
Neuroanatomical Alterations in the CNTNAP2 Mouse Model of Autism Spectrum Disorder




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Care and Housing
2.2. Genotyping
2.3. Retrograde Tract-Tracing
2.4. Histology
2.5. Imaging and Quantification
3. Results
3.1. Genotyping
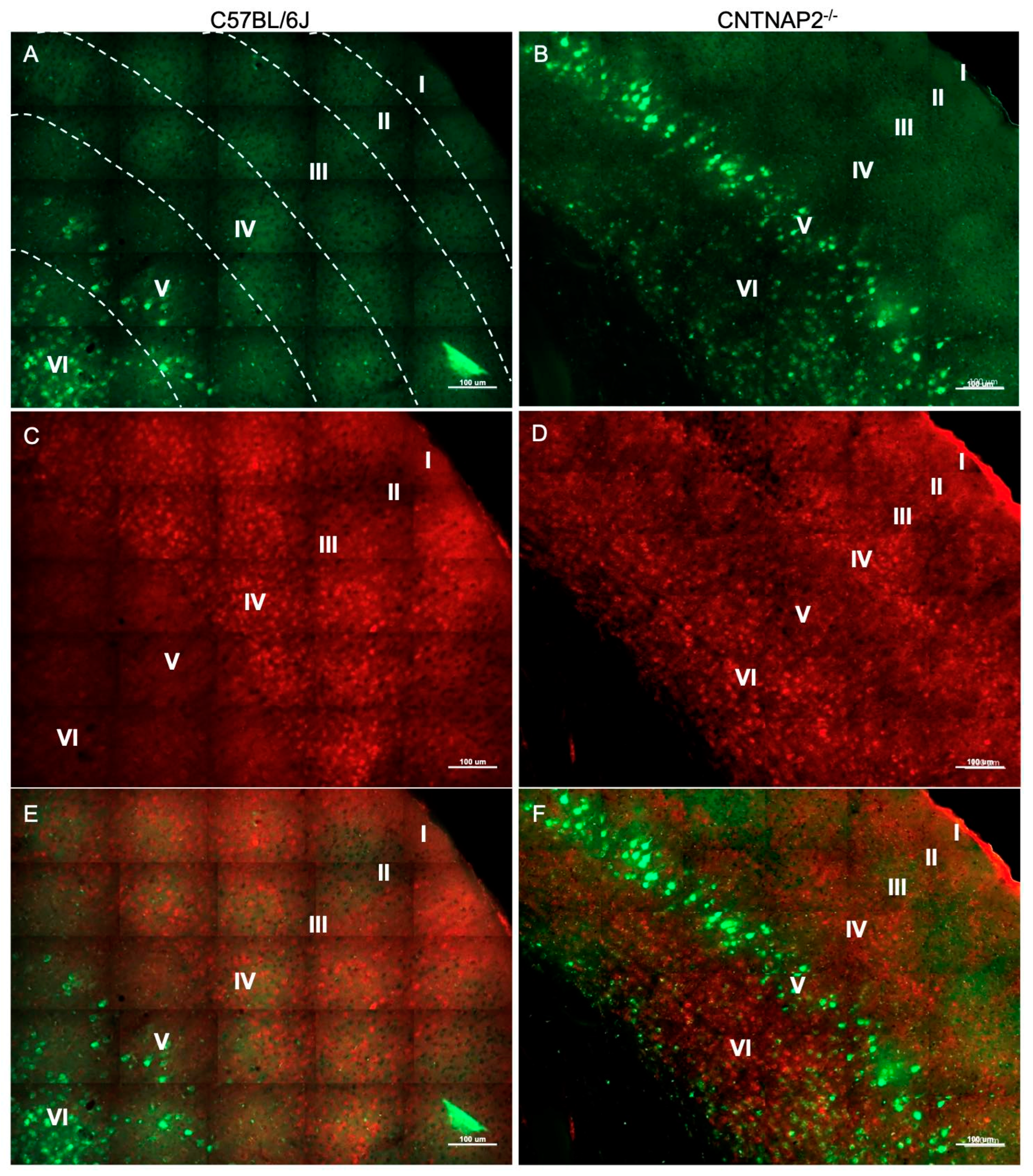
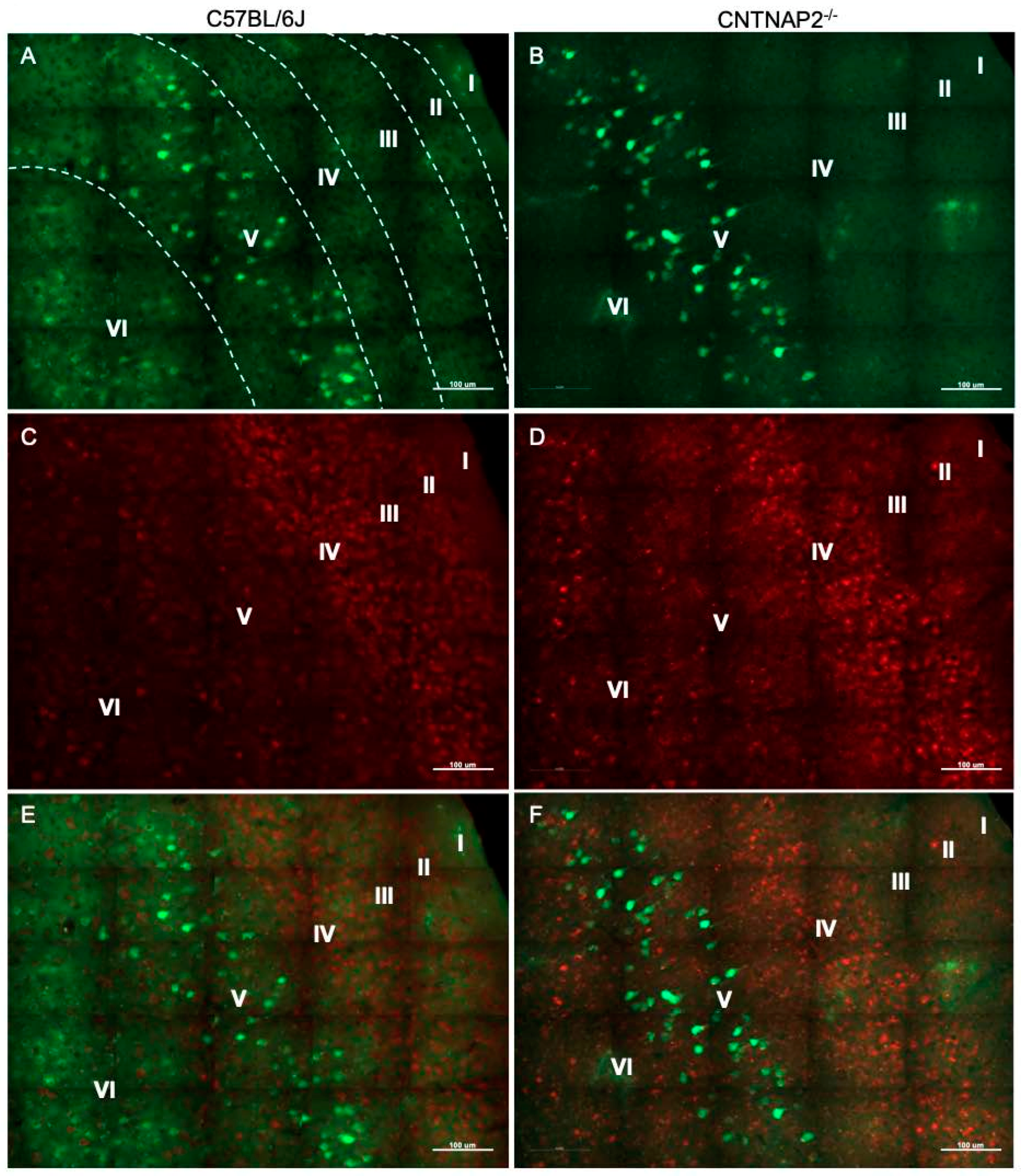
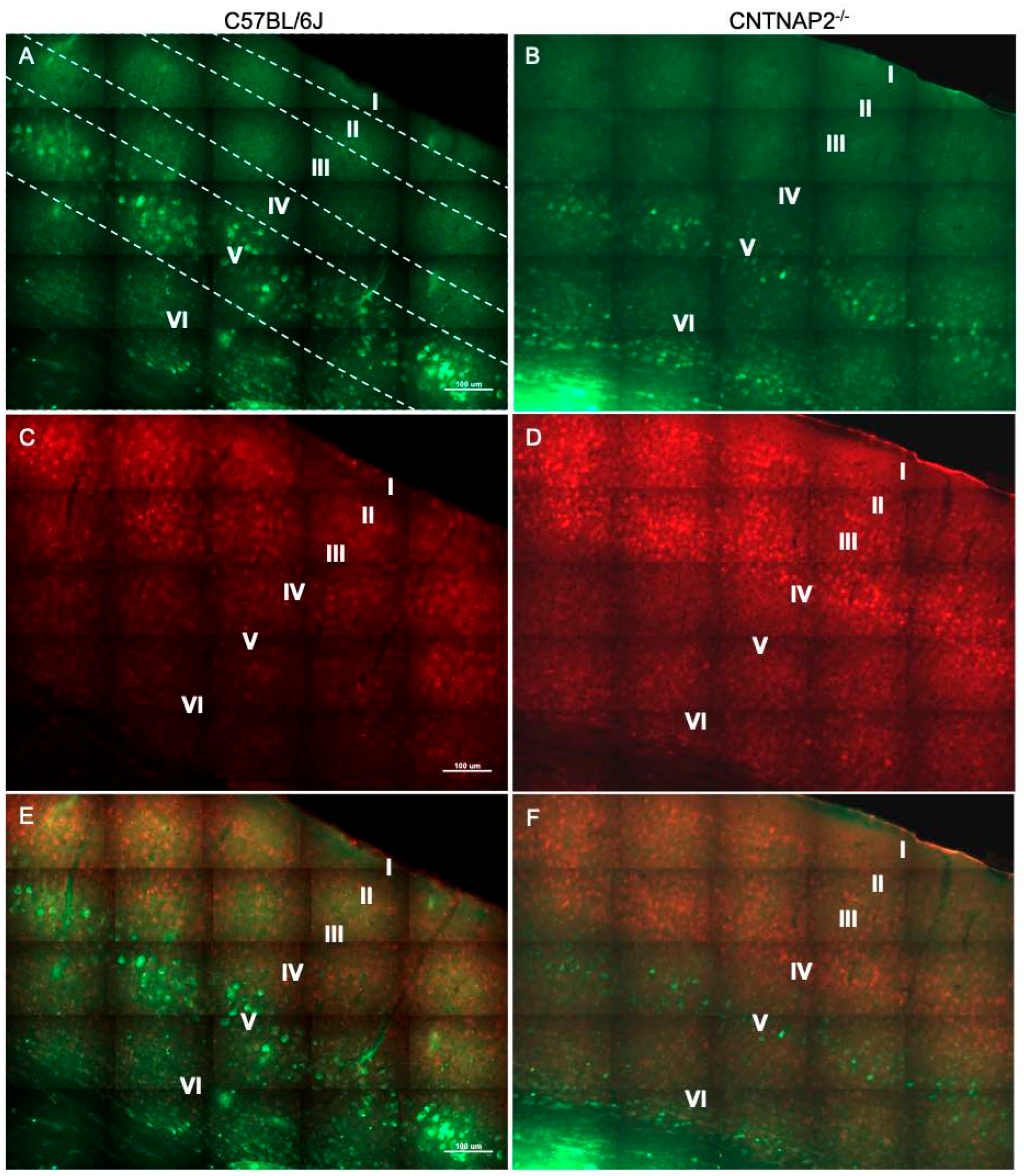
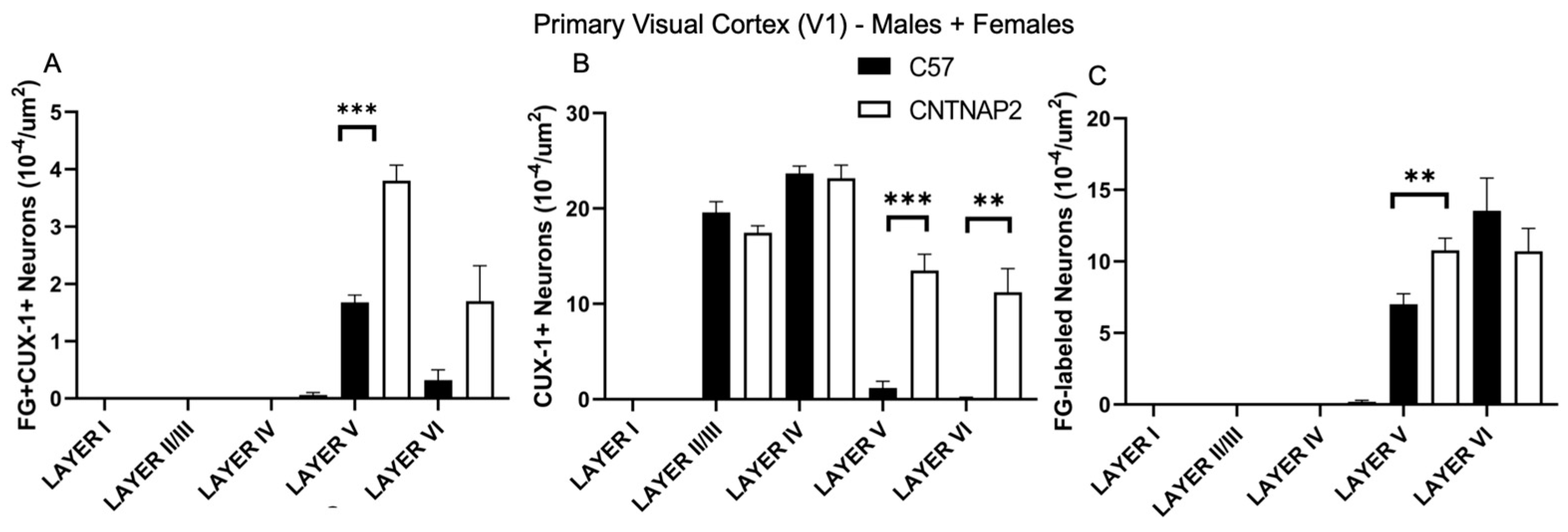
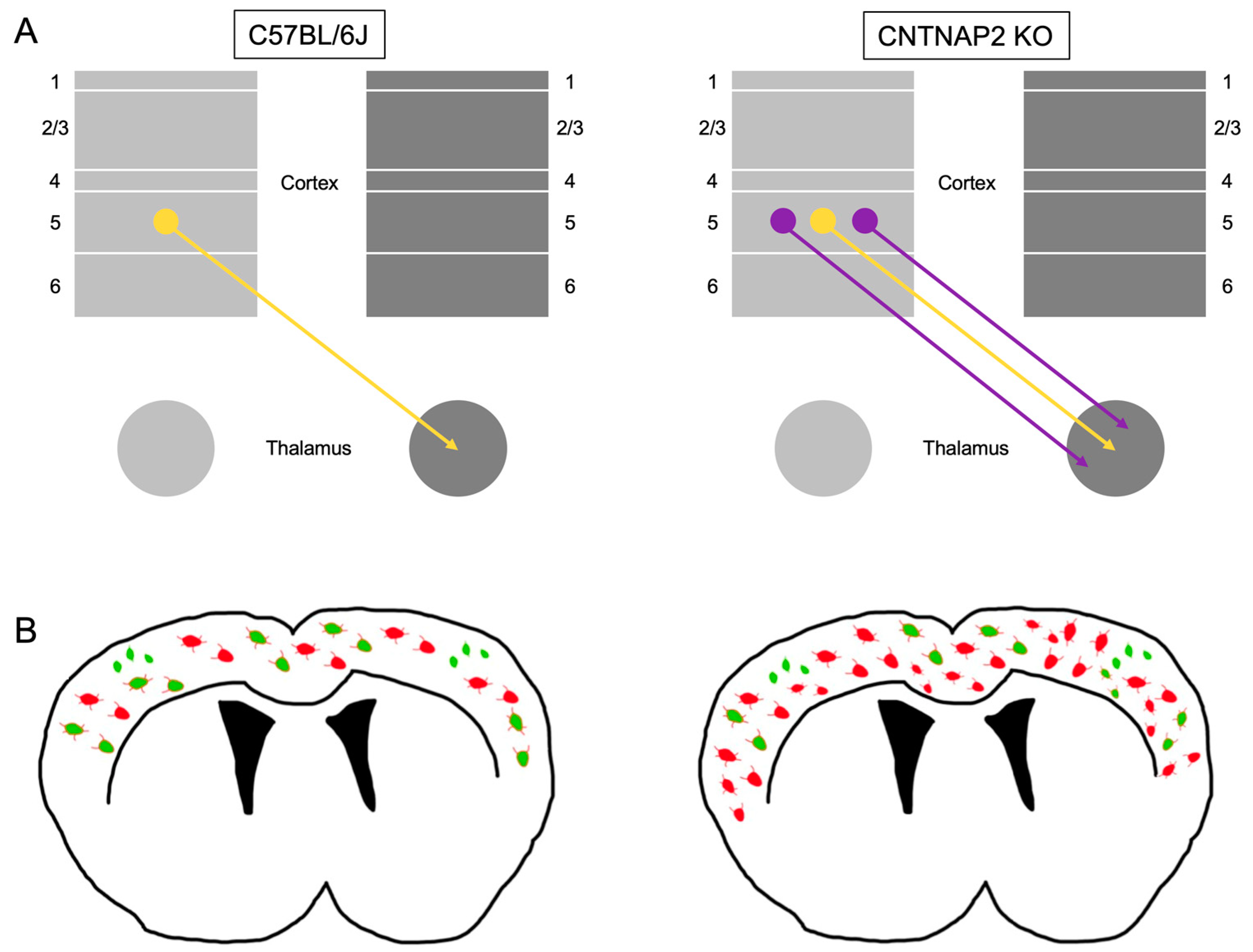
3.2. Ectopic Cortical Neurons Project to Sensory Thalamic Nuclei in CNTNAP2 KO Mice
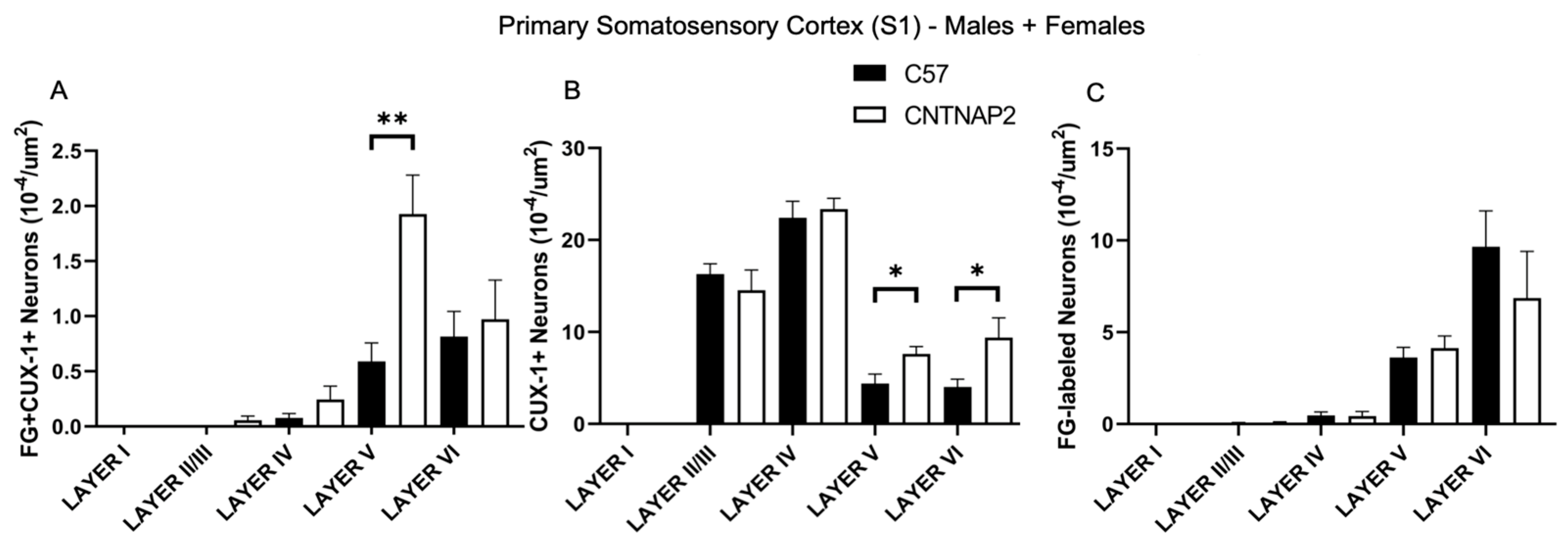
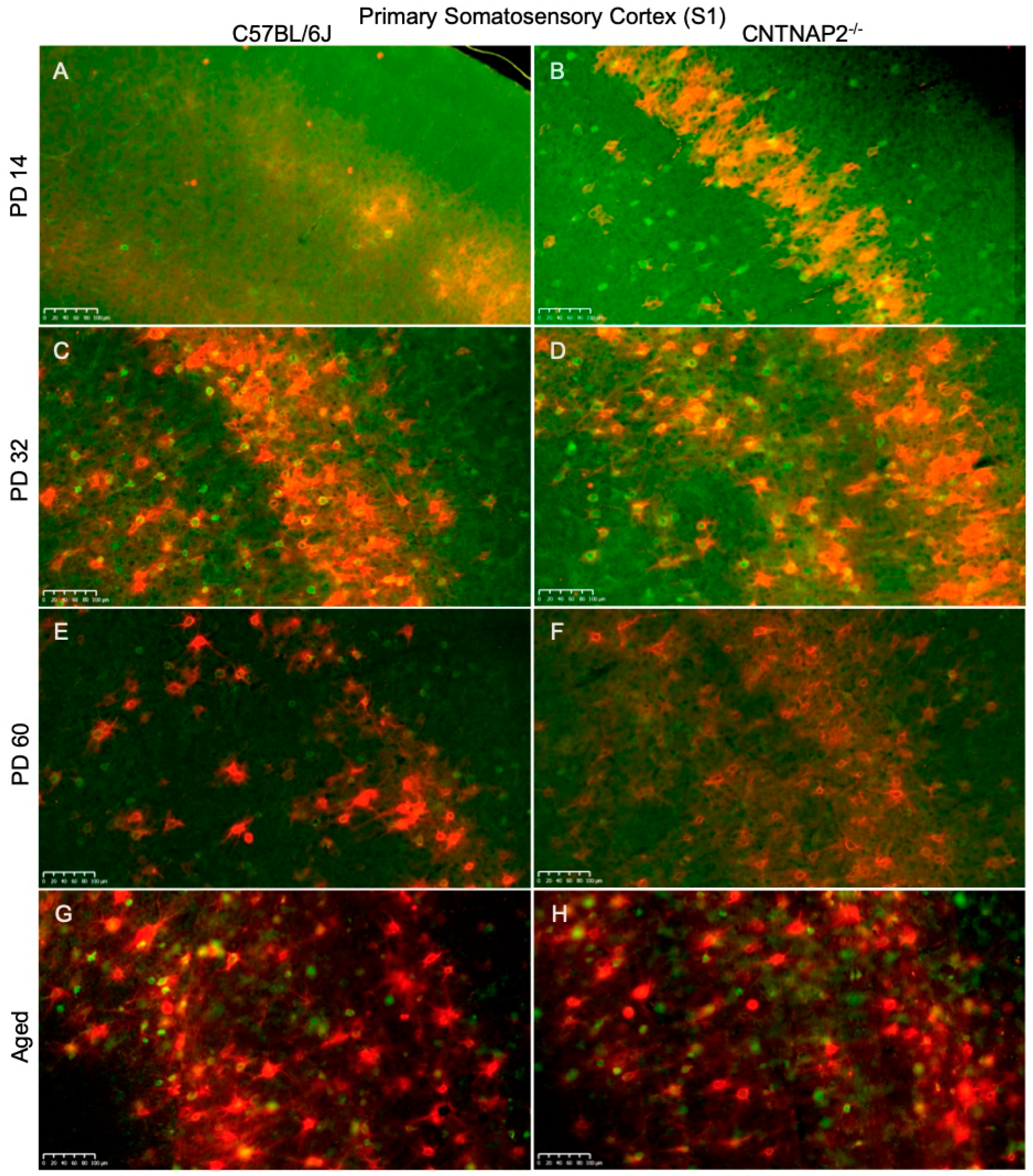
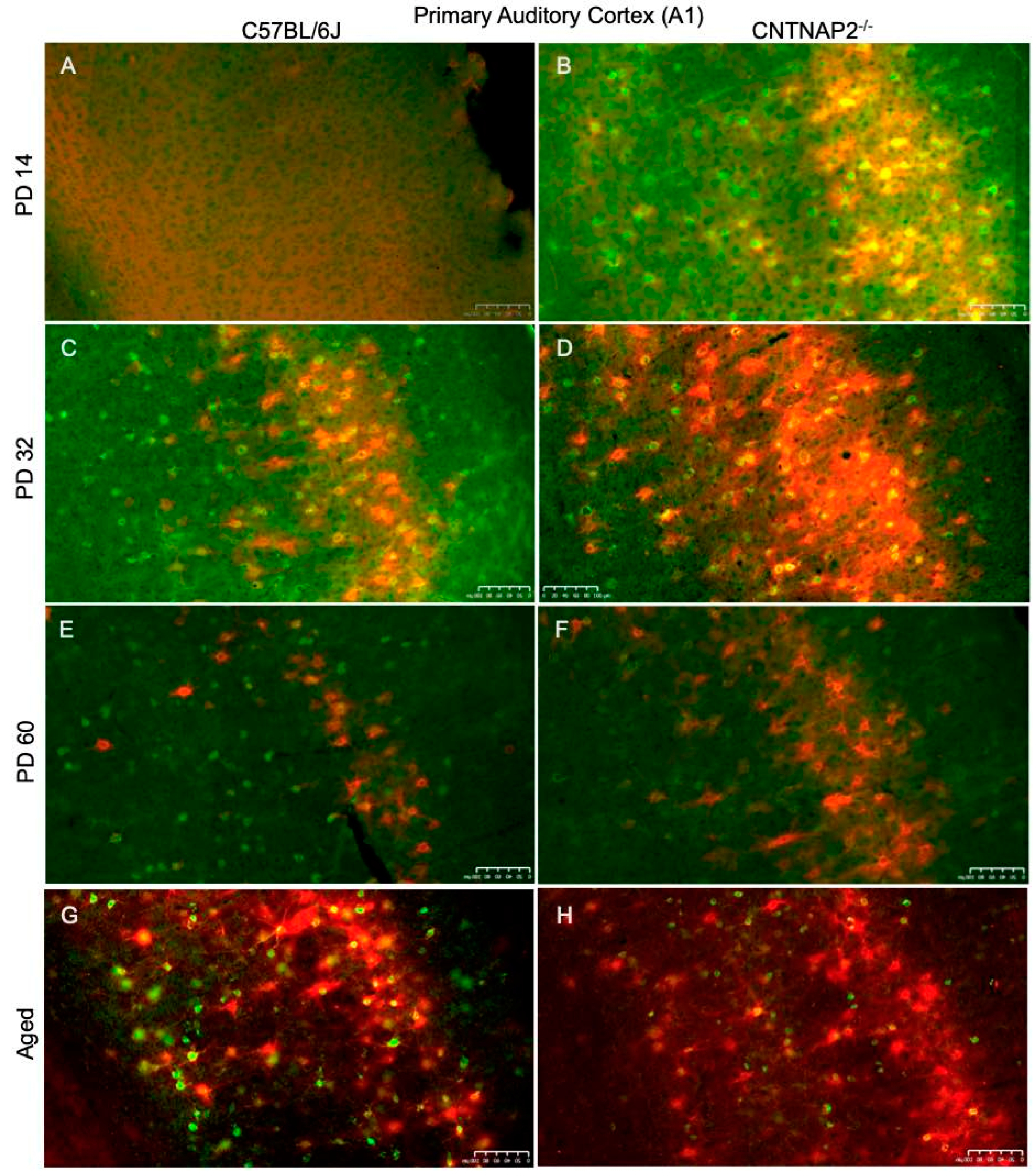
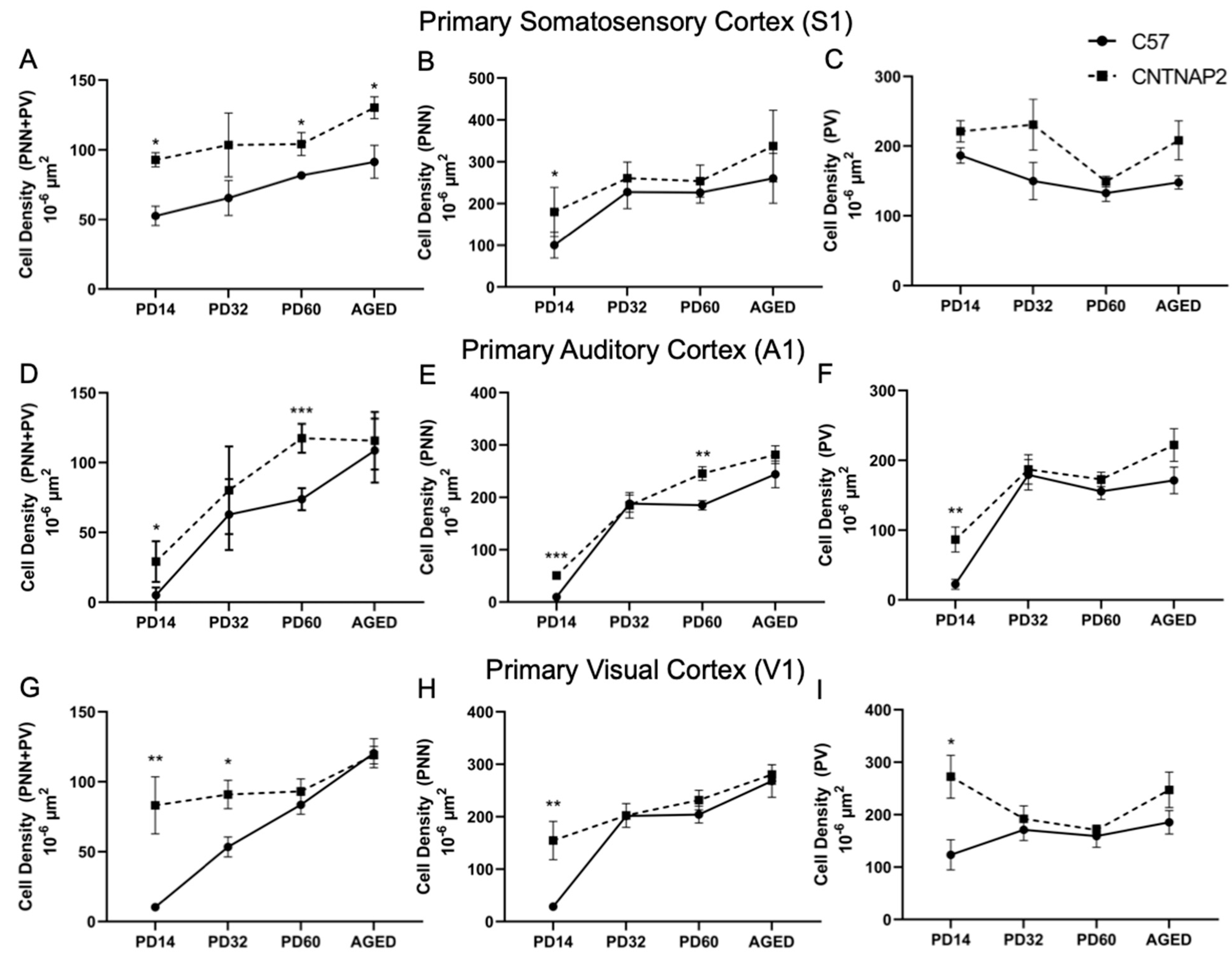
3.3. Perineuronal Nets (PNNs) and Parvalbumin (PV)-Positive Interneurons Exhibit Altered Development with Age in the Primary Sensory Cortical Regions (S1, A1, V1) of CNTNAP2 KO Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kassim, A.; Mohamed, N.H.B. The global prevalence and diagnosis of autism spectrum disorder (ASD) among young children. Southeast Asia Psychol. J. 2019, 7, 26–45. [Google Scholar]
- Lord, C.; Charman, T.; Cusack, J.; Guillaume, D.; Frazier, T.; Jones, R.M.; Pickles, A.; Lounds, T.J.; Jeremy, V.-V. Autism spectrum disorder (Primer). Nat. Rev. Dis. Prim. 2020, 6, 5. [Google Scholar] [CrossRef]
- Zeidan, J.; Fombonne, E.; Scorah, J.; Ibrahim, A.; Durkin, M.S.; Saxena, S.; Yusuf, A.; Shih, A.; Elsabbagh, M. Global prevalence of autism: A systematic review update. Autism Res. 2022, 15, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G.; et al. Prevalence of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 sites, United States, 2016. MMWR Surveill. Summ. 2020, 69, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zablotsky, B.; Black, L.I.; Maenner, M.J.; Schieve, L.A.; Danielson, M.L.; Bitsko, R.H.; Blumberg, S.J.; Kogan, M.D.; Boyle, C.A. Prevalence and trends of developmental disabilities among children in the United States: 2009–2017. Pediatrics 2019, 144, e20190811. [Google Scholar] [CrossRef]
- Maenner, M.J.; Shaw, K.A.; Bakian, A.V.; Bilder, D.A.; Durkin, M.S.; Esler, A.; Furnier, S.M.; Hallas, L.; Hall-Lande, J.; Hudson, A.; et al. Prevalence and characteristics of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 sites, United States, 2018. MMWR Surveill. Summ. 2021, 70, 1–16. [Google Scholar] [CrossRef]
- GuiFeng, X.; Strathearn, L.; BuYun, L.; Wei, B. Prevalence of autism spectrum disorder among US children and adolescents, 2014-2016. JAMA J. Am. Med. Assoc. 2018, 319, 81–82. [Google Scholar]
- Baranek, G.T. Autism during infancy: A retrospective video analysis of sensory-motor and social behaviors at 9–12 months of age. J. Autism Dev. Disord. 1999, 29, 213–224. [Google Scholar] [CrossRef]
- Lord, C.; Cook, E.H.; Leventhal, B.L.; Amaral, D.G. Autism spectrum disorders. Neuron 2000, 28, 355–363. [Google Scholar] [CrossRef]
- Wetherby, A.M.; Woods, J.; Allen, L.; Cleary, J.; Dickinson, H.; Lord, C. Early indicators of autism spectrum disorders in the second year of life. J. Autism Dev. Disord. 2004, 34, 473–493. [Google Scholar] [CrossRef]
- Osterling, J.; Dawson, G. Early recognition of children with autism: A study of first birthday home videotapes. J. Autism Dev. Disord. 1994, 24, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Richler, J.; Bishop, S.L.; Kleinke, J.R.; Lord, C. Restricted and repetitive behaviors in young children with autism spectrum disorders. J. Autism Dev. Disord. 2007, 37, 73–85. [Google Scholar] [CrossRef]
- King, B.H.; Hollander, E.; Sikich, L.; McCracken, J.T.; Scahill, L.; Bregman, J.D.; Donnelly, C.L.; Anagnostou, E.; Dukes, K.; Sullivan, L. Lack of efficacy of citalopram in children with autism spectrum disorders and high levels of repetitive behavior: Citalopram ineffective in children with autism. Arch. Gen. Psychiatry 2009, 66, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, R.C.; Carter, A.S. State-level trends in the prevalence of Autism Spectrum Disorder (ASD) from 2000 to 2012: A reanalysis of findings from the autism and developmental disabilities network. J. Autism Dev. Disord. 2018, 48, 3086–3092. [Google Scholar] [CrossRef] [PubMed]
- Eissa, N.; Al-Houqani, M.; Sadeq, A.; Ojha, S.K.; Sasse, A.; Sadek, B. Current enlightenment about etiology and pharmacological treatment of autism spectrum disorder. Front. Neurosci. 2018, 12, 304. [Google Scholar] [CrossRef]
- Chahin, S.S.; Apple, R.W.; Kuo, K.H.; Dickson, C.A. Autism spectrum disorder: Psychological and functional assessment, and behavioral treatment approaches. Transl. Pediatr. 2020, 9, S66. [Google Scholar] [CrossRef]
- Levitt, P.; Campbell, D.B. The genetic and neurobiologic compass points toward common signaling dysfunctions in autism spectrum disorders. J. Clin. Investig. 2009, 119, 747–754. [Google Scholar] [CrossRef]
- Muhle, R.; Trentacoste, S.V.; Rapin, I. The genetics of autism. Pediatrics 2004, 113, e472–e486. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef]
- Ronemus, M.; Iossifov, I.; Levy, D.; Wigler, M. The role of de novo mutations in the genetics of autism spectrum disorders. Nat. Rev. Genet. 2014, 15, 133–141. [Google Scholar] [CrossRef]
- Stubbs, G.; Henley, K.; Green, J. Autism: Will vitamin D supplementation during pregnancy and early childhood reduce the recurrence rate of autism in newborn siblings? Med. Hypotheses 2016, 88, 74–78. [Google Scholar] [CrossRef]
- Castelbaum, L.; Sylvester, C.M.; Zhang, Y.; Yu, Q.; Constantino, J.N. On the nature of monozygotic twin concordance and discordance for autistic trait severity: A quantitative analysis. Behav. Genet. 2020, 50, 263–272. [Google Scholar] [CrossRef]
- Arking, D.E.; Cutler, D.J.; Brune, C.W.; Teslovich, T.M.; West, K.; Ikeda, M.; Rea, A.; Guy, M.; Lin, S.; Cook, E.H., Jr. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am. J. Hum. Genet. 2008, 82, 160–164. [Google Scholar] [CrossRef]
- Peñagarikano, O.; Geschwind, D.H. What does CNTNAP2 reveal about autism spectrum disorder? Trends Mol. Med. 2012, 18, 156–163. [Google Scholar] [CrossRef]
- Alarcón, M.; Abrahams, B.S.; Stone, J.L.; Duvall, J.A.; Perederiy, J.V.; Bomar, J.M.; Sebat, J.; Wigler, M.; Martin, C.L.; Ledbetter, D.H. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 2008, 82, 150–159. [Google Scholar] [CrossRef]
- Strauss, K.A.; Puffenberger, E.G.; Huentelman, M.J.; Gottlieb, S.; Dobrin, S.E.; Parod, J.M.; Stephan, D.A.; Morton, D.H. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N. Engl. J. Med. 2006, 354, 1370–1377. [Google Scholar] [CrossRef]
- Poliak, S.; Salomon, D.; Elhanany, H.; Sabanay, H.; Kiernan, B.; Pevny, L.; Stewart, C.L.; Xu, X.; Chiu, S.-Y.; Shrager, P. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J. Cell Biol. 2003, 162, 1149–1160. [Google Scholar] [CrossRef]
- Rodenas-Cuadrado, P.; Ho, J.; Vernes, S.C. Shining a light on CNTNAP2: Complex functions to complex disorders. Eur. J. Hum. Genet. 2014, 22, 171–178. [Google Scholar] [CrossRef]
- Anderson, G.R.; Galfin, T.; Xu, W.; Aoto, J.; Malenka, R.C.; Südhof, T.C. Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc. Natl. Acad. Sci. USA 2012, 109, 18120–18125. [Google Scholar] [CrossRef]
- Penagarikano, O.; Abrahams, B.S.; Herman, E.I.; Winden, K.D.; Gdalyahu, A.; Dong, H.; Sonnenblick, L.I.; Gruver, R.; Almajano, J.; Bragin, A.; et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 2011, 147, 235–246. [Google Scholar] [CrossRef]
- Toma, C.; Hervas, A.; Torrico, B.; Balmaña, N.; Salgado, M.; Maristany, M.; Vilella, E.; Martínez-Leal, R.; Planelles, M.I.; Cusco, I. Analysis of two language-related genes in autism: A case–control association study of: FOXP2: And: CNTNAP2. Psychiatr. Genet. 2013, 23, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Scott-Van Zeeland, A.A.; Abrahams, B.S.; Alvarez-Retuerto, A.I.; Sonnenblick, L.I.; Rudie, J.D.; Ghahremani, D.; Mumford, J.A.; Poldrack, R.A.; Dapretto, M.; Geschwind, D.H.; et al. Altered functional connectivity in frontal lobe circuits is associated with variation in the autism risk gene CNTNAP2. Sci. Transl. Med. 2010, 2, 56ra80. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.L.; Jahanshad, N.; Rudie, J.D.; Brown, J.A.; Johnson, K.; McMahon, K.L.; de Zubicaray, G.I.; Montgomery, G.; Martin, N.G.; Wright, M.J. Altered structural brain connectivity in healthy carriers of the autism risk gene, CNTNAP2. Brain Connect. 2011, 1, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.; Kitamura, H.; Endo, T.; Hasegawa, N.; Someya, T. Reduced thalamic volume observed across different subgroups of autism spectrum disorders. Psychiatry Res. Neuroimaging 2010, 184, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Treiber, J.M.; Shukla, D.K.; Shih, P.; Müller, R.-A. Impaired thalamocortical connectivity in autism spectrum disorder: A study of functional and anatomical connectivity. Brain 2013, 136, 1942–1955. [Google Scholar] [CrossRef]
- Tsatsanis, K.D.; Rourke, B.P.; Klin, A.; Volkmar, F.R.; Cicchetti, D.; Schultz, R.T. Reduced thalamic volume in high-functioning individuals with autism. Biol. Psychiatry 2003, 53, 121–129. [Google Scholar] [CrossRef]
- Friedman, S.; Shaw, D.; Artru, A.; Richards, T.; Gardner, J.; Dawson, G.; Posse, S.; Dager, S. Regional brain chemical alterations in young children with autism spectrum disorder. Neurology 2003, 60, 100–107. [Google Scholar] [CrossRef]
- Cheon, K.-A.; Kim, Y.-S.; Oh, S.-H.; Park, S.-Y.; Yoon, H.-W.; Herrington, J.; Nair, A.; Koh, Y.-J.; Jang, D.-P.; Kim, Y.-B. Involvement of the anterior thalamic radiation in boys with high functioning autism spectrum disorders: A diffusion tensor imaging study. Brain Res. 2011, 1417, 77–86. [Google Scholar] [CrossRef]
- Mizuno, A.; Villalobos, M.E.; Davies, M.M.; Dahl, B.C.; Müller, R.-A. Partially enhanced thalamocortical functional connectivity in autism. Brain Res. 2006, 1104, 160–174. [Google Scholar] [CrossRef]
- Gandhi, T.; Lee, C.C. Neural mechanisms underlying repetitive behaviors in rodent models of autism spectrum disorders. Front. Cell. Neurosci. 2021, 14, 592710. [Google Scholar] [CrossRef]
- Reichova, I.; Sherman, S.M. Somatosensory corticothalamic projections: Distinguishing drivers from modulators. J. Neurophysiol. 2004, 92, 2185–2197. [Google Scholar] [CrossRef]
- Lee, C.C.; Sherman, S.M. Drivers and modulators in the central auditory pathways. Front. Neurosci. 2010, 4, 14. [Google Scholar] [CrossRef]
- Theyel, B.B.; Llano, D.A.; Sherman, S.M. The corticothalamocortical circuit drives higher-order cortex in the mouse. Nat. Neurosci. 2010, 13, 84–88. [Google Scholar] [CrossRef]
- Amunts, K.; Schleicher, A.; Zilles, K. Cytoarchitecture of the cerebral cortex—More than localization. Neuroimage 2007, 37, 1061–1065. [Google Scholar] [CrossRef]
- Amaral, D.G.; Schumann, C.M.; Nordahl, C.W. Neuroanatomy of autism. Trends Neurosci. 2008, 31, 137–145. [Google Scholar] [CrossRef]
- Casanova, M.; Van Kooten, I.; Switala, A.; Van Engeland, H.; Heinsen, H.; Steinbusch, H.; Hof, P.; Schmitz, C. Abnormalities of cortical minicolumnar organization in the prefrontal lobes of autistic patients. Clin. Neurosci. Res. 2006, 6, 127–133. [Google Scholar] [CrossRef]
- Lee, C.C. Thalamic and cortical pathways supporting auditory processing. Brain Lang. 2013, 126, 22–28. [Google Scholar] [CrossRef]
- Lee, C.C. Exploring functions for the non-lemniscal auditory thalamus. Front. Neural Circuits 2015, 9, 69. [Google Scholar] [CrossRef]
- Adeyelu, T.; Gandhi, T.; Lee, C.C. Crossed Connections From Insular Cortex to the Contralateral Thalamus. Front. Neural Circuits 2021, 15, 710925. [Google Scholar] [CrossRef]
- Canitano, R.; Pallagrosi, M. Autism spectrum disorders and schizophrenia spectrum disorders: Excitation/inhibition imbalance and developmental trajectories. Front. Psychiatry 2017, 8, 69. [Google Scholar] [CrossRef]
- Rubenstein, J.; Merzenich, M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Culotta, L.; Penzes, P. Exploring the mechanisms underlying excitation/inhibition imbalance in human iPSC-derived models of ASD. Mol. Autism 2020, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Manent, J.-B.; Represa, A. Neurotransmitters and brain maturation: Early paracrine actions of GABA and glutamate modulate neuronal migration. Neuroscientist 2007, 13, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, E.; Ariza, J.; Rogers, H.; Noctor, S.C.; Martínez-Cerdeño, V. The number of parvalbumin-expressing interneurons is decreased in the prefrontal cortex in autism. Cereb. Cortex 2017, 27, 1931–1943. [Google Scholar]
- Selby, L.; Zhang, C.; Sun, Q.-Q. Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neurosci. Lett. 2007, 412, 227–232. [Google Scholar] [CrossRef]
- Chao, H.-T.; Chen, H.; Samaco, R.C.; Xue, M.; Chahrour, M.; Yoo, J.; Neul, J.L.; Gong, S.; Lu, H.-C.; Heintz, N. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010, 468, 263–269. [Google Scholar] [CrossRef]
- Ito-Ishida, A.; Ure, K.; Chen, H.; Swann, J.W.; Zoghbi, H.Y. Loss of MeCP2 in parvalbumin-and somatostatin-expressing neurons in mice leads to distinct Rett syndrome-like phenotypes. Neuron 2015, 88, 651–658. [Google Scholar] [CrossRef]
- Goel, A.; Cantu, D.A.; Guilfoyle, J.; Chaudhari, G.R.; Newadkar, A.; Todisco, B.; de Alba, D.; Kourdougli, N.; Schmitt, L.M.; Pedapati, E. Impaired perceptual learning in a mouse model of Fragile X syndrome is mediated by parvalbumin neuron dysfunction and is reversible. Nat. Neurosci. 2018, 21, 1404–1411. [Google Scholar] [CrossRef]
- Kalinowska, M.; van der Lei, M.B.; Kitiashvili, M.; Mamcarz, M.; Oliveira, M.M.; Longo, F.; Klann, E. Deletion of Fmr1 in parvalbumin-expressing neurons results in dysregulated translation and selective behavioral deficits associated with fragile X syndrome. Mol. Autism 2022, 13, 29. [Google Scholar] [CrossRef]
- Marín, O. Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 107–120. [Google Scholar] [CrossRef]
- Lauber, E.; Filice, F.; Schwaller, B. Parvalbumin neurons as a hub in autism spectrum disorders. J. Neurosci. Res. 2018, 96, 360–361. [Google Scholar] [CrossRef]
- Lunden, J.W.; Durens, M.; Phillips, A.W.; Nestor, M.W. Cortical interneuron function in autism spectrum condition. Pediatr. Res. 2019, 85, 146–154. [Google Scholar] [CrossRef]
- Nomura, T. Interneuron dysfunction and inhibitory deficits in autism and fragile X syndrome. Cells 2021, 10, 2610. [Google Scholar] [CrossRef]
- Contractor, A.; Ethell, I.M.; Portera-Cailliau, C. Cortical interneurons in autism. Nat. Neurosci. 2021, 24, 1648–1659. [Google Scholar] [CrossRef]
- Sohal, V.S.; Zhang, F.; Yizhar, O.; Deisseroth, K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 2009, 459, 698–702. [Google Scholar] [CrossRef]
- Whittington, M.A.; Cunningham, M.O.; LeBeau, F.E.; Racca, C.; Traub, R.D. Multiple origins of the cortical gamma rhythm. Dev. Neurobiol. 2011, 71, 92–106. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, M.; Czéh, B.; Flügge, G.; Zhang, W. Stress impairs GABAergic network function in the hippocampus by activating nongenomic glucocorticoid receptors and affecting the integrity of the parvalbumin-expressing neuronal network. Neuropsychopharmacology 2010, 35, 1693–1707. [Google Scholar] [CrossRef]
- Cabungcal, J.-H.; Steullet, P.; Kraftsik, R.; Cuenod, M.; Do, K.Q. Early-life insults impair parvalbumin interneurons via oxidative stress: Reversal by N-acetylcysteine. Biol. Psychiatry 2013, 73, 574–582. [Google Scholar] [CrossRef]
- Cabungcal, J.-H.; Steullet, P.; Morishita, H.; Kraftsik, R.; Cuenod, M.; Hensch, T.K.; Do, K.Q. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 9130–9135. [Google Scholar] [CrossRef]
- Beurdeley, M.; Spatazza, J.; Lee, H.H.; Sugiyama, S.; Bernard, C.; Di Nardo, A.A.; Hensch, T.K.; Prochiantz, A. Otx2 binding to perineuronal nets persistently regulates plasticity in the mature visual cortex. J. Neurosci. 2012, 32, 9429–9437. [Google Scholar] [CrossRef]
- Kwok, J.C.; Dick, G.; Wang, D.; Fawcett, J.W. Extracellular matrix and perineuronal nets in CNS repair. Dev. Neurobiol. 2011, 71, 1073–1089. [Google Scholar] [CrossRef]
- Morawski, M.; Brückner, M.K.; Riederer, P.; Brückner, G.; Arendt, T. Perineuronal nets potentially protect against oxidative stress. Exp. Neurol. 2004, 188, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Suttkus, A.; Rohn, S.; Jäger, C.; Arendt, T.; Morawski, M. Neuroprotection against iron-induced cell death by perineuronal nets-an in vivo analysis of oxidative stress. Am. J. Neurodegener. Dis. 2012, 1, 122. [Google Scholar] [PubMed]
- Reichelt, A.C.; Hare, D.J.; Bussey, T.J.; Saksida, L.M. Perineuronal nets: Plasticity, protection, and therapeutic potential. Trends Neurosci. 2019, 42, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.W.; Oohashi, T.; Pizzorusso, T. The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nat. Rev. Neurosci. 2019, 20, 451–465. [Google Scholar] [CrossRef]
- Fawcett, J.W.; Fyhn, M.; Jendelova, P.; Kwok, J.C.; Ruzicka, J.; Sorg, B.A. The extracellular matrix and perineuronal nets in memory. Mol. Psychiatry 2022, 27, 3192–3203. [Google Scholar] [CrossRef]
- Gandhi, T.; Liu, C.-C.; Adeyelu, T.T.; Canepa, C.R.; Lee, C.C. Behavioral regulation by perineuronal nets in the prefrontal cortex of the CNTNAP2 mouse model of autism spectrum disorder. Front. Behav. Neurosci. 2023, 17, 1114789. [Google Scholar] [CrossRef]
- Lauber, E.; Filice, F.; Schwaller, B. Dysregulation of parvalbumin expression in the Cntnap2−/− mouse model of autism spectrum disorder. Front. Mol. Neurosci. 2018, 11, 262. [Google Scholar] [CrossRef]
- Lander, C.; Kind, P.; Maleski, M.; Hockfield, S. A family of activity-dependent neuronal cell-surface chondroitin sulfate proteoglycans in cat visual cortex. J. Neurosci. 1997, 17, 1928–1939. [Google Scholar] [CrossRef]
- Pizzorusso, T.; Medini, P.; Berardi, N.; Chierzi, S.; Fawcett, J.W.; Maffei, L. Reactivation of ocular dominance plasticity in the adult visual cortex. Science 2002, 298, 1248–1251. [Google Scholar] [CrossRef]
- Vogt, D.; Cho, K.K.; Shelton, S.M.; Paul, A.; Huang, Z.J.; Sohal, V.S.; Rubenstein, J.L. Mouse Cntnap2 and human CNTNAP2 ASD alleles cell autonomously regulate PV+ cortical interneurons. Cereb. Cortex 2018, 28, 3868–3879. [Google Scholar] [CrossRef]
- Nishimura, Y.; Martin, C.L.; Vazquez-Lopez, A.; Spence, S.J.; Alvarez-Retuerto, A.I.; Sigman, M.; Steindler, C.; Pellegrini, S.; Schanen, N.C.; Warren, S.T. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum. Mol. Genet. 2007, 16, 1682–1698. [Google Scholar] [CrossRef]
- Schellenberg, G.; Dawson, G.; Sung, Y.; Estes, A.; Munson, J.; Rosenthal, E.; Rothstein, J.; Flodman, P.; Smith, M.; Coon, H. Evidence for multiple loci from a genome scan of autism kindreds. Mol. Psychiatry 2006, 11, 1049–1060. [Google Scholar] [CrossRef]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef]
- Szatmari, P.; Paterson, A.; Zwaigenbaum, L.; Roberts, W.; Brian, J.; Liu, X.-Q.; Vincent, J.; Skaug, J.; Thompson, A.; Senman, L. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007, 39, 319. [Google Scholar]
- Yang, M.S.; Gill, M. A review of gene linkage, association and expression studies in autism and an assessment of convergent evidence. Int. J. Dev. Neurosci. 2007, 25, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Fuccillo, M.V. Striatal circuits as a common node for autism pathophysiology. Front. Neurosci. 2016, 10, 27. [Google Scholar] [CrossRef]
- Rothwell, P.E.; Fuccillo, M.V.; Maxeiner, S.; Hayton, S.J.; Gokce, O.; Lim, B.K.; Fowler, S.C.; Malenka, R.C.; Südhof, T.C. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell 2014, 158, 198–212. [Google Scholar] [CrossRef]
- Sherman, S.M.; Guillery, R. Distinct functions for direct and transthalamic corticocortical connections. J. Neurophysiol. 2011, 106, 1068–1077. [Google Scholar] [CrossRef]
- Courchesne, E. Brain development in autism: Early overgrowth followed by premature arrest of growth. Ment. Retard. Dev. Disabil. Res. Rev. 2004, 10, 106–111. [Google Scholar] [CrossRef]
- Courchesne, E.; Pierce, K. Brain overgrowth in autism during a critical time in development: Implications for frontal pyramidal neuron and interneuron development and connectivity. Int. J. Dev. Neurosci. 2005, 23, 153–170. [Google Scholar] [CrossRef]
- Courchesne, E.; Redcay, E.; Kennedy, D.P. The autistic brain: Birth through adulthood. Curr. Opin. Neurol. 2004, 17, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Minshew, N.J.; Williams, D.L. The new neurobiology of autism: Cortex, connectivity, and neuronal organization. Arch. Neurol. 2007, 64, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Casanova, M.F.; Buxhoeveden, D.P.; Switala, A.E.; Roy, E. Minicolumnar pathology in autism. Neurology 2002, 58, 428–432. [Google Scholar] [CrossRef]
- Casanova, M.F.; Buxhoeveden, D.; Gomez, J. Disruption in the inhibitory architecture of the cell minicolumn: Implications for autisim. Neuroscientist 2003, 9, 496–507. [Google Scholar] [CrossRef]
- Bailey, A.; Luthert, P.; Dean, A.; Harding, B.; Janota, I.; Montgomery, M.; Rutter, M.; Lantos, P. A clinicopathological study of autism. Brain A J. Neurol. 1998, 121, 889–905. [Google Scholar] [CrossRef]
- Bauman, M.L.; Kemper, T.L. Neuroanatomic observations of the brain in autism: A review and future directions. Int. J. Dev. Neurosci. 2005, 23, 183–187. [Google Scholar] [CrossRef]
- Bauman, M.L.; Kemper, T.L. Structural brain anatomy in autism: What is the evidence. Neurobiol. Autism 2005, 2, 121–135. [Google Scholar]
- Kemper, T.L.; Bauman, M. Neuropathology of infantile autism. J. Neuropathol. Exp. Neurol. 1998, 57, 645–652. [Google Scholar] [CrossRef]
- Kemper, T. Neuroanatomic studies of dyslexia and autism. In Disorders of the Developing Nervous System: Changing Views of Their Origin, Diagnosis and Treatment; Liss: New York, NY, USA, 1988. [Google Scholar]
- Kemper, T.L.; Bauman, M.L. The contribution of neuropathologic studies to the understanding of autism. Neurol. Clin. 1993, 11, 175–187. [Google Scholar] [CrossRef]
- Bauman, M.L.; Kemper, T.L. The neuropathology of the autism spectrum disorders: What have we learned. In Autism: Neural Basis and Treatment Possibilities: Novartis Foundation Symposium 251; John Wiley & Sons, Ltd.: Chichester, UK, 2003; pp. 112–122. [Google Scholar]
- Simms, M.L.; Kemper, T.L.; Timbie, C.M.; Bauman, M.L.; Blatt, G.J. The anterior cingulate cortex in autism: Heterogeneity of qualitative and quantitative cytoarchitectonic features suggests possible subgroups. Acta Neuropathol. 2009, 118, 673–684. [Google Scholar] [CrossRef]
- Palmen, S.J.; van Engeland, H.; Hof, P.R.; Schmitz, C. Neuropathological findings in autism. Brain 2004, 127, 2572–2583. [Google Scholar] [CrossRef]
- Casanova, M.F.; van Kooten, I.A.; Switala, A.E.; van Engeland, H.; Heinsen, H.; Steinbusch, H.W.; Hof, P.R.; Trippe, J.; Stone, J.; Schmitz, C. Minicolumnar abnormalities in autism. Acta Neuropathol. 2006, 112, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Pardo, C.A.; Eberhart, C.G. The neurobiology of autism. Brain Pathol. 2007, 17, 434–447. [Google Scholar] [CrossRef]
- Grandin, T.; Johnson, C. Animals in Translation: Using the Mysteries of Autism to Decode Animal Behavior; Houghton Mifflin Harcourt: Boston, MA, USA, 2006. [Google Scholar]
- Baron-Cohen, S. The cognitive neuroscience of autism. J. Neurol. Neurosurg. Psychiatry 2004, 75, 945–948. [Google Scholar] [CrossRef]
- Lee, C.C.; Sherman, S.M. Synaptic properties of thalamic and intracortical inputs to layer 4 of the first-and higher-order cortical areas in the auditory and somatosensory systems. J. Neurophysiol. 2008, 100, 317–326. [Google Scholar] [CrossRef]
- Lee, C.C.; Sherman, S.M. Topography and physiology of ascending streams in the auditory tectothalamic pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 372–377. [Google Scholar] [CrossRef]
- Sherman, S.M.; Guillery, R. The role of the thalamus in the flow of information to the cortex. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2002, 357, 1695–1708. [Google Scholar] [CrossRef]
- Lee, C.C.; Lam, Y.-W.; Sherman, S.M. Intracortical convergence of layer 6 neurons. Neuroreport 2012, 23, 736. [Google Scholar] [CrossRef]
- Cho, K.K.; Hoch, R.; Lee, A.T.; Patel, T.; Rubenstein, J.L.; Sohal, V.S. Gamma rhythms link prefrontal interneuron dysfunction with cognitive inflexibility in Dlx5/6+/− mice. Neuron 2015, 85, 1332–1343. [Google Scholar] [CrossRef]
- Fader, S.M.; Imaizumi, K.; Yanagawa, Y.; Lee, C.C. Wisteria floribunda agglutinin-labeled perineuronal nets in the mouse inferior colliculus, thalamic reticular nucleus and auditory cortex. Brain Sci. 2016, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Manseau, F.; Marinelli, S.; Méndez, P.; Schwaller, B.; Prince, D.A.; Huguenard, J.R.; Bacci, A. Desynchronization of neocortical networks by asynchronous release of GABA at autaptic and synaptic contacts from fast-spiking interneurons. PLoS Biol. 2010, 8, e1000492. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Brooks, C.B.; Shrestha, A.; Ogundele, O.M.; Lee, C.C. Perineuronal nets in the prefrontal cortex of a schizophrenia mouse model: Assessment of neuroanatomical, electrophysiological, and behavioral contributions. Int. J. Mol. Sci. 2021, 22, 11140. [Google Scholar] [CrossRef] [PubMed]
- Donato, F.; Rompani, S.B.; Caroni, P. Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature 2013, 504, 272–276. [Google Scholar] [CrossRef]
- Liu, H.; Gao, P.-F.; Xu, H.-W.; Liu, M.-M.; Yu, T.; Yao, J.-P.; Yin, Z.-Q. Perineuronal nets increase inhibitory GABAergic currents during the critical period in rats. Int. J. Ophthalmol. 2013, 6, 120. [Google Scholar]
- Morawski, M.; Reinert, T.; Meyer-Klaucke, W.; Wagner, F.E.; Tröger, W.; Reinert, A.; Jäger, C.; Brückner, G.; Arendt, T. Ion exchanger in the brain: Quantitative analysis of perineuronally fixed anionic binding sites suggests diffusion barriers with ion sorting properties. Sci. Rep. 2015, 5, 16471. [Google Scholar] [CrossRef]
- Sugiyama, S.; Prochiantz, A.; Hensch, T.K. From brain formation to plasticity: Insights on Otx2 homeoprotein. Dev. Growth Differ. 2009, 51, 369–377. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandhi, T.; Canepa, C.R.; Adeyelu, T.T.; Adeniyi, P.A.; Lee, C.C. Neuroanatomical Alterations in the CNTNAP2 Mouse Model of Autism Spectrum Disorder. Brain Sci. 2023, 13, 891. https://doi.org/10.3390/brainsci13060891
Gandhi T, Canepa CR, Adeyelu TT, Adeniyi PA, Lee CC. Neuroanatomical Alterations in the CNTNAP2 Mouse Model of Autism Spectrum Disorder. Brain Sciences. 2023; 13(6):891. https://doi.org/10.3390/brainsci13060891
Chicago/Turabian StyleGandhi, Tanya, Cade R. Canepa, Tolulope T. Adeyelu, Philip A. Adeniyi, and Charles C. Lee. 2023. "Neuroanatomical Alterations in the CNTNAP2 Mouse Model of Autism Spectrum Disorder" Brain Sciences 13, no. 6: 891. https://doi.org/10.3390/brainsci13060891
APA StyleGandhi, T., Canepa, C. R., Adeyelu, T. T., Adeniyi, P. A., & Lee, C. C. (2023). Neuroanatomical Alterations in the CNTNAP2 Mouse Model of Autism Spectrum Disorder. Brain Sciences, 13(6), 891. https://doi.org/10.3390/brainsci13060891