

Involvement of Nrf2 Activation and NF-kB Pathway Inhibition in the Antioxidant and Anti-Inflammatory Effects of Hesperetin in Activated BV-2 Microglial Cells



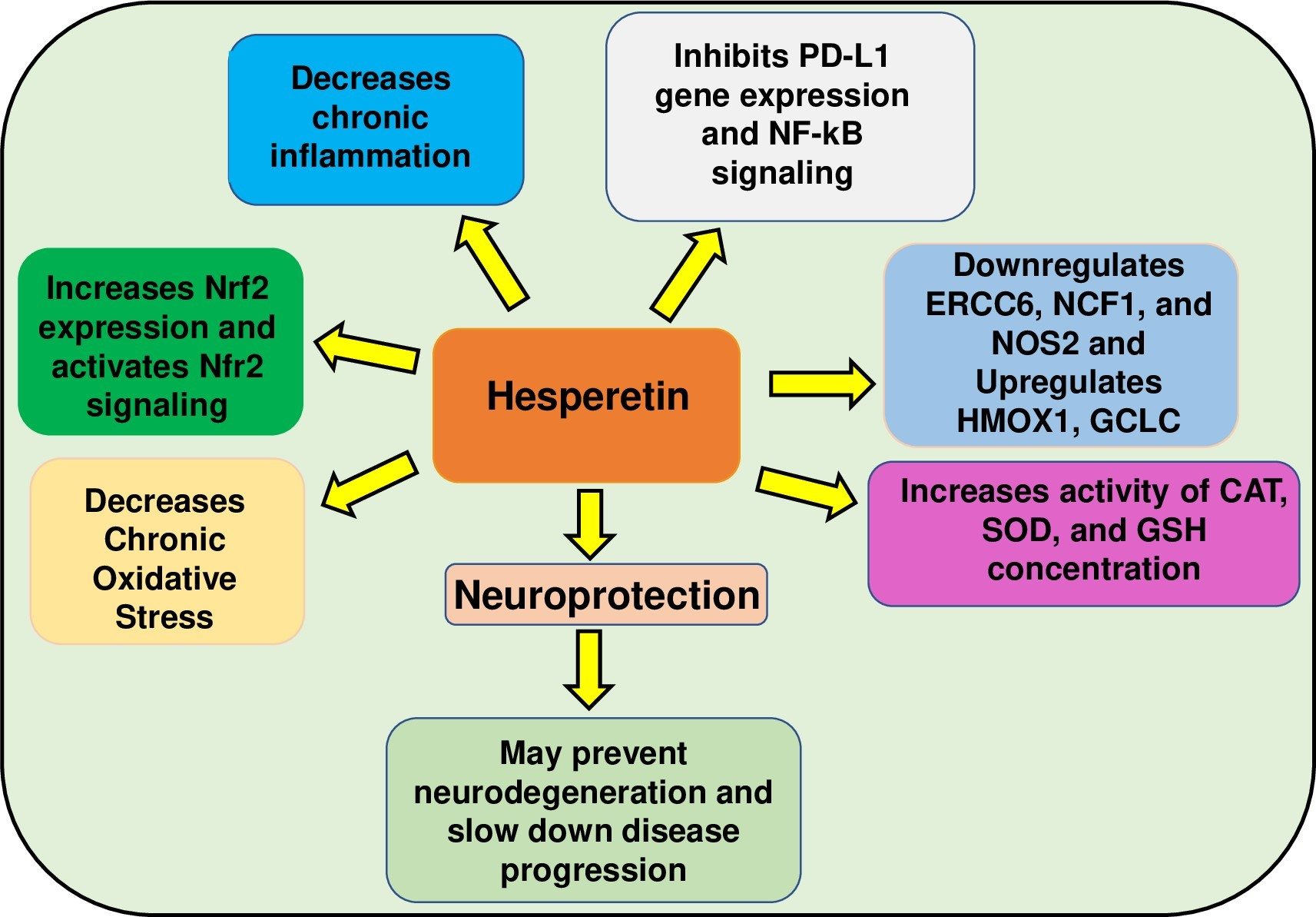
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Chemicals, and Reagents
2.2. Cell Culture
2.3. Cell Viability
2.4. Nitric Oxide (Griess) Assay
2.5. Catalase, Superoxide Dismutase, and Glutathione Assays
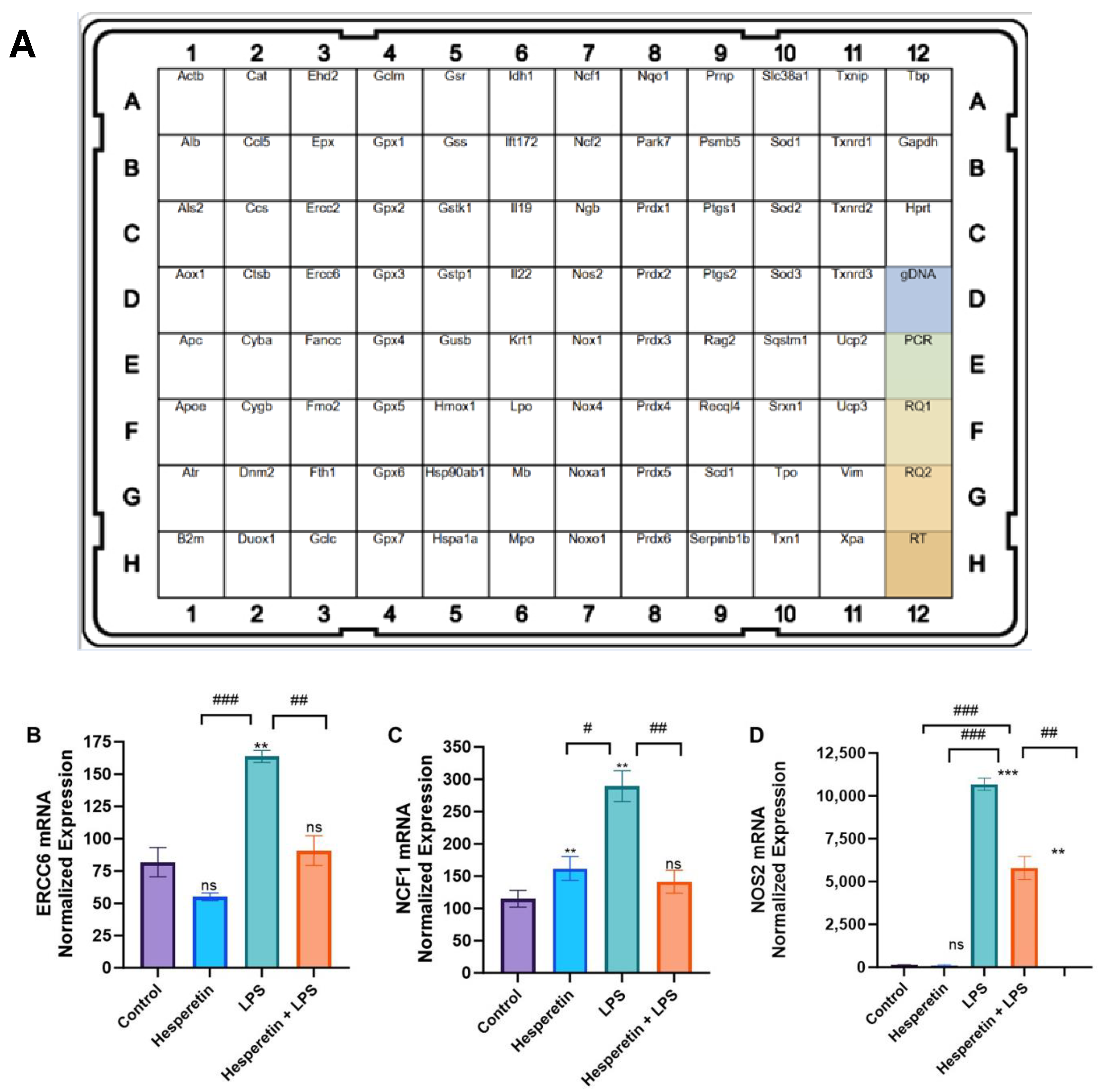
2.6. Real-Time Polymerase Chain Reaction (RT-PCR) Arrays
2.7. Real-Time Polymerase Chain Reaction (RT-PCR) with Individual Primers
2.8. Capillary Western Analysis
2.9. Data Analysis
3. Results
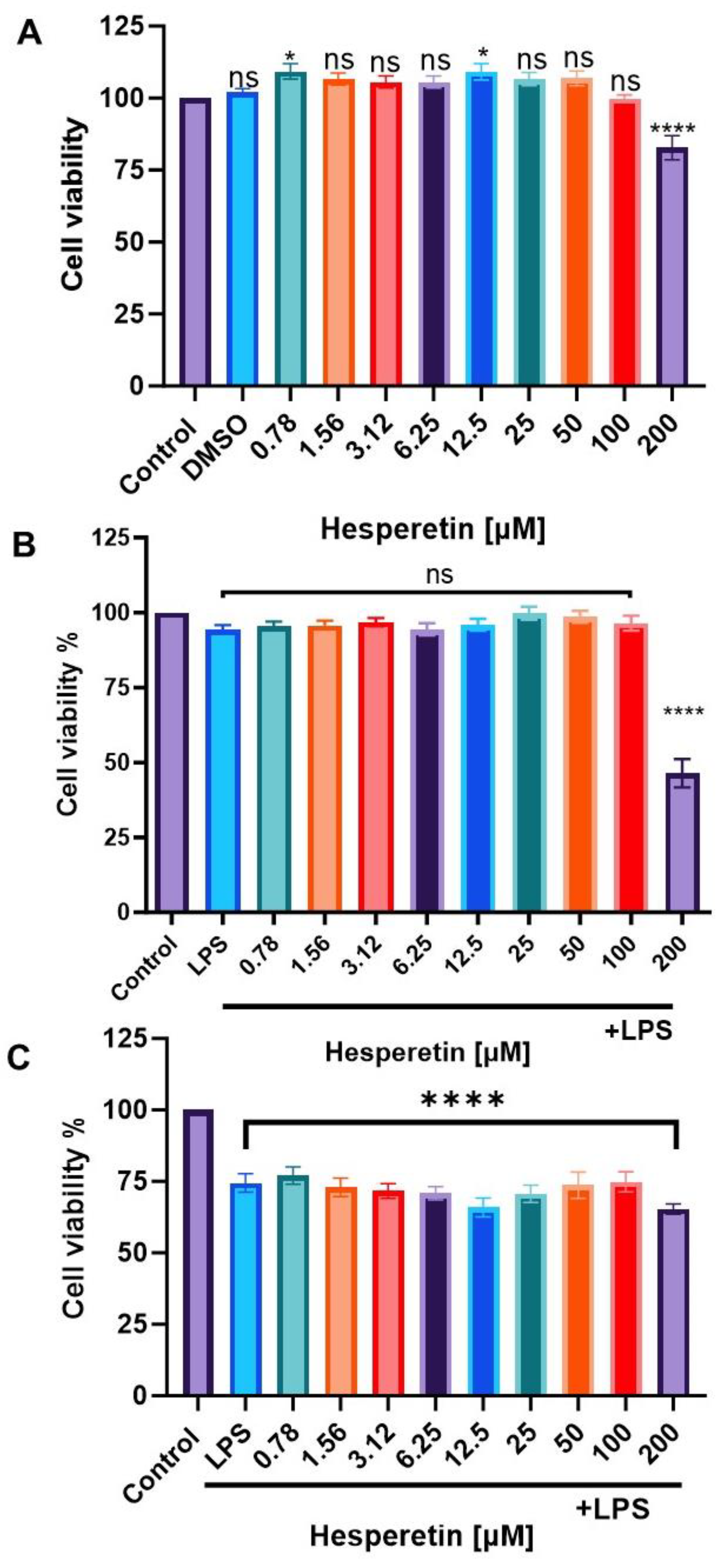
3.1. Effect of Hesperetin and LPS on Cell Viability
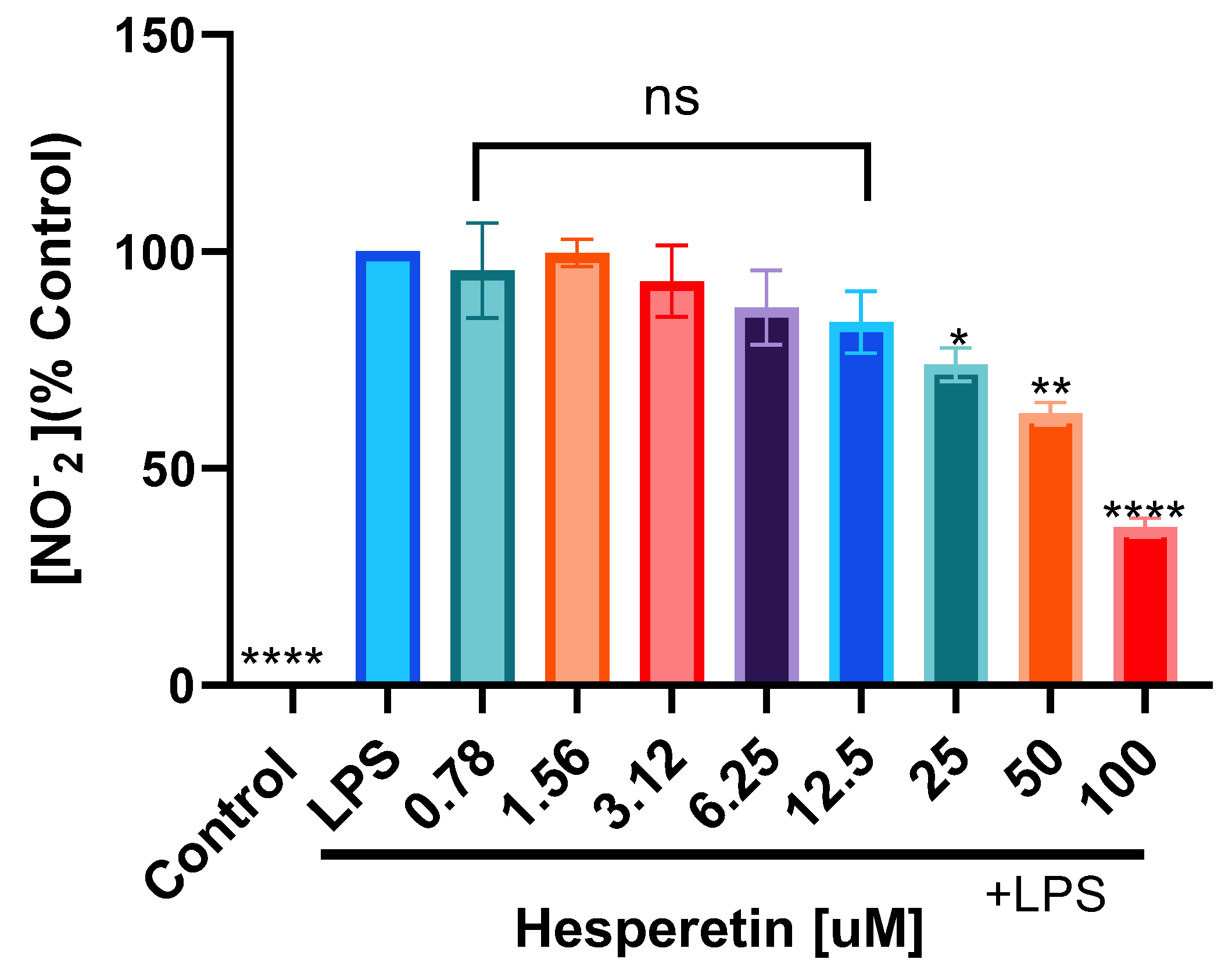
3.2. Hesperetin’s Effect on Nitric Oxide Production
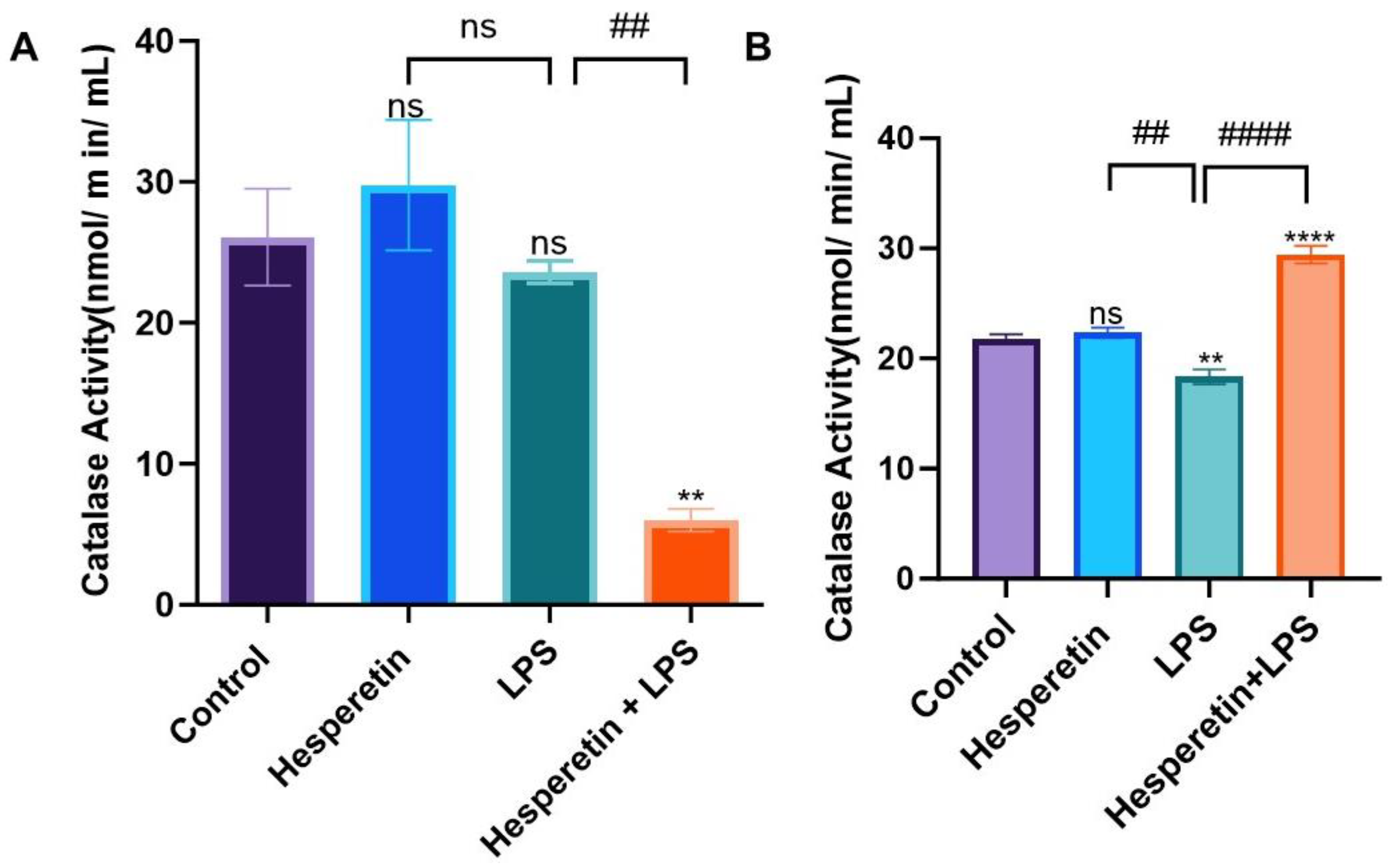
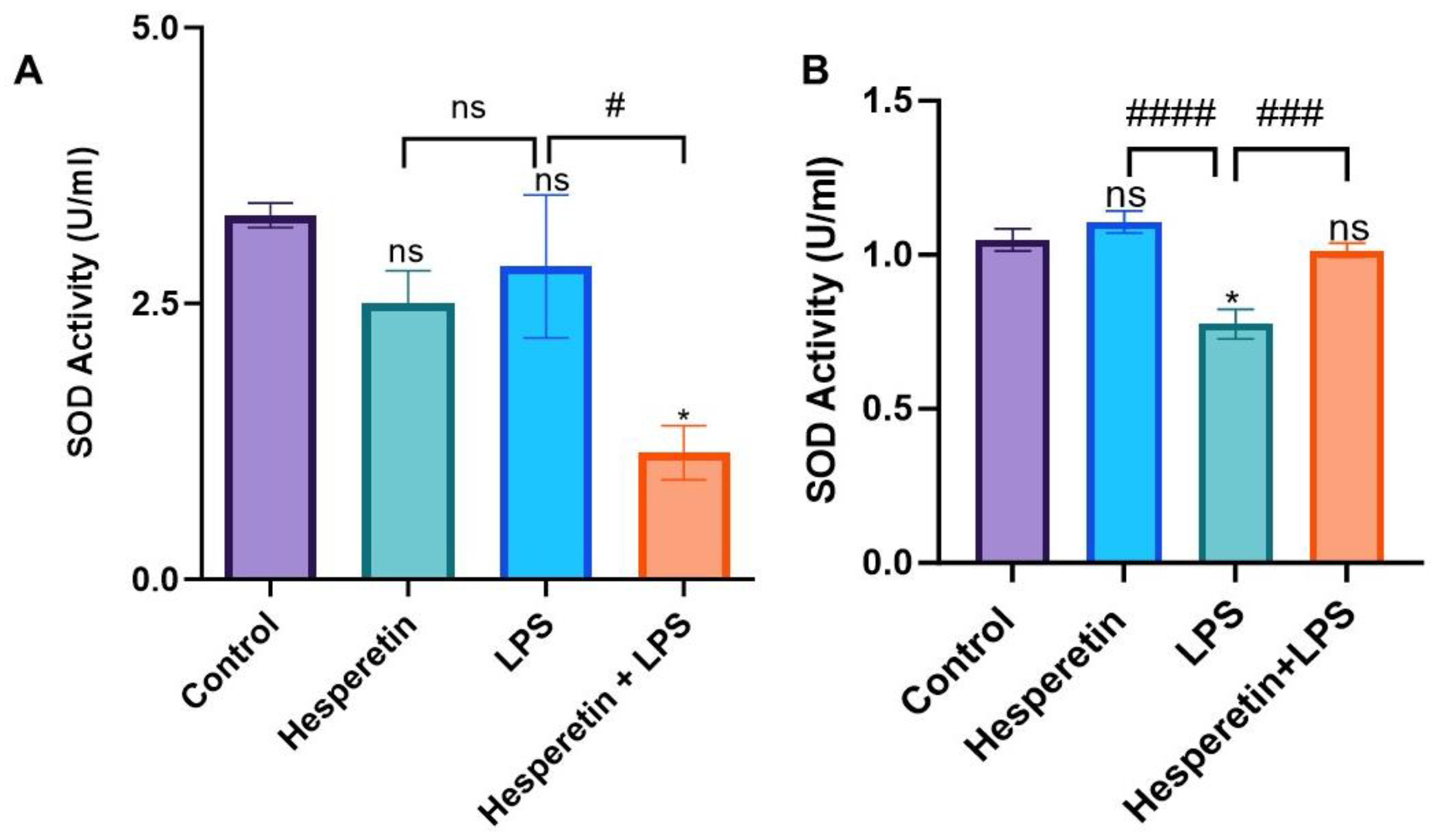
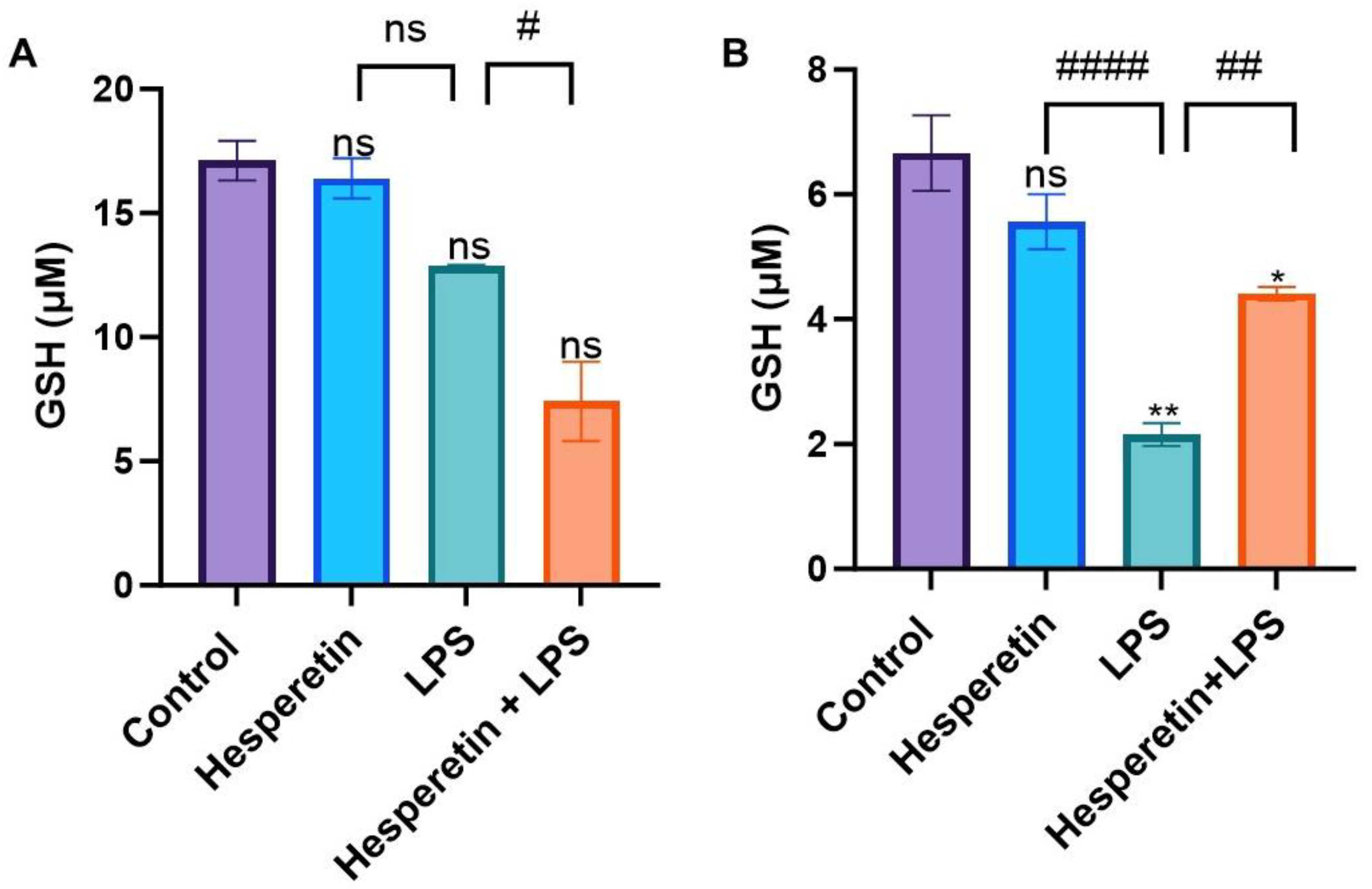
3.3. Hesperetin’s Effect on Antioxidant Mediators
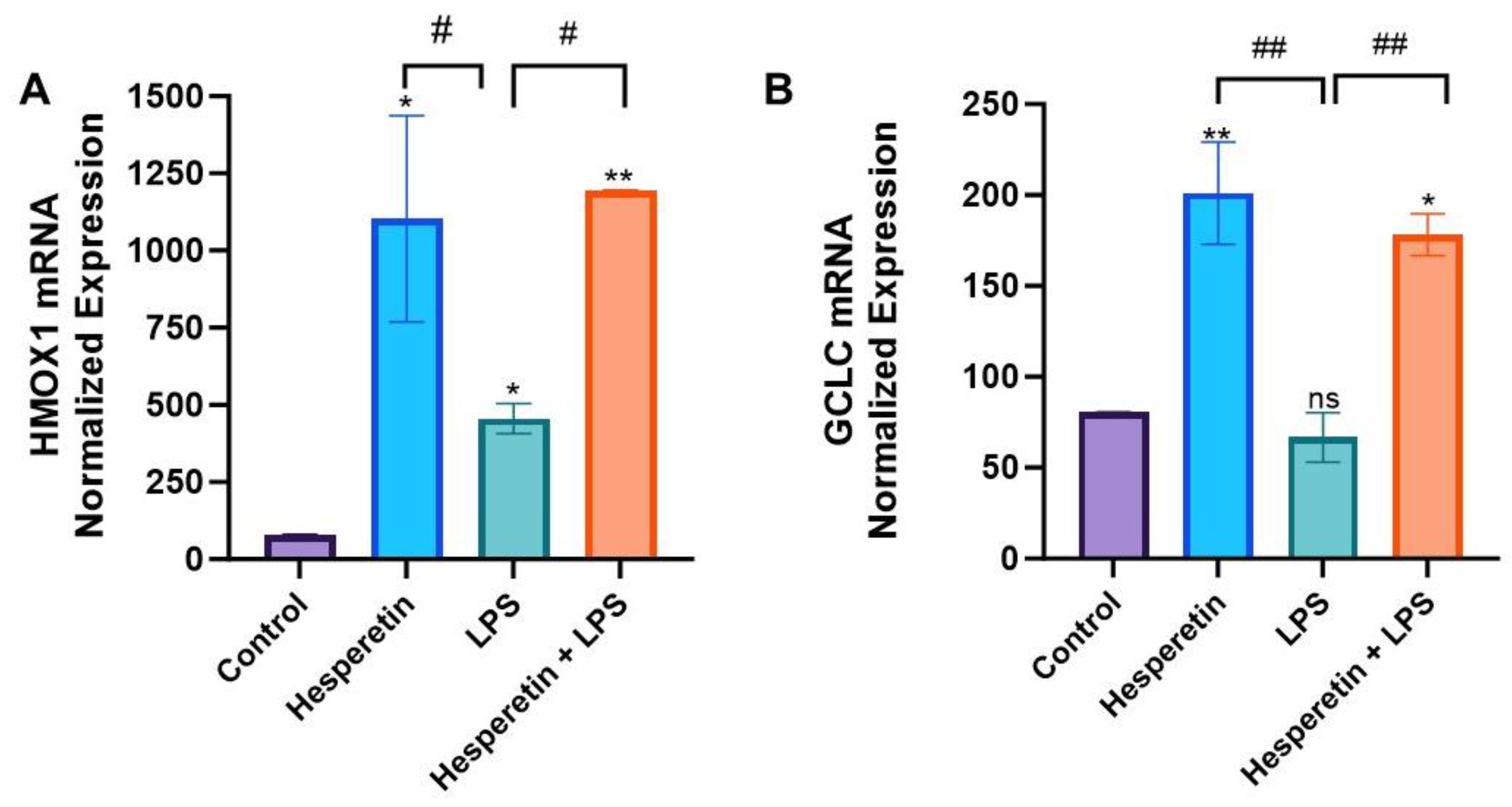
3.4. Hesperetin’s Effects on Oxidative Stress Gene Expression
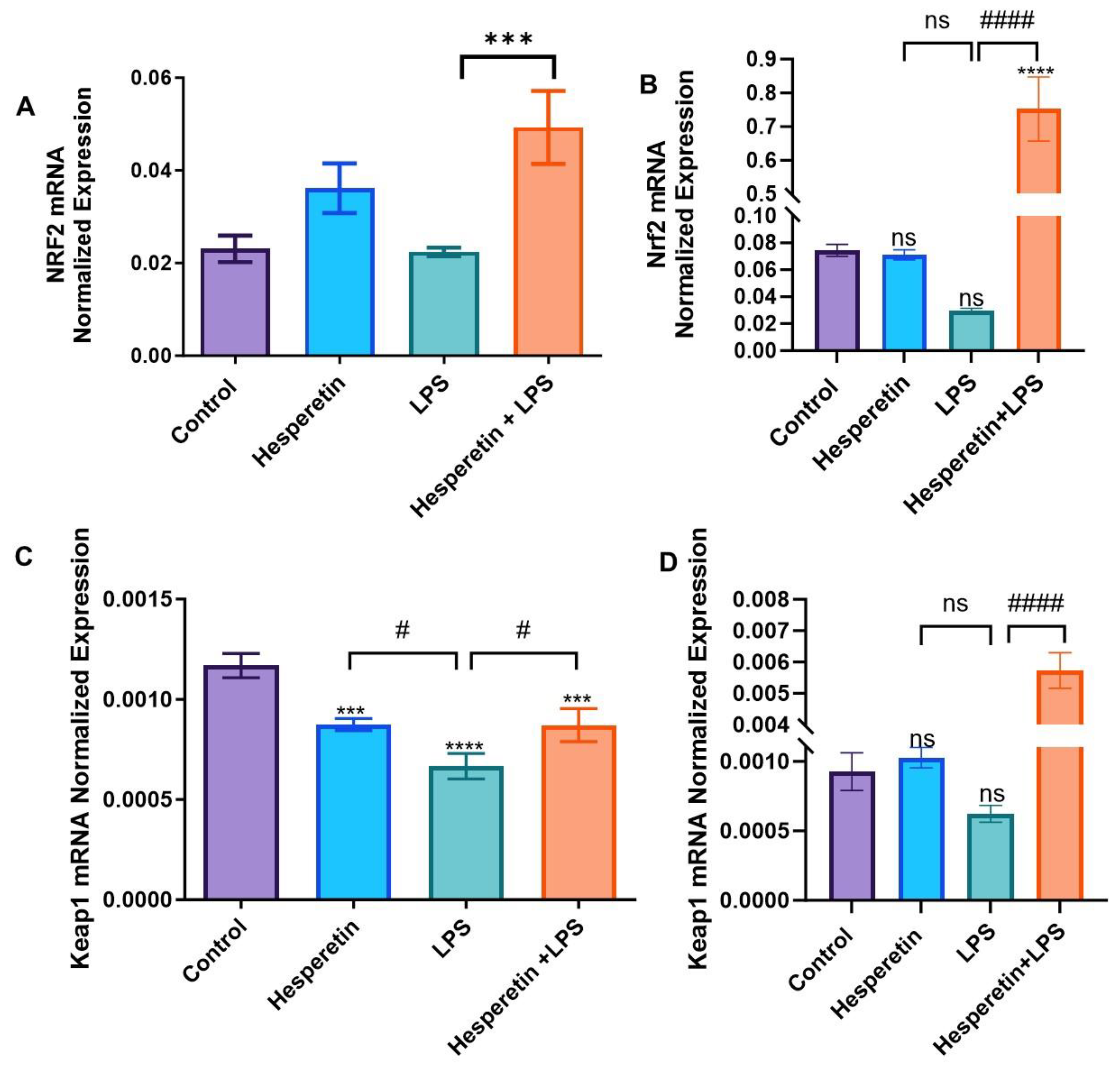
3.5. Hesperetin Modulation of Keap1/Nrf2 Signaling
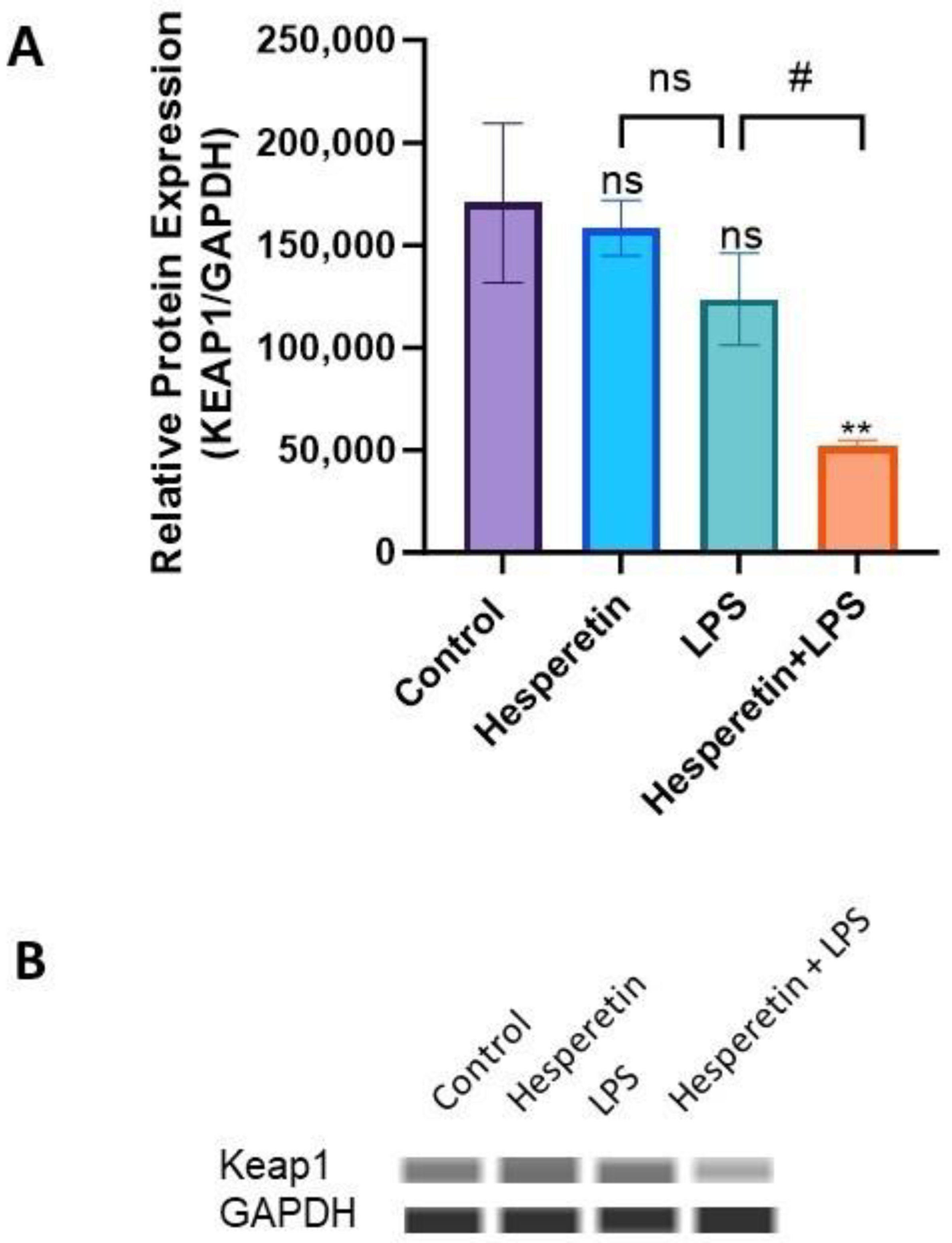
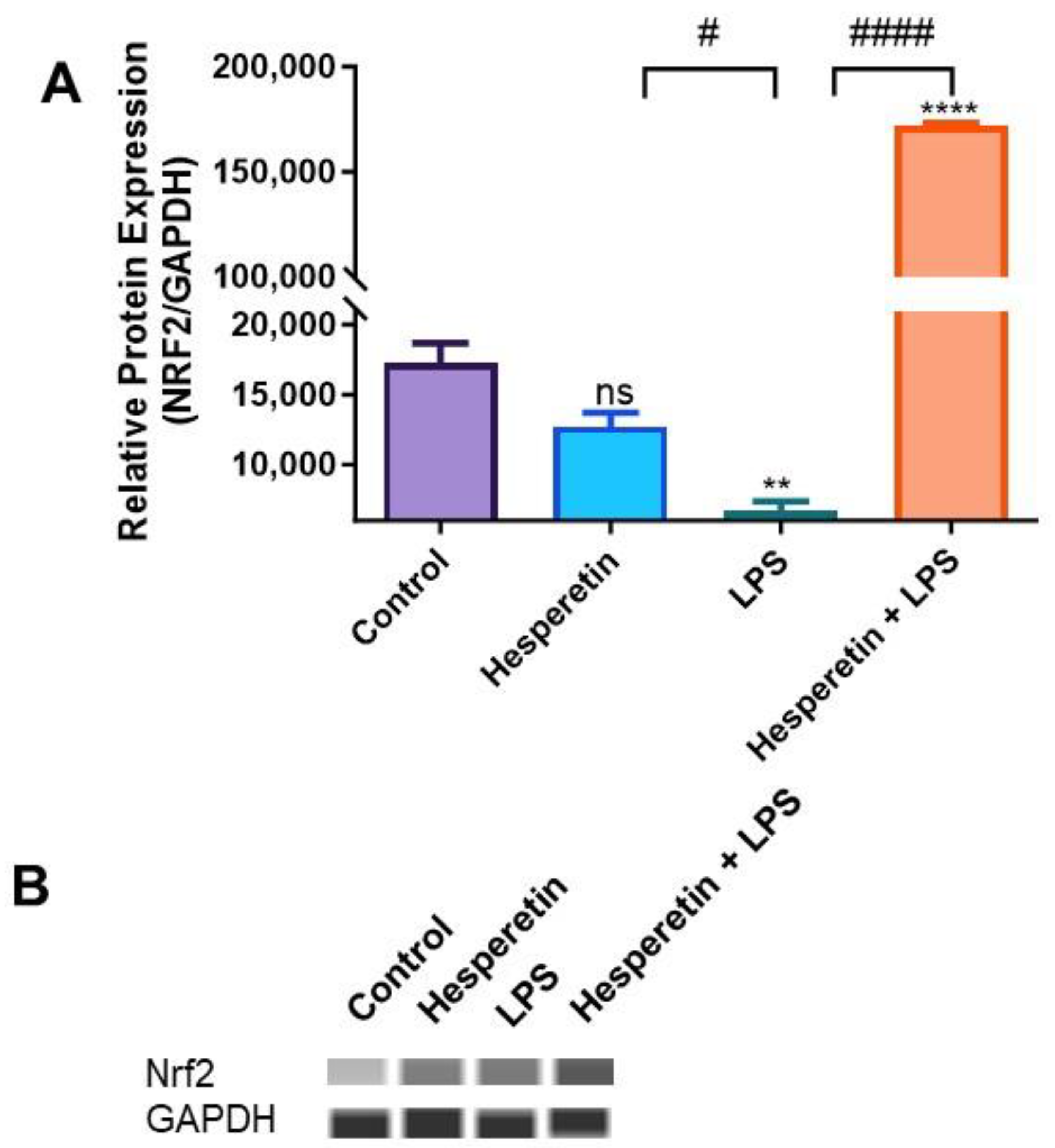
3.6. Hesperetin’s Effect on Protein Expression of Keap1 and Nrf2
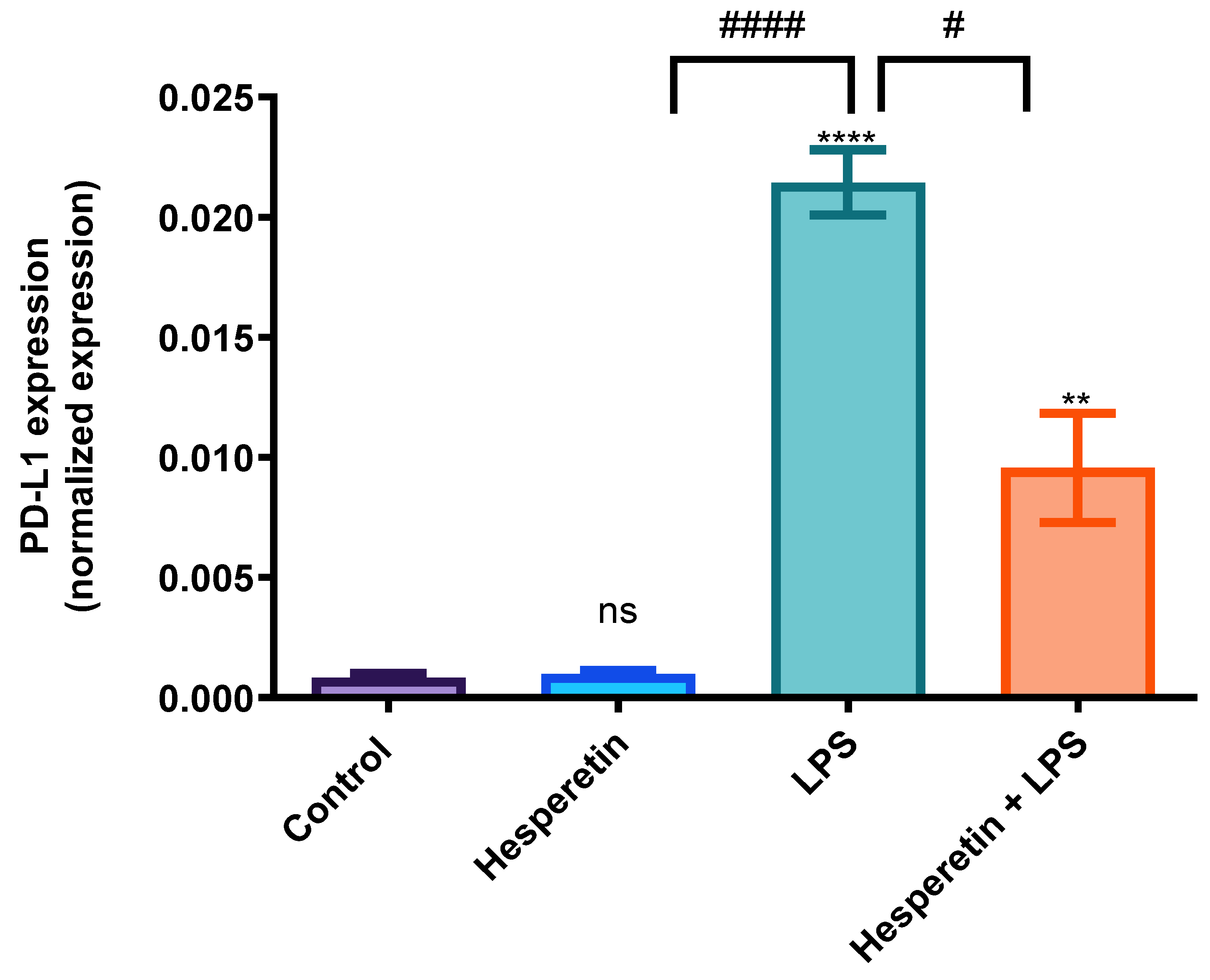
3.7. Hesperetin’s Suppression of PD-L1 Gene
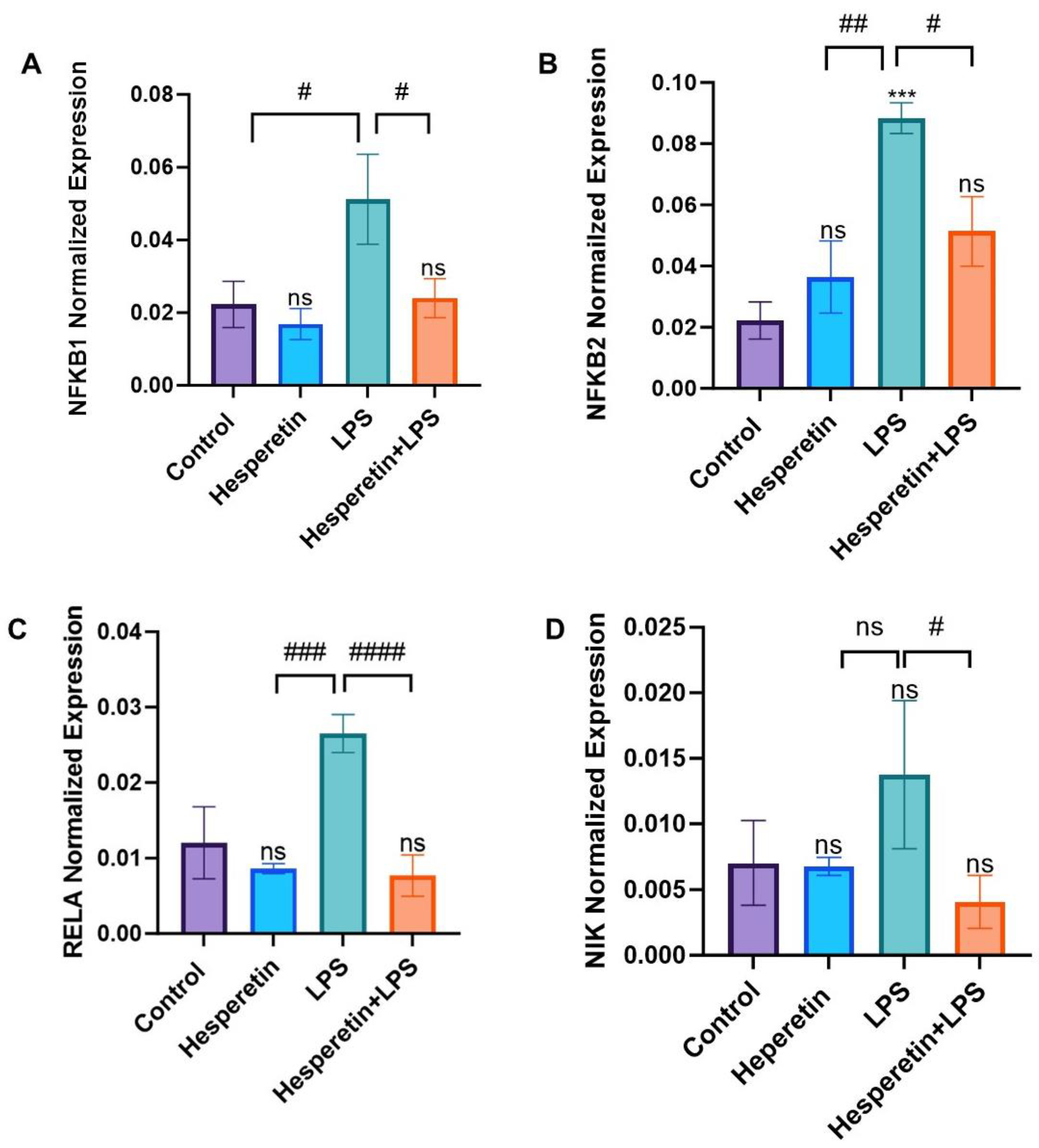
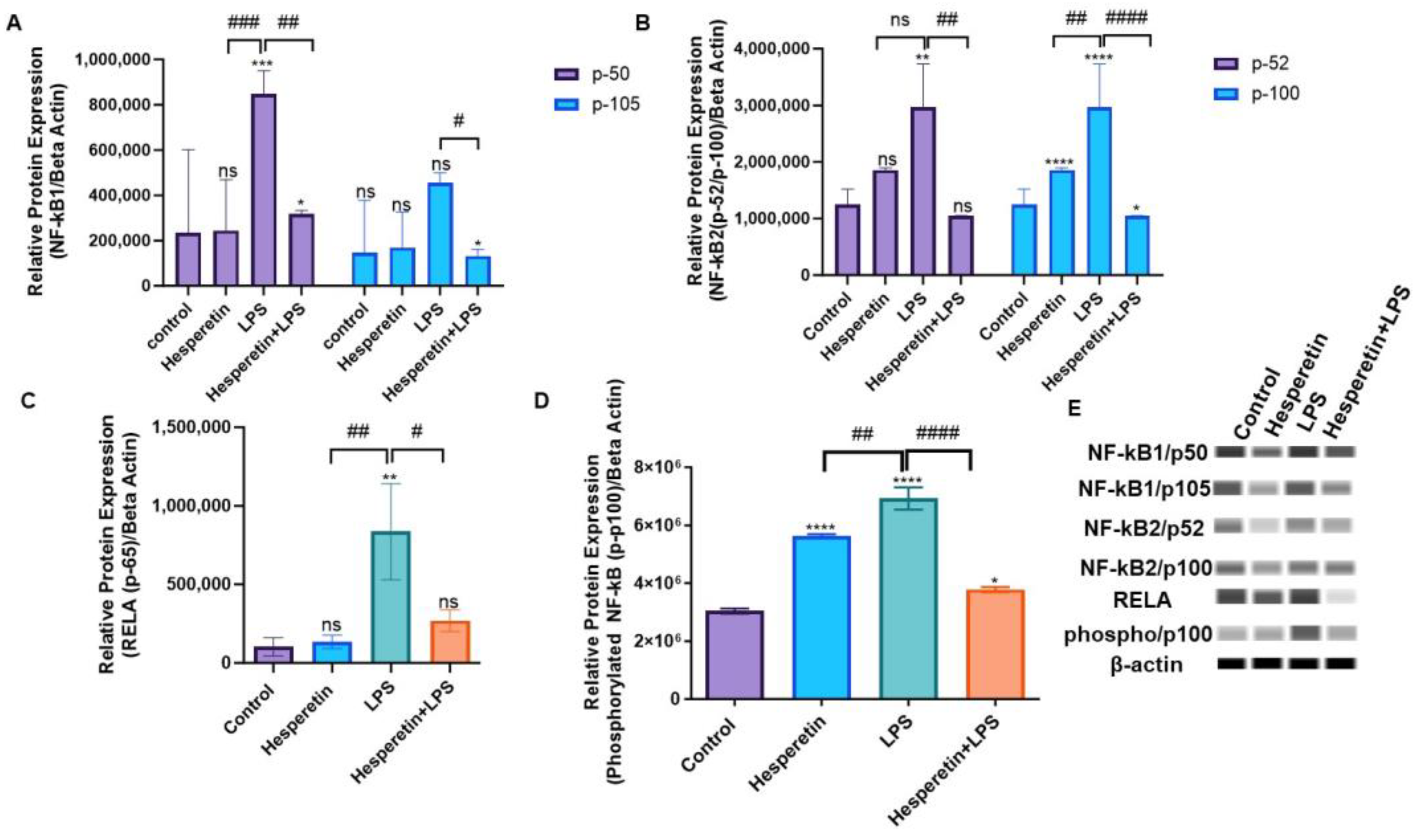
3.8. Hesperetin’s Effects on NF-kB Signaling-Associated Genes and Proteins
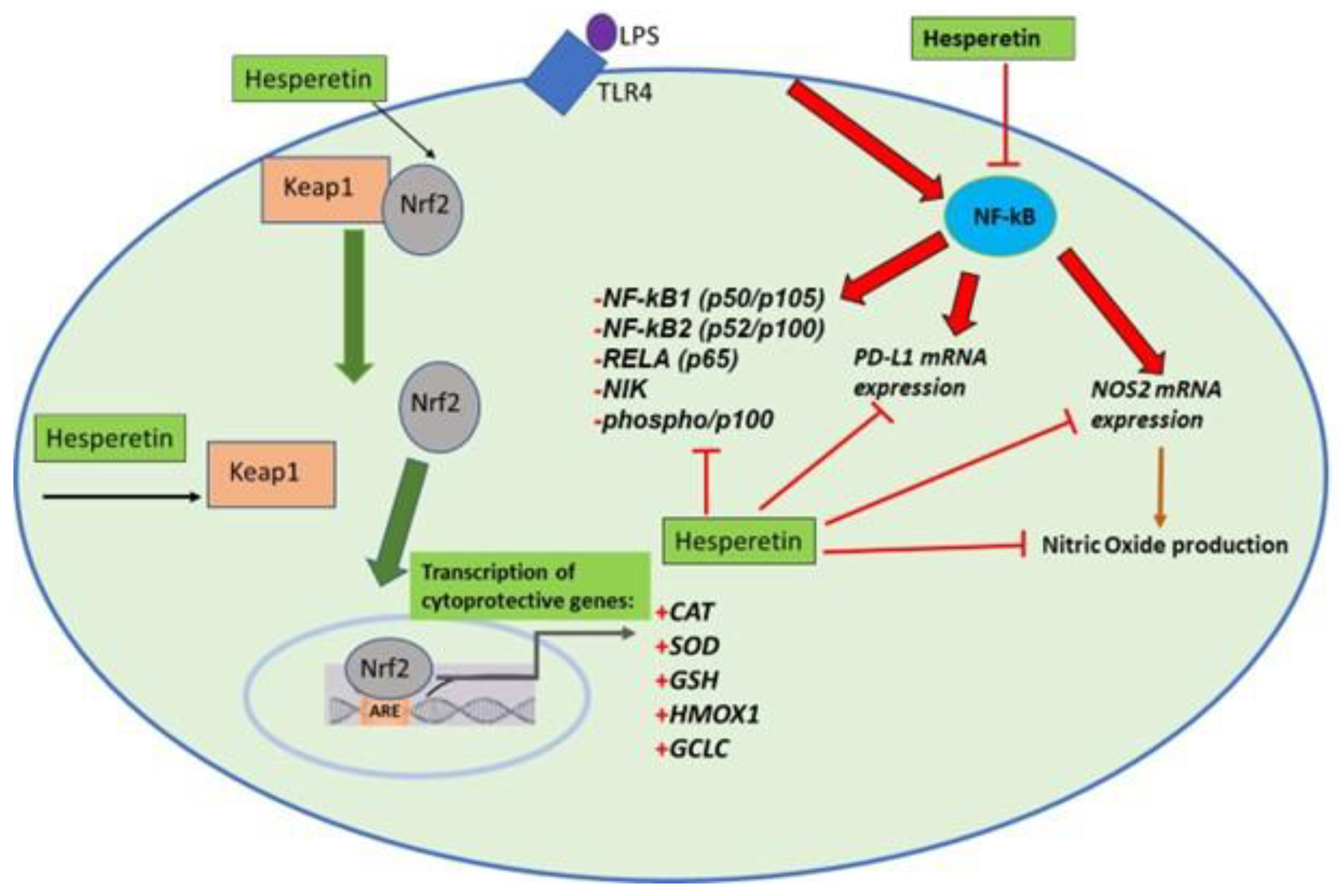
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schetters, S.T.T.; Gomez-Nicola, D.; Garcia-Vallejo, J.J.; Van Kooyk, Y. Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front. Immunol. 2018, 8, 1905. [Google Scholar] [CrossRef]
- Khan, A.; Ikram, M.; Hahm, J.R.; Kim, M.O. Antioxidant and Anti-Inflammatory Effects of Citrus Flavonoid Hesperetin: Special Focus on Neurological Disorders. Antioxidants 2020, 9, 609. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O. Hesperetin, a Citrus Flavonoid, Attenuates LPS-Induced Neuroinflammation, Apoptosis and Memory Impairments by Modulating TLR4/NF-κB Signaling. Nutrients 2019, 11, 648. [Google Scholar] [CrossRef] [PubMed]
- Carmona, J.J.; Michan, S. Biology of Healthy Aging and Longevity. Rev. Investig. Clin. 2016, 68, 7–16. [Google Scholar]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4+T Cells in Neurodegenerative Diseases. Front. Cell. Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Hong, J.-S. Role of Microglia in Inflammation-Mediated Neurodegenerative Diseases: Mechanisms and Strategies for Therapeutic Intervention. Experiment 2003, 304, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Boje, K.M.; Arora, P.K. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992, 587, 250–256. [Google Scholar] [CrossRef]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.-I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and Electrophilic Stresses Activate Nrf2 through Inhibition of Ubiquitination Activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef]
- Joshi, G.; Johnson, J.A. The Nrf2-ARE Pathway: A Valuable Therapeutic Target for the Treatment of Neurodegenerative Diseases. Recent Pat. CNS Drug Discov. 2012, 7, 218–229. [Google Scholar] [CrossRef]
- Evans, J.A.; Mendonca; Soliman, K.F.A. Neuroprotective Effects and Therapeutic Potential of the Citrus Flavonoid Hes-peretin in Neurodegenerative Diseases. Nutrients 2022, 14, 2228. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kensler, T.W. The Role of Keap1 in Cellular Protective Responses. Chem. Res. Toxicol. 2005, 18, 1779–1791. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary me-tabolism. Trends. Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.-C.; Li, X.; Henzl, M.T.; Beamer, L.J.; Hannink, M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. [Google Scholar] [CrossRef]
- Azam, S.; Haque, E.; Balakrishnan, R.; Kim, I.-S.; Choi, D.-K. The Ageing Brain: Molecular and Cellular Basis of Neurodegeneration. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Davies, K.J.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free. Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef]
- Schmidlin, C.J.; Dodson, M.B.; Madhavan, L.; Zhang, D.D. Redox regulation by NRF2 in aging and disease. Free. Radic. Biol. Med. 2019, 134, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Clarke, J.R.; Bomfim, T.R.; De Felice, F.G. Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer’s disease. Alzheimer’s Dement 2014, 10 (Suppl. S1), S76–S83. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P. Neurovascular dysfunction, inflammation, and endothelial activation: Implications for the pathogenesis of Alz-heimer’s disease. J. Neuroinflamm. 2011, 8, 26. [Google Scholar] [CrossRef]
- Ren, X.; Akiyoshi, K.; Vandenbark, A.A.; Hurn, P.D.; Offner, H. Programmed Death-1 Pathway Limits Central Nervous System Inflammation and Neurologic Deficits in Murine Experimental Stroke. Stroke 2011, 42, 2578–2583. [Google Scholar] [CrossRef]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyro-sine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef]
- Kummer, M.P.; Ising, C.; Kummer, C.; Sarlus, H.; Griep, A.; Vieira-Saecker, A.; Schwartz, S.; Halle, A.; Brückner, M.; Händler, K.; et al. Microglial PD-1 stimulation by astrocytic PD-L1 suppresses neuroinflammation and Alzheimer’s disease pathology. Embo J. 2021, 40, e108662. [Google Scholar] [CrossRef]
- Hayden, M.S.; West, A.P.; Ghosh, S. NF-kappaB, and the immune response. Oncogene 2006, 25, 6758–6780. [Google Scholar] [CrossRef]
- Sun, S.C.; Chang, J.H.; Jin, J. Regulation of nuclear factor-κB in autoimmunity. Trends Immunol. 2013, 34, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef]
- de Andrade Teles, R.B.; Diniz, T.C.; Costa Pinto, T.C.; de Oliveira Junior, R.G.; Gama, E.S.M.; de Lavor, E.M.; Fernandes, A.W.C.; de Oliveira, A.P.; de Almeida Ribeiro, F.P.R.; da Silva, A.A.M.; et al. Flavonoids as Therapeutic Agents in Alzheimer’s and Parkinson’s Diseases: A Systematic Review of Preclinical Evidences. Oxid. Med. Cell. Longev. 2018, 2018, 7043213. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, G.B.; Smagghe, G.; Grootaert, C.; Zotti, M.; Raes, K.; Camp, J.V. Flavonoid interactions during digestion, absorption, distribution, and metabolism: A sequentials structure–activity/property relationship-based approach in the study of bioavaila-bility and bioactivity. Drug Metab. Rev. 2015, 47, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Parhiz, H.; Roohbakhsh, A.; Soltani, F.; Rezaee, R.; Iranshahi, M. Antioxidant and anti-inflammatory properties of the citrus flavonoid’s hesperidin and hesperetin: An updated review of their molecular mechanisms and experimental models. Phyto-Ther. Res. 2015, 29, 323–331. [Google Scholar] [CrossRef]
- Huang, S.-M.; Tsai, S.-Y.; Lin, J.-A.; Wu, C.-H.; Yen, G.-C. Cytoprotective effects of hesperetin and hesperidin against amyloid β-induced impairment of glucose transport through downregulation of neuronal autophagy. Mol. Nutr. Food Res. 2012, 56, 601–609. [Google Scholar] [CrossRef]
- Choi, E.J. Antioxidative effects of hesperetin against 7,12-dimethylbenz(a)anthracene-induced oxidative stress in mice. Life Sci. 2008, 82, 1059–1064. [Google Scholar] [CrossRef]
- Blasi, E.; Barluzzi, R.; Bocchini, V.; Mazzolla, R.; Bistoni, F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 1990, 27, 229–237. [Google Scholar] [CrossRef]
- Erlund, I.; Meririnne, E.; Alfthan, G.; Aro, A. Plasma Kinetics and Urinary Excretion of the Flavanones Naringenin and Hesperetin in Humans after Ingestion of Orange Juice and Grapefruit Juice. J. Nutr. 2001, 131, 235–241. [Google Scholar] [CrossRef]
- Kwon, J.Y.; Jung, U.J.; Kim, D.W.; Kim, S.; Moon, G.J.; Hong, J.; Jeon, M.-T.; Shin, M.; Chang, J.H.; Kim, S.R. Beneficial Effects of Hesperetin in a Mouse Model of Temporal Lobe Epilepsy. J. Med. Food 2018, 21, 1306–1309. [Google Scholar] [CrossRef]
- Ikram, M.; Muhammad, T.; Rehman, S.U.; Khan, A.; Jo, M.G.; Ali, T.; Kim, M.O. Hesperetin Confers Neuroprotection by Regulating Nrf2/TLR4/NF-κB Signaling in an Aβ Mouse Model. Mol. Neurobiol. 2019, 56, 6293–6309. [Google Scholar] [CrossRef]
- Cho, J. Antioxidant and neuroprotective effects of hesperidin and its aglycone hesperetin. Arch. Pharmacal Res. 2006, 29, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Mccord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef] [PubMed]
- Tavassolifar, M.J.; Vodjgani, M.; Salehi, Z.; Izad, M. The Influence of Reactive Oxygen Species in the Immune System and Pathogenesis of Multiple Sclerosis. Autoimmune Dis. 2020, 2020, 5793817. [Google Scholar] [CrossRef]
- Day, B.J. Catalase and glutathione peroxidase mimics. Biochem. Pharmacol. 2009, 77, 285–296. [Google Scholar] [CrossRef]
- Feng, W.; Rosca, M.; Fan, Y.; Hu, Y.; Feng, P.; Lee, H.-G.; Monnier, V.M.; Fan, X. Gclc deficiency in mouse CNS causes mitochondrial damage and neurodegeneration. Hum. Mol. Genet. 2017, 26, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.X.; Iqbal, K. Hyperphosphorylation of microtubule-associated protein tau: A promising therapeutic target for Alz-heimer’s disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef]
- Lee, J.-M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related Factor-2-dependent Genes Conferring Protection against Oxidative Stress in Primary Cortical Astrocytes Using Oligonucleotide Microarray Analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef]
- Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.M.; Pronzato, M.A.; Furfaro, A.L. Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce neurodegeneration? Int. J. Mol. Sci. 2018, 19, 2260. [Google Scholar] [CrossRef]
- Muhammad, T.; Ali, T.; Ikram, M.; Khan, A.; Alam, S.I.; Kim, M.O. Melatonin rescue oxidative stress-mediated neuroin-flammation/neurodegeneration and memory impairment in scopolamine-induced amnesia mice model. J. Neuro-Immune Pharmacol. 2019, 14, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, B.; Tong, K.I.; Kobayashi, A.; Yamamoto, M.; Yokoyama, S. Structural insights into the similar modes of Nrf2 transcription factor recognition by the cytoplasmic repressor Keap1. J. Synchrotron Radiat. 2008, 15, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Magesh, S.; Chen, Y.; Hu, L. Small Molecule Modulators of Keap1-Nrf2-ARE Pathway as Potential Preventive and Therapeutic Agents. Med. Res. Rev. 2012, 32, 687–726. [Google Scholar] [CrossRef]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregu-lation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-Antioxidant Response Element Signaling Pathway and Its Activation by Oxidative Stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V.W. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496. [Google Scholar] [CrossRef] [PubMed]
- Lipp, M.; Brandt, C.; Dehghani, F.; Kwidzinski, E.; Bechmann, I. PD-L1 (B7-H1) regulation in zones of axonal degeneration. Neurosci. Lett. 2007, 425, 156–161. [Google Scholar] [CrossRef]
- Sun, E.; Motolani, A.; Campos, L.; Lu, T. The Pivotal Role of NF-kB in the Pathogenesis and Therapeutics of Alzheimer&rsquo’s Disease. Int. J. Mol. Sci. 2022, 23, 8972. [Google Scholar]
- Pan, H.; Wang, H.; Wang, X.; Zhu, L.; Mao, L. The absence of Nrf2 enhances NF-κB-dependent inflammation following scratch injury in mouse primary cultured astrocytes. Mediat. Inflamm. 2012, 2012, 217580. [Google Scholar] [CrossRef]
- Henn, A.; Lund, S.; Hedtjärn, M.; Schrattenholz, A.; Pörzgen, P.; Leist, M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 2009, 26, 83–94. [Google Scholar] [CrossRef]
- Horvath, R.J.; Nutile-McMenemy, N.; Alkaitis, M.S.; DeLeo, J.A. Differential migration, LPS-induced cytokine, chemokine, and NO expression in immortalized BV-2 and HAPI cell lines and primary microglial cultures. J. Neurochem. 2008, 107, 557–569. [Google Scholar] [CrossRef] [PubMed]















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evans, J.A.; Mendonca, P.; Soliman, K.F.A. Involvement of Nrf2 Activation and NF-kB Pathway Inhibition in the Antioxidant and Anti-Inflammatory Effects of Hesperetin in Activated BV-2 Microglial Cells. Brain Sci. 2023, 13, 1144. https://doi.org/10.3390/brainsci13081144
Evans JA, Mendonca P, Soliman KFA. Involvement of Nrf2 Activation and NF-kB Pathway Inhibition in the Antioxidant and Anti-Inflammatory Effects of Hesperetin in Activated BV-2 Microglial Cells. Brain Sciences. 2023; 13(8):1144. https://doi.org/10.3390/brainsci13081144
Chicago/Turabian StyleEvans, Jasmine A., Patricia Mendonca, and Karam F. A. Soliman. 2023. "Involvement of Nrf2 Activation and NF-kB Pathway Inhibition in the Antioxidant and Anti-Inflammatory Effects of Hesperetin in Activated BV-2 Microglial Cells" Brain Sciences 13, no. 8: 1144. https://doi.org/10.3390/brainsci13081144
APA StyleEvans, J. A., Mendonca, P., & Soliman, K. F. A. (2023). Involvement of Nrf2 Activation and NF-kB Pathway Inhibition in the Antioxidant and Anti-Inflammatory Effects of Hesperetin in Activated BV-2 Microglial Cells. Brain Sciences, 13(8), 1144. https://doi.org/10.3390/brainsci13081144