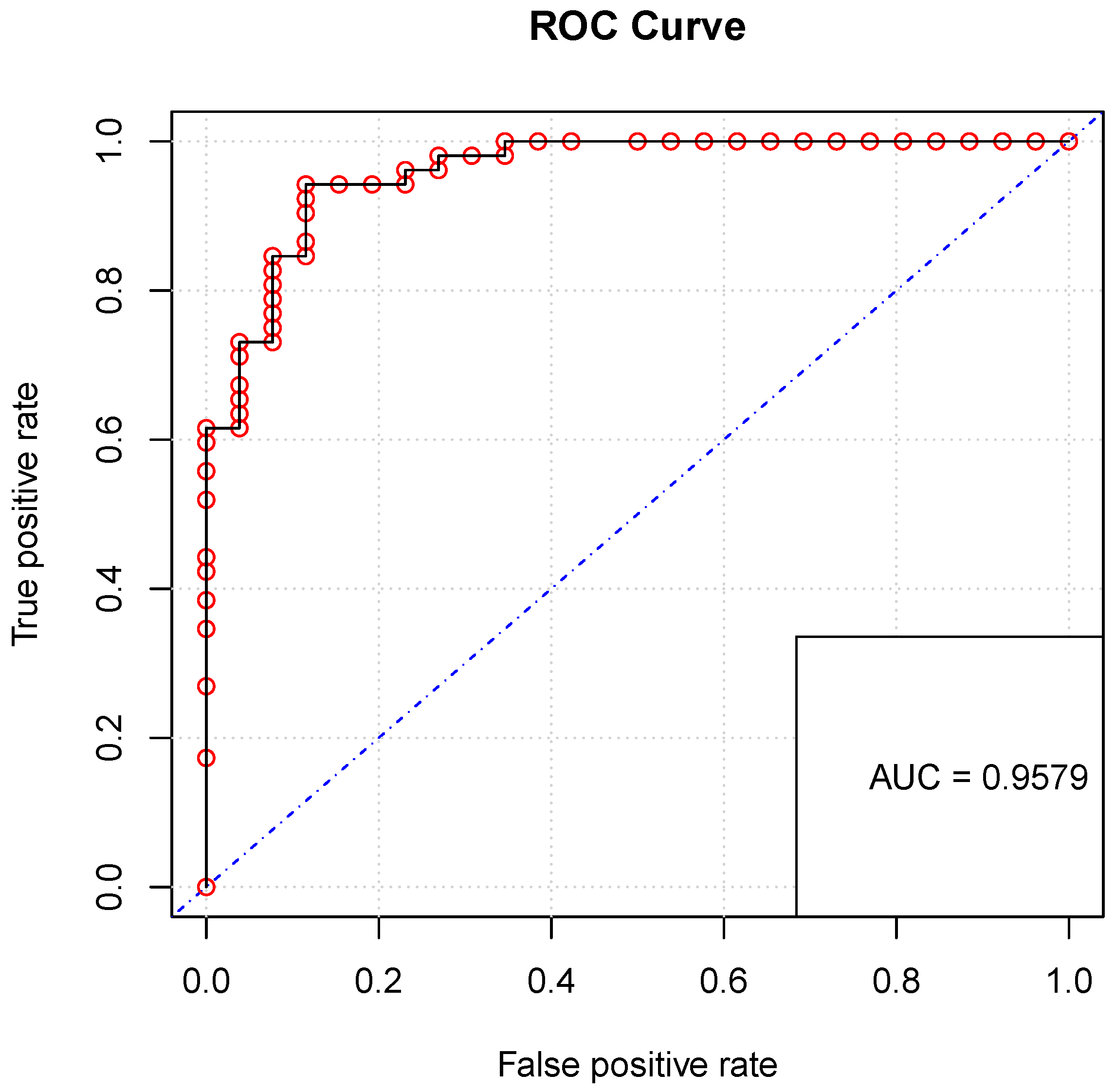
4.3. Validation
5. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SCA | Spinocerebellar Ataxias |
| MRI | Magnetic Resonance Image |
| ADM | Anteroposterior Diameter of the Midbrain |
| MJD | Machado-Joseph Disease |
| CIRAH | Centro para la Investigación y Rehabilitación de Ataxias Hereditarias |
| CONAHCYT | Consejo Nacional de Humanidades, Ciencias y Tecnologías |
References
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar ataxia. Nat. Rev. Dis. Prim. 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Klaes, A.; Reckziegel, E.; Franca, M.C., Jr.; Rezende, T.J.R.; Vedolin, L.M.; Jardim, L.B.; Saute, J.A. MR Imaging in Spinocerebellar Ataxias: A Systematic Review. Am. J. Neuroradiol. 2016, 37, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Reetz, K.; Rodríguez-Labrada, R.; Dogan, I.; Mirzazade, S.; Romanzetti, S.; Schulz, J.B.; Cruz-Rivas, E.M.; Alvarez-Cuesta, J.A.; Aguilera Rodríguez, R.; Gonzalez Zaldivar, Y.; et al. Brain atrophy measures in preclinical and manifest spinocerebellar ataxia type 2. Ann. Clin. Transl. Neurol. 2018, 5, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Velázquez-Pérez, L.; Medrano-Montero, J.; Rodríguez-Labrada, R.; Canales-Ochoa, N.; Campins Alí, J.; Carrillo Rodes, F.J.; Rodríguez Graña, T.; Hernández Oliver, M.O.; Aguilera Rodríguez, R.; Domínguez Barrios, Y.; et al. Hereditary Ataxias in Cuba: A Nationwide Epidemiological and Clinical Study in 1001 Patients. Cerebellum 2020, 19, 252–264. [Google Scholar] [CrossRef]
- Velázquez-Pérez, L.C.; Rodríguez-Labrada, R.; Fernandez-Ruiz, J. Spinocerebellar Ataxia Type 2: Clinicogenetic Aspects, Mechanistic Insights, and Management Approaches. Front. Neurol. 2017, 8, 472. [Google Scholar] [CrossRef]
- Rodríguez-Labrada, R.; Martins, A.C.; Magaña, J.J.; Vazquez-Mojena, Y.; Medrano-Montero, J.; Fernandez-Ruíz, J.; Cisneros, B.; Teive, H.; McFarland, K.N.; Saraiva-Pereira, M.L.; et al. Founder Effects of Spinocerebellar Ataxias in the American Continents and the Caribbean. Cerebellum 2020, 19, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Paulson, H.L.; Shakkottai, V.G.; Clark, H.B.; Orr, H.T. Polyglutamine spinocerebellar ataxias—From genes to potential treatments. Nat. Rev. Neurosci. 2017, 18, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.C.; Choi, S.I.; Du, A.X.; Cuzzocreo, J.L.; Ying, H.S.; Landman, B.A.; Perlman, S.L.; Baloh, R.W.; Zee, D.S.; Toga, A.W.; et al. MRI Shows a Region-Specific Pattern of Atrophy in Spinocerebellar Ataxia Type 2. Cerebellum 2012, 11, 272–279. [Google Scholar] [CrossRef]
- Pilotto, F.; Saxena, S. Epidemiology of inherited cerebellar ataxias and challenges in clinical research. Clin. Transl. Neurosci. 2018, 2, 2514183X18785258. [Google Scholar] [CrossRef]
- Perlman, S. Hereditary Ataxia Overview; 28 October 1998 [Updated 16 June 2022]. GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 1993–2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1138/ (accessed on 21 November 2023).
- Moriarty, A.; Cook, A.; Hunt, H.; Adams, M.E.; Cipolotti, L.; Giunti, P. A longitudinal investigation into cognition and disease progression in spinocerebellar ataxia types 1, 2, 3, 6, and 7. Orphanet J. Rare Dis. 2016, 11, 82. [Google Scholar] [CrossRef]
- Meira, A.T.; Arruda, W.O.; Ono, S.E.; de Carvalho Neto, A.; Raskin, S.; Camargo, C.H.F.; Teive, H.A.G. Neuroradiological Findings in the Spinocerebellar Ataxias. Tremor Other Hyperkinetic Movements 2019, 9, 516. [Google Scholar] [CrossRef]
- Ashizawa, T.; Öz, G.; Paulson, H.L. Author Correction: Spinocerebellar ataxias: Prospects and challenges for therapy development. Nat. Rev. Neurol. 2018, 14, 749. [Google Scholar] [CrossRef] [PubMed]
- Mascalchi, M.; Vella, A. Neuroimaging Biomarkers in SCA2 Gene Carriers. Int. J. Mol. Sci. 2020, 21, 1020. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, R.; Lee, J.Y.; Lee, K.M. Clinical, Imaging, and Laboratory Markers of Premanifest Spinocerebellar Ataxia 1, 2, 3, and 6: A Systematic Review. J. Clin Neurol. 2021, 17, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Wilke, C.; Haas, E.; Reetz, K.; Faber, J.; Garcia-Moreno, H.; Santana, M.M.; van de Warrenburg, B.; Hengel, H.; Lima, M.; Filla, A.; et al. Neurofilaments in spinocerebellar ataxia type 3: Blood biomarkers at the preataxic and ataxic stage in humans and mice. EMBO Mol. Med. 2020, 12, e11803. [Google Scholar] [CrossRef] [PubMed]
- Öz, G.; Harding, I.H.; Krahe, J.; Reetz, K. MR imaging and spectroscopy in degenerative ataxias: Toward multimodal, multisite, multistage monitoring of neurodegeneration. Curr. Opin. Neurol. 2020, 33, 451–461. [Google Scholar] [CrossRef]
- Monte, T.L.; Reckziegel, E.d.R.; Augustin, M.C.; Locks-Coelho, L.D.; Santos, A.S.P.; Furtado, G.V.; de Mattos, E.P.; Pedroso, J.L.; Barsottini, O.P.; Vargas, F.R.; et al. The progression rate of spinocerebellar ataxia type 2 changes with stage of disease. Orphanet J. Rare Dis. 2018, 13, 20. [Google Scholar] [CrossRef]
- Silva, R.N.d.; Vallortigara, J.; Greenfield, J.; Hunt, B.; Giunti, P.; Hadjivassiliou, M. Diagnosis and management of progressive ataxia in adults. Pract. Neurol. 2019, 19, 196–207. [Google Scholar] [CrossRef]
- Mascalchi, M.; Diciotti, S.; Giannelli, M.; Ginestroni, A.; Soricelli, A.; Nicolai, E.; Aiello, M.; Tessa, C.; Galli, L.; Dotti, M.T.; et al. Progression of Brain Atrophy in Spinocerebellar Ataxia Type 2: A Longitudinal Tensor-Based Morphometry Study. PLoS ONE 2014, 9, e89410. [Google Scholar] [CrossRef]
- Hara, D.; Maki, F.; Tanaka, S.; Sasaki, R.; Hasegawa, Y. MRI-based cerebellar volume measurements correlate with the International Cooperative Ataxia Rating Scale score in patients with spinocerebellar degeneration or multiple system atrophy. Cerebellum Ataxias 2016, 3, 14. [Google Scholar] [CrossRef]
- Cocozza, S.; Pontillo, G.; De Michele, G.; Di Stasi, M.; Guerriero, E.; Perillo, T.; Pane, C.; De Rosa, A.; Ugga, L.; Brunetti, A. Conventional MRI findings in hereditary degenerative ataxias: A pictorial review. Neuroradiology 2021, 63, 983–999. [Google Scholar] [CrossRef] [PubMed]
- Coarelli, G.; Brice, A.; Durr, A. Recent Advances in Understanding Dominant Spinocerebellar Ataxias from Clinical and Genetic Points of View. F1000Research 2018, 7, 1781. [Google Scholar] [CrossRef] [PubMed]
- Straub, S.; Mangesius, S.; Emmerich, J.; Indelicato, E.; Nachbauer, W.; Degenhardt, K.S.; Ladd, M.E.; Boesch, S.; Gizewski, E.R. Toward quantitative neuroimaging biomarkers for Friedreich’s ataxia at 7 Tesla: Susceptibility mapping, diffusion imaging, R2 and R1 relaxometry. J. Neurosci. Res. 2020, 98, 2219–2231. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Ahn, J.H.; Cho, Y.; Kim, J.S.; Youn, J.; Cho, J.W. Differential value of brain magnetic resonance imaging in multiple system atrophy cerebellar phenotype and spinocerebellar ataxias. Sci. Rep. 2019, 9, 17329. [Google Scholar] [CrossRef] [PubMed]
- Adanyeguh, I.M.; Perlbarg, V.; Henry, P.G.; Rinaldi, D.; Petit, E.; Valabregue, R.; Brice, A.; Durr, A.; Mochel, F. Autosomal dominant cerebellar ataxias: Imaging biomarkers with high effect sizes. NeuroImage Clin. 2018, 19, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Altamarino, L.S.; Toglia, J.U. Cerebellar atrophy: Pneumoencephalography and computerized tomography correlation. Neuroradiology 1978, 16, 179–180. [Google Scholar] [CrossRef] [PubMed]
- Steele, J.R.; Hoffman, J. Brainstem evaluation with CT cisternography. Am. J. Roentgenol. 1981, 136, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Abe, S.; Miyasaka, K.; Tashiro, K.; Takei, H.; Isu, T.; Tsuru, M. Evaluation of the Brainstem with High-Resolution CT in Cerebellar Atrophic Processes. AJNR Am. J. Neuroradiol. 1983, 4, 446–449. [Google Scholar]
- Koller, W.C.; Glatt, S.L.; Perlik, S.; Huckman, M.S.; Fox, J.H. Cerebellar atrophy demonstrated by computed tomography. Neurology 1981, 31, 405–412. [Google Scholar] [CrossRef]
- Allen, J.H.; True Martin, J.; William McLain, L. Computed Tomography in Cerebellar Atrophic Processes. Radiology 1979, 130, 379–382. [Google Scholar] [CrossRef]
- Ramos, A.; Quintana, F.; Díez, C.; Leno, C.; Berciano, J. CT Findings in Spinocerebellar Degeneration. AJNR Am. J. Neuroradiol. 1987, 8, 635–640. [Google Scholar] [PubMed]
- Kumar, S.; Chand, R.; Gururaj, A.; Jeans, W. CT Features of Olivopontocerebellar Atrophy in Children. Acta Radiol. 1995, 36, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Sharma, B.R.; Bhatta, M.; Poudel, N. Measurement of Anteroposterior diameters of normal brainstem by Magnetic Resonance Imaging. J. Gandaki Med-Coll.-Nepal 2019, 12, 53–58. [Google Scholar] [CrossRef]
- Metwally, M.I.; Basha, M.A.A.; AbdelHamid, G.A.; Nada, M.G.; Ali, R.R.; frere, R.A.F.; Elshetry, A.S.F. Neuroanatomical MRI study: Reference values for the measurements of brainstem, cerebellar vermis, and peduncles. Br. J. Radiol. 2021, 94, 20201353. [Google Scholar] [CrossRef] [PubMed]
- Martínez Guerrero, J.; Paz-Gutiérrez, J.; Vega-Gaxiola, S.B. Ataxia Espinocerebelosa Tipo 2. Arch. Neurociencias 2016, 21, 73–79. [Google Scholar] [CrossRef]
- Jandeaux, C.; Kuchcinski, G.; Ternynck, C.; Riquet, A.; Leclerc, X.; Pruvo, J.P.; Soto-Ares, G. Biometry of the Cerebellar Vermis and Brain Stem in Children: MR Imaging Reference Data from Measurements in 718 Children. AJNR Am. J. Neuroradiol. 2019, 40, 1835–1841. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, J.; Batista, C.; Vargas-De-León, C.; Rodes, F.; Guzman-Martinez, M.; Fitz, S. Search for A Midbrain Anteroposterior Diameter Threshold to Study Brain Atrophy in Spinocerebellar Ataxia Type 2. In Proceedings of the 8th World Congress on Electrical Engineering and Computer Systems and Science, EECSS 2022, Prague, Czech Republic, 28–30 July 2022; Benedicenti, L., Liu, Z., Eds.; Avestia Publishing: Orléans, ON, Canada, 2022. [Google Scholar] [CrossRef]
- Öz, G.; Cocozza, S.; Henry, P.; Lenglet, C.; Deistung, A.; Faber, J.; Schwarz, A.; Timmann, D.; Van Dijk, K.; Harding, I.; et al. MR Imaging in Ataxias: Consensus Recommendations by the Ataxia Global Initiative Working Group on MRI Biomarkers. Cerebellum 2023, 1–15. [Google Scholar] [CrossRef]
- Ye, C.; Yang, Z.; Ying, S.H.; Prince, J.L. Segmentation of the Cerebellar Peduncles Using a Random Forest Classifier and a Multi-object Geometric Deformable Model: Application to Spinocerebellar Ataxia Type 6. Neuroinformatics 2015, 13, 367–381. [Google Scholar] [CrossRef]
- Cabeza-Ruiz, R.; Velázquez-Pérez, L.; Linares-Barranco, A.; Pérez-Rodríguez, R. Convolutional Neural Networks for Segmenting Cerebellar Fissures from Magnetic Resonance Imaging. Sensors 2022, 22, 1345. [Google Scholar] [CrossRef]
- Ravanfar, P.; Loi, S.M.; Syeda, W.T.; Van Rheenen, T.E.; Bush, A.I.; Desmond, P.; Cropley, V.L.; Lane, D.J.R.; Opazo, C.M.; Moffat, B.A.; et al. Systematic Review: Quantitative Susceptibility Mapping (QSM) of Brain Iron Profile in Neurodegenerative Diseases. Front. Neurosci. 2021, 15, 41. [Google Scholar] [CrossRef]
- Xie, F.; Weihua, L.; Lirong, O.; Wang, X.; Xing, W. Quantitative susceptibility mapping in spinocerebellar ataxia type 3/Machado–Joseph disease (SCA3/MJD). Acta Radiol. 2020, 61, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Coarelli, G.; Darios, F.; Petit, E.; Dorgham, K.; Adanyeguh, I.; Petit, E.; Brice, A.; Mochel, F.; Durr, A. Plasma neurofilament light chain predicts cerebellar atrophy and clinical progression in spinocerebellar ataxia. Neurobiol. Dis. 2021, 153, 105311. [Google Scholar] [CrossRef]
- Liang, X.; Jiang, H.; Chen, C.; Zhou, G.; Wang, J.; Zhang, S.; Lei, L.; Wang, X.; Tang, B. The correlation between magnetic resonance imaging features of the brainstem and cerebellum and clinical features of spinocerebellar ataxia 3/Machado-Joseph disease. Neurol. India 2009, 57, 578. [Google Scholar] [CrossRef] [PubMed]
- MedCalc Software Ltd. Diagnostic Test Evaluation Calculator. Version 22.014. 2023. Available online: https://www.medcalc.org/calc/diagnostic_test.php (accessed on 11 November 2023).
- Selvadurai, L.P.; Corben, L.A.; Delatycki, M.B.; Storey, E.; Egan, G.F.; Georgiou-Karistianis, N.; Harding, I.H. Multiple mechanisms underpin cerebral and cerebellar white matter deficits in Friedreich ataxia: The IMAGE-FRDA study. Hum. Brain Mapp. 2020, 41, 1920–1933. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, S.; Zeng, N.; Duan, W.; Chen, Z.; Liu, Y.; Li, W.; Guo, Y.; Chen, H.; Li, X.; et al. Lung Radiomics Features Selection for COPD Stage Classification Based on Auto-Metric Graph Neural Network. Diagnostics 2022, 12, 2274. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.h.; Shi, C.h.; Zhou, L.n.; Li, Y.s.; Yang, J.; Liu, Y.t.; Mao, C.y.; Luo, H.y.; Xu, G.w.; Xu, Y.m. Metabolic Profiling Reveals Biochemical Pathways and Potential Biomarkers of Spinocerebellar Ataxia 3. Front. Mol. Neurosci. 2019, 12, 159. [Google Scholar] [CrossRef]
- Yap, K.H.; Abdul Manan, H.; Yahya, N.; Azmin, S.; Mohamed Mukari, S.A.; Mohamed Ibrahim, N. Magnetic Resonance Imaging and Its Clinical Correlation in Spinocerebellar Ataxia Type 3: A Systematic Review. Front. Neurosci. 2022, 16, 859651. [Google Scholar] [CrossRef]
- Rezende, T.J.R.; Martinez, A.R.M.; Faber, I.; Girotto Takazaki, K.A.; Martins, M.P.; de Lima, F.D.; Lopes-Cendes, I.; Cendes, F.; França, M.C. Developmental and neurodegenerative damage in Friedreich’s ataxia. Eur. J. Neurol. 2019, 26, 483–489. [Google Scholar] [CrossRef]
- Mascalchi, M.; Vella, A. Chapter Four-Neuroimaging Applications in Chronic Ataxias. In Imaging in Movement Disorders: Imaging Applications in Non-Parkinsonian and Other Movement Disorders; Politis, M., Ed.; International Review of Neurobiology; Academic Press: Cambridge, MA, USA, 2018; Volume 143, pp. 109–162. [Google Scholar] [CrossRef]
- Garali, I.; Adanyeguh, I.M.; Ichou, F.; Perlbarg, V.; Seyer, A.; Colsch, B.; Moszer, I.; Guillemot, V.; Durr, A.; Mochel, F.; et al. A strategy for multimodal data integration: Application to biomarkers identification in spinocerebellar ataxia. Briefings Bioinform. 2018, 19, 1356–1369. [Google Scholar] [CrossRef]
- Buijsen, R.A.; Toonen, L.J.; Gardiner, S.L.; van Roon-Mom, W.M. Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias. Neurotherapeutics 2019, 16, 263–286. [Google Scholar] [CrossRef]
- Dogan, I.; Romanzetti, S.; Didszun, C.; Mirzazade, S.; Timmann, D.; Saft, C.; Schöls, L.; Synofzik, M.; Giordano, I.A.; Klockgether, T.; et al. Structural characteristics of the central nervous system in Friedreich ataxia: An in vivo spinal cord and brain MRI study. J. Neurol. Neurosurg. Psychiatry 2019, 90, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Faber, J.; Schaprian, T.; Berkan, K.; Reetz, K.; França, M.C.; de Rezende, T.J.R.; Hong, J.; Liao, W.; van de Warrenburg, B.; van Gaalen, J.; et al. Regional Brain and Spinal Cord Volume Loss in Spinocerebellar Ataxia Type 3. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 2273–2281. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Reimold, M.; Globas, C.; Hauser, T.K.; Walter, U.; Machulla, H.J.; Rolfs, A.; Schöls, L. PET and MRI reveal early evidence of neurodegeneration in spinocerebellar ataxia type 17. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2012, 53, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Lindig, T.; Bender, B.; Kumar, V.J.; Hauser, T.K.; Grodd, W.; Brendel, B.; Just, J.; Synofzik, M.; Klose, U.; Scheffler, K.; et al. Pattern of Cerebellar Atrophy in Friedreich’s Ataxia-Using the SUIT Template. Cerebellum 2019, 18, 435–447. [Google Scholar] [CrossRef]
- Li, Q.F.; Dong, Y.; Yang, L.; Xie, J.J.; Ma, Y.; Du, Y.C.; Cheng, H.L.; Ni, W.; Wu, Z.Y. Neurofilament light chain is a promising serum biomarker in spinocerebellar ataxia type 3. Mol. Neurodegener. 2019, 14, 39. [Google Scholar] [CrossRef]

{kind=link}
| Feature | SCA2 Patients | Control Patients | p-Value |
|---|---|---|---|
| n = 66 | n = 33 | ||
| Sex (Male) 1 | 28 (33.73) | 11 (28.20) | 0.15128 |
| Age (Years) 2 | 45.06 (10.25) | 36.06 (9.14) | <0.0001 |
| SARA scale 1 | |||
| Light Stadium (13–14) | 44 (66.7) | NA | NA |
| Stage Moderate (15–26) | 20 (30.3) | NA | NA |
| Severe stage (27–34) | 2 (3.0) | NA | NA |
| ADM 2 | 16.08 (1.28) | 19.13 (0.94) | <0.001 |
| B | ES | 95% CI | p-Value | |
|---|---|---|---|---|
| Intercept | 35.814 | 8.358 | (22.212, 55.659) | <0.001 |
| ADM | −1.972 | 0.461 | (−3.067, −1.219) | <0.001 |
| Diagnosis of Spinocerebellar Ataxia Type 2 | |||
|---|---|---|---|
| Present | Absent | ||
| SCA2 atrophy risk score | Positive | 14 | 1 |
| Negative | 0 | 6 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez-Cuesta, J.A.; Mora-Batista, C.; Reyes-Carreto, R.; Carrillo-Rodes, F.J.; Fitz, S.J.T.; González-Zaldivar, Y.; Vargas-De-León, C. On the Cut-Off Value of the Anteroposterior Diameter of the Midbrain Atrophy in Spinocerebellar Ataxia Type 2 Patients. Brain Sci. 2024, 14, 53. https://doi.org/10.3390/brainsci14010053
Álvarez-Cuesta JA, Mora-Batista C, Reyes-Carreto R, Carrillo-Rodes FJ, Fitz SJT, González-Zaldivar Y, Vargas-De-León C. On the Cut-Off Value of the Anteroposterior Diameter of the Midbrain Atrophy in Spinocerebellar Ataxia Type 2 Patients. Brain Sciences. 2024; 14(1):53. https://doi.org/10.3390/brainsci14010053
Chicago/Turabian StyleÁlvarez-Cuesta, José Alberto, Camilo Mora-Batista, Ramón Reyes-Carreto, Frank Jesus Carrillo-Rodes, Sergio J. Torralbaz Fitz, Yanetza González-Zaldivar, and Cruz Vargas-De-León. 2024. "On the Cut-Off Value of the Anteroposterior Diameter of the Midbrain Atrophy in Spinocerebellar Ataxia Type 2 Patients" Brain Sciences 14, no. 1: 53. https://doi.org/10.3390/brainsci14010053
APA StyleÁlvarez-Cuesta, J. A., Mora-Batista, C., Reyes-Carreto, R., Carrillo-Rodes, F. J., Fitz, S. J. T., González-Zaldivar, Y., & Vargas-De-León, C. (2024). On the Cut-Off Value of the Anteroposterior Diameter of the Midbrain Atrophy in Spinocerebellar Ataxia Type 2 Patients. Brain Sciences, 14(1), 53. https://doi.org/10.3390/brainsci14010053