
From Theory to Practice: Implementing the WHO 2021 Classification of Adult Diffuse Gliomas in Neuropathology Diagnosis


Abstract
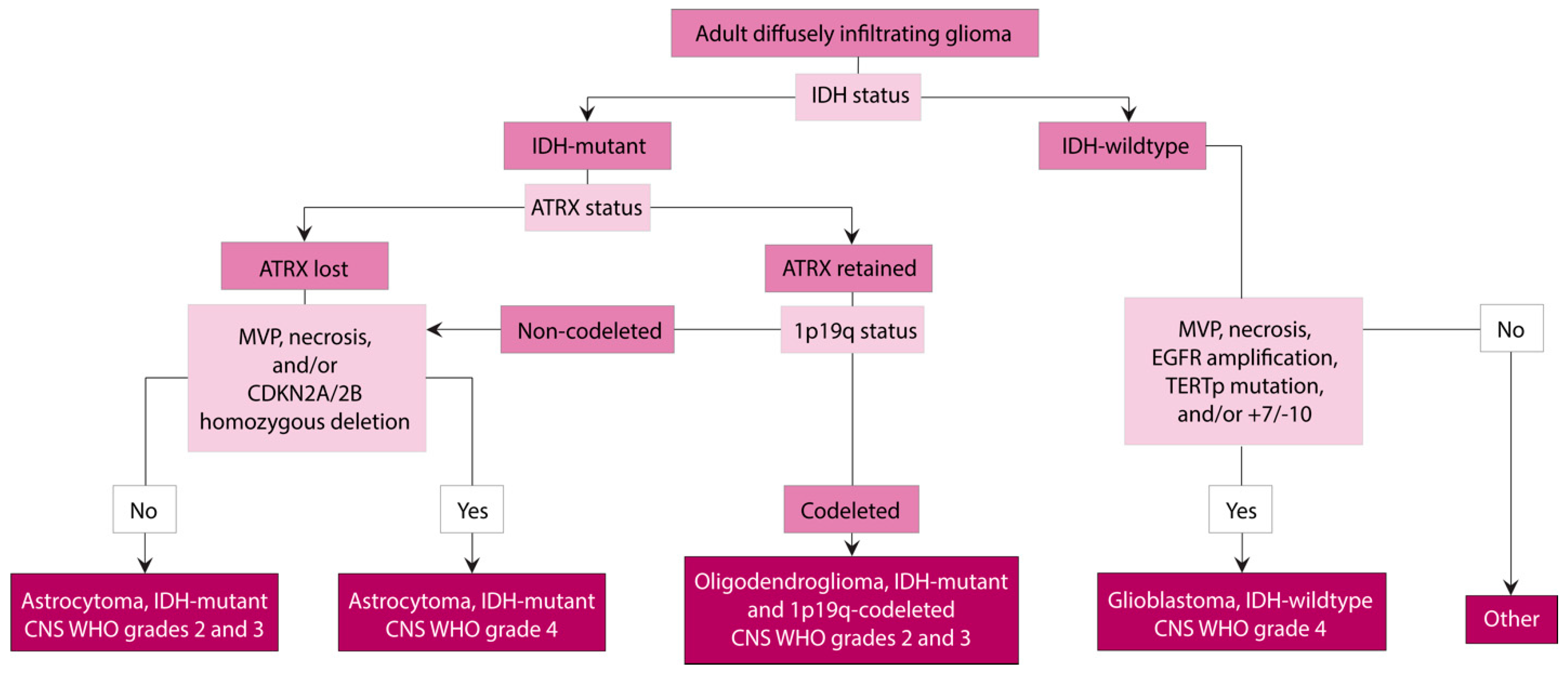
1. Introduction
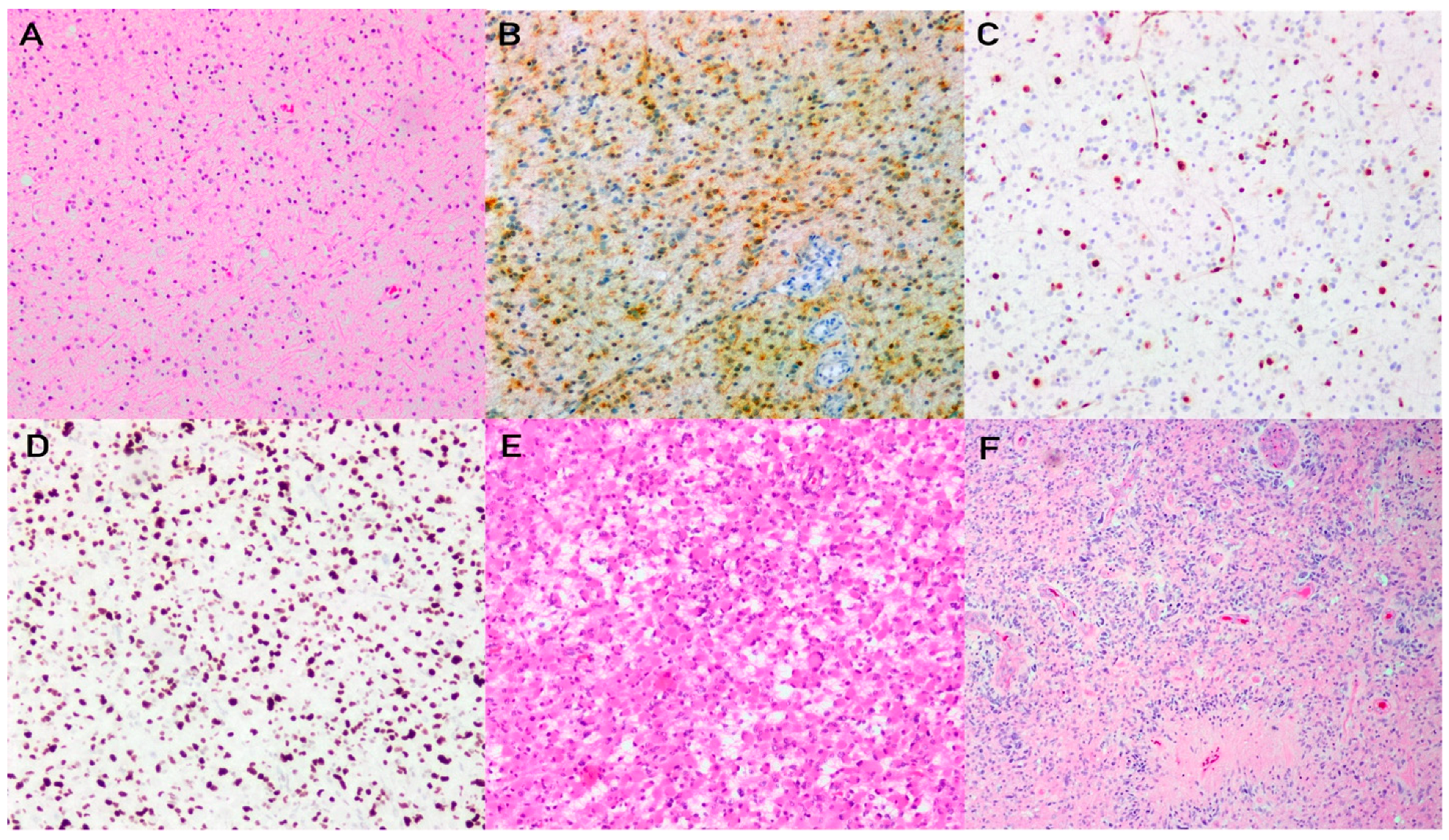
2. Astrocytoma, IDH-Mutant
2.1. Pathophysiology
2.2. Histology and Diagnostic Workup
2.3. Grading and Molecular Integration for Diagnosis and Prognosis
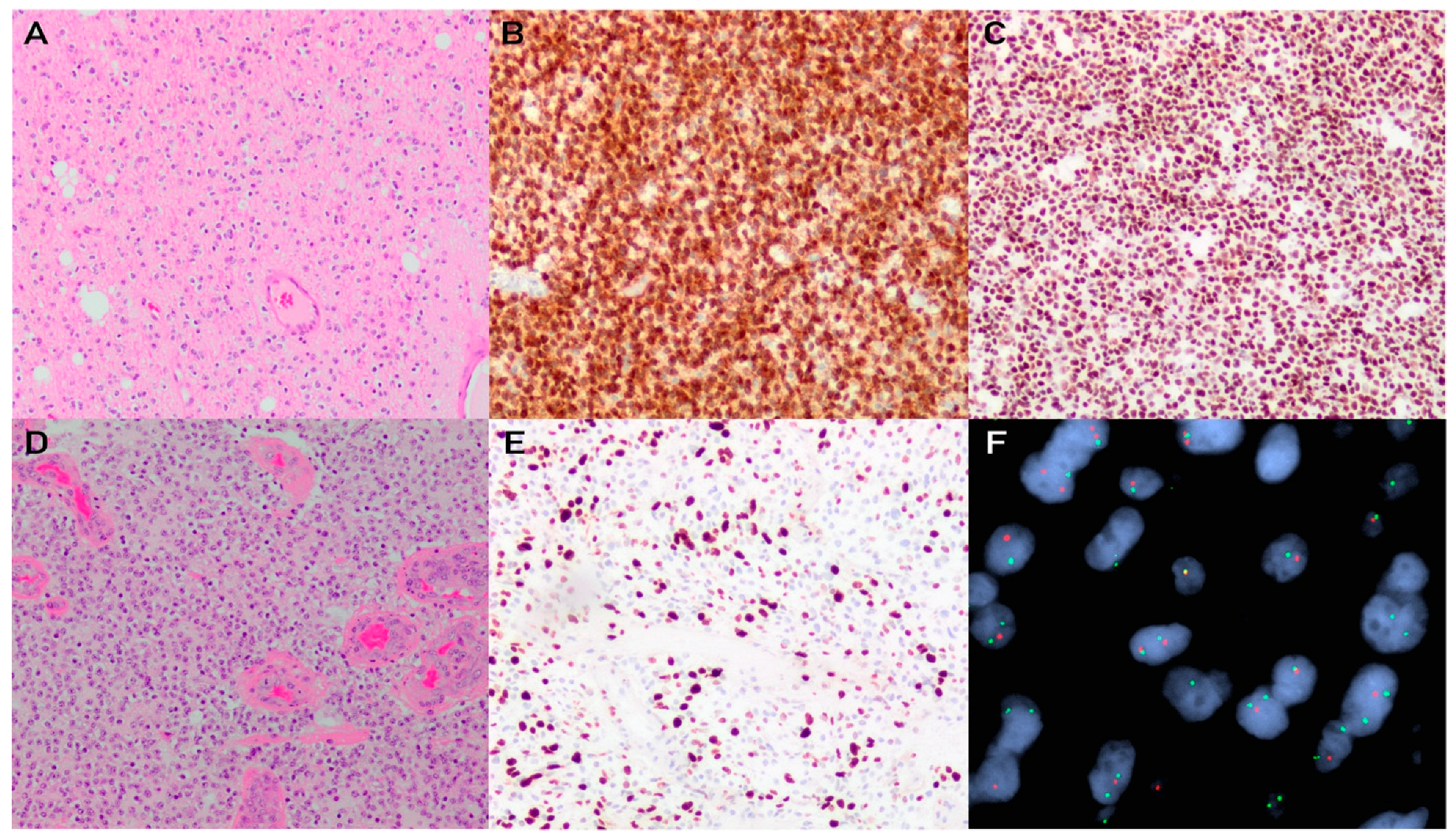
3. Oligodendroglioma, IDH-Mutant and 1p/19q-Codeleted
3.1. Pathophysiology
3.2. Histology and Diagnostic Workup
3.3. Grading and Molecular Integration for Diagnosis and Prognosis
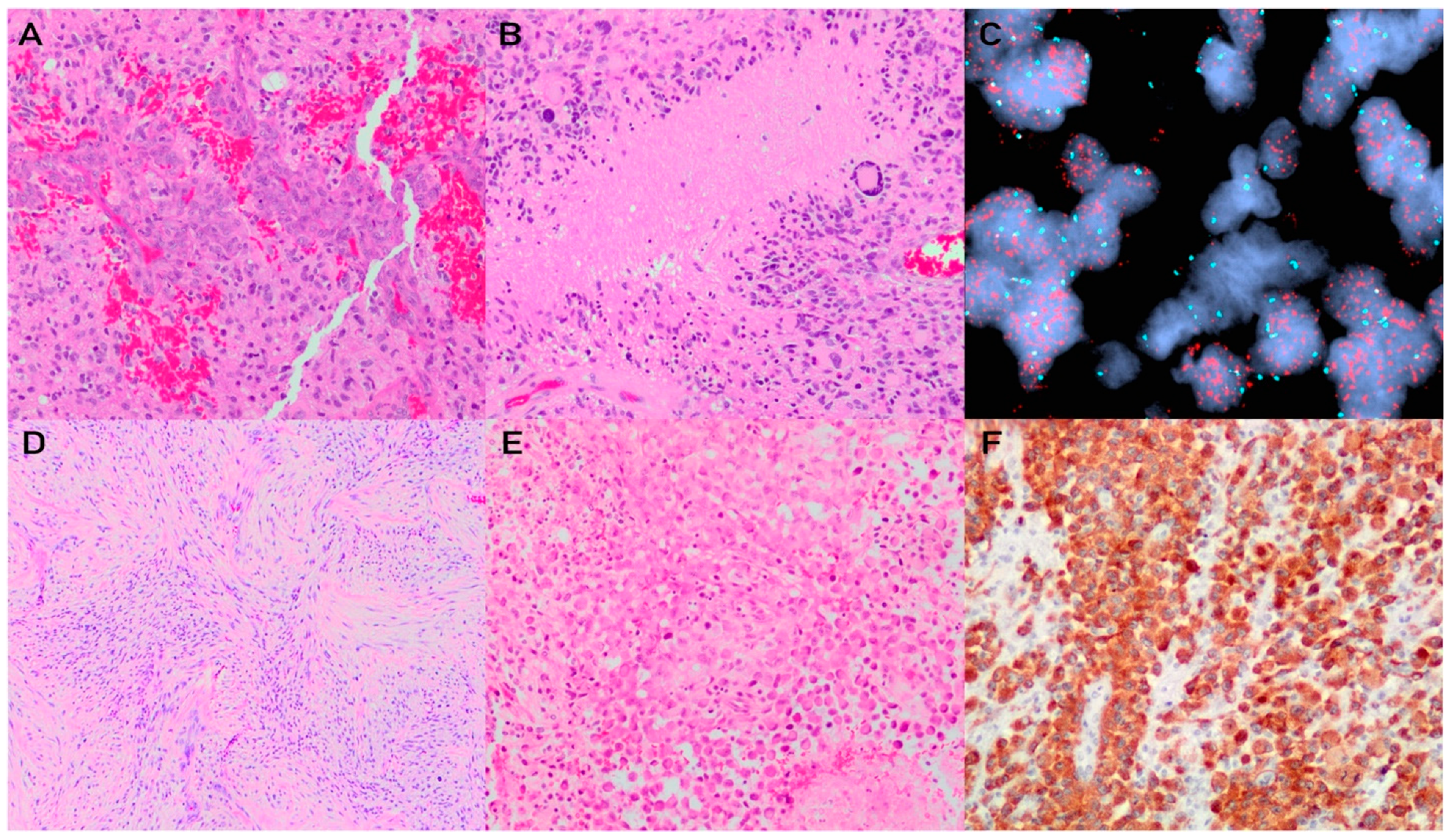
4. Glioblastoma, IDH-Wildtype
4.1. Pathophysiology
4.2. Histology and Diagnostic Workup
4.3. Grading and Molecular Integration for Diagnosis and Prognosis
5. Standard of Care Molecular Testing of Adult Diffuse Glioma: Application in Neuropathology Practice
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro Oncol. 2020, 22, iv1–iv96. [Google Scholar] [CrossRef] [PubMed]
- Horbinski, C.; Ligon, K.L.; Brastianos, P.; Huse, J.T.; Venere, M.; Chang, S.; Buckner, J.; Cloughesy, T.; Jenkins, R.B.; Giannini, C.; et al. The medical necessity of advanced molecular testing in the diagnosis and treatment of brain tumor patients. Neuro Oncol. 2019, 21, 1498–1508. [Google Scholar] [CrossRef] [PubMed]
- Suwala, A.K.; Stichel, D.; Schrimpf, D.; Kloor, M.; Wefers, A.K.; Reinhardt, A.; Maas, S.L.N.; Kratz, C.P.; Schweizer, L.; Hasselblatt, M.; et al. Primary mismatch repair deficient IDH-mutant astrocytoma (PMMRDIA) is a distinct type with a poor prognosis. Acta Neuropathol. 2021, 141, 85–100. [Google Scholar] [CrossRef]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020, 580, 517–523. [Google Scholar] [CrossRef]
- Board, W.C.o.T.E. World Health Organization Classification of Tumours of the Central Nervous System, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2007. [Google Scholar]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network; Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef]
- Barresi, V.; Eccher, A.; Simbolo, M.; Cappellini, R.; Ricciardi, G.K.; Calabria, F.; Cancedda, M.; Mazzarotto, R.; Bonetti, B.; Pinna, G.; et al. Diffuse gliomas in patients aged 55 years or over: A suggestion for IDH mutation testing. Neuropathology 2020, 40, 68–74. [Google Scholar] [CrossRef]
- Stockhammer, F.; Misch, M.; Helms, H.J.; Lengler, U.; Prall, F.; von Deimling, A.; Hartmann, C. IDH1/2 mutations in WHO grade II astrocytomas associated with localization and seizure as the initial symptom. Seizure 2012, 21, 194–197. [Google Scholar] [CrossRef]
- Carrillo, J.A.; Lai, A.; Nghiemphu, P.L.; Kim, H.J.; Phillips, H.S.; Kharbanda, S.; Moftakhar, P.; Lalaezari, S.; Yong, W.; Ellingson, B.M.; et al. Relationship between tumor enhancement, edema, IDH1 mutational status, MGMT promoter methylation, and survival in glioblastoma. AJNR Am. J. Neuroradiol. 2012, 33, 1349–1355. [Google Scholar] [CrossRef]
- Banan, R.; Stichel, D.; Bleck, A.; Hong, B.; Lehmann, U.; Suwala, A.; Reinhardt, A.; Schrimpf, D.; Buslei, R.; Stadelmann, C.; et al. Infratentorial IDH-mutant astrocytoma is a distinct subtype. Acta Neuropathol. 2020, 140, 569–581. [Google Scholar] [CrossRef]
- Poetsch, L.; Bronnimann, C.; Loiseau, H.; Frenel, J.S.; Siegfried, A.; Seizeur, R.; Gauchotte, G.; Cappellen, D.; Carpentier, C.; Figarella-Branger, D.; et al. Characteristics of IDH-mutant gliomas with non-canonical IDH mutation. J. Neurooncol. 2021, 151, 279–286. [Google Scholar] [CrossRef]
- Matsumura, N.; Ikota, H.; Yamazaki, T.; Nakata, S.; Nobusawa, S.; Hirato, J.; Yoshimoto, Y.; Yokoo, H. Cerebellar high-grade astrocytoma with IDH mutations in the elderly: A report of two cases. Neuropathology 2018, 38, 411–416. [Google Scholar] [CrossRef]
- Buckner, J.C.; Shaw, E.G.; Pugh, S.L.; Chakravarti, A.; Gilbert, M.R.; Barger, G.R.; Coons, S.; Ricci, P.; Bullard, D.; Brown, P.D.; et al. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N. Engl. J. Med. 2016, 374, 1344–1355. [Google Scholar] [CrossRef]
- Bell, E.H.; Zhang, P.; Shaw, E.G.; Buckner, J.C.; Barger, G.R.; Bullard, D.E.; Mehta, M.P.; Gilbert, M.R.; Brown, P.D.; Stelzer, K.J.; et al. Comprehensive Genomic Analysis in NRG Oncology/RTOG 9802: A Phase III Trial of Radiation Versus Radiation Plus Procarbazine, Lomustine (CCNU), and Vincristine in High-Risk Low-Grade Glioma. J. Clin. Oncol. 2020, 38, 3407–3417. [Google Scholar] [CrossRef] [PubMed]
- Reuss, D.E.; Mamatjan, Y.; Schrimpf, D.; Capper, D.; Hovestadt, V.; Kratz, A.; Sahm, F.; Koelsche, C.; Korshunov, A.; Olar, A.; et al. IDH mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: A grading problem for WHO. Acta Neuropathol. 2015, 129, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.H.; Poisson, L.M.; Brat, D.J.; Zhou, Y.; Cooper, L.; Snuderl, M.; Thomas, C.; Franceschi, A.M.; Griffith, B.; Flanders, A.E.; et al. T2-FLAIR Mismatch, an Imaging Biomarker for IDH and 1p/19q Status in Lower-grade Gliomas: A TCGA/TCIA Project. Clin. Cancer Res. 2017, 23, 6078–6085. [Google Scholar] [CrossRef] [PubMed]
- Maynard, J.; Okuchi, S.; Wastling, S.; Busaidi, A.A.; Almossawi, O.; Mbatha, W.; Brandner, S.; Jaunmuktane, Z.; Koc, A.M.; Mancini, L.; et al. World Health Organization Grade II/III Glioma Molecular Status: Prediction by MRI Morphologic Features and Apparent Diffusion Coefficient. Radiology 2020, 296, 111–121. [Google Scholar] [CrossRef]
- Zhao, K.; Sun, G.; Wang, Q.; Xue, Z.; Liu, G.; Xia, Y.; Yao, A.; Zhao, Y.; You, N.; Yang, C.; et al. The Diagnostic Value of Conventional MRI and CT Features in the Identification of the IDH1-Mutant and 1p/19q Co-Deletion in WHO Grade II Gliomas. Acad. Radiol. 2021, 28, e189–e198. [Google Scholar] [CrossRef]
- Juratli, T.A.; Tummala, S.S.; Riedl, A.; Daubner, D.; Hennig, S.; Penson, T.; Zolal, A.; Thiede, C.; Schackert, G.; Krex, D.; et al. Radiographic assessment of contrast enhancement and T2/FLAIR mismatch sign in lower grade gliomas: Correlation with molecular groups. J. Neurooncol. 2019, 141, 327–335. [Google Scholar] [CrossRef]
- Ilkhanizadeh, S.; Lau, J.; Huang, M.; Foster, D.J.; Wong, R.; Frantz, A.; Wang, S.; Weiss, W.A.; Persson, A.I. Glial progenitors as targets for transformation in glioma. Adv. Cancer Res. 2014, 121, 1–65. [Google Scholar] [CrossRef]
- Zong, H.; Parada, L.F.; Baker, S.J. Cell of origin for malignant gliomas and its implication in therapeutic development. Cold Spring Harb. Perspect. Biol. 2015, 7, a020610. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, N.; Jiang, Y.; Xie, Y.; Bolouri, H.; Kastemar, M.; Olofsson, T.; Holland, E.C.; Uhrbom, L. Oncogenic signaling is dominant to cell of origin and dictates astrocytic or oligodendroglial tumor development from oligodendrocyte precursor cells. J. Neurosci. 2014, 34, 14644–14651. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Mak, T.W. Oncogenic isocitrate dehydrogenase mutations: Mechanisms, models, and clinical opportunities. Cancer Discov. 2013, 3, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Makarov, V.; Taranda, J.; Wang, Y.; Fabius, A.W.M.; Wu, W.; Zheng, Y.; El-Amine, N.; Haddock, S.; Nanjangud, G.; et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet. 2018, 50, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Meyer, J.; Balss, J.; Capper, D.; Mueller, W.; Christians, A.; Felsberg, J.; Wolter, M.; Mawrin, C.; Wick, W.; et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: A study of 1,010 diffuse gliomas. Acta Neuropathol. 2009, 118, 469–474. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Balss, J.; Meyer, J.; Mueller, W.; Korshunov, A.; Hartmann, C.; von Deimling, A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008, 116, 597–602. [Google Scholar] [CrossRef]
- Liu, X.Y.; Gerges, N.; Korshunov, A.; Sabha, N.; Khuong-Quang, D.A.; Fontebasso, A.M.; Fleming, A.; Hadjadj, D.; Schwartzentruber, J.; Majewski, J.; et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol. 2012, 124, 615–625. [Google Scholar] [CrossRef]
- Shirahata, M.; Ono, T.; Stichel, D.; Schrimpf, D.; Reuss, D.E.; Sahm, F.; Koelsche, C.; Wefers, A.; Reinhardt, A.; Huang, K.; et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 2018, 136, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, A.; Casalini, B.; Chavez, L.; Hielscher, T.; Sill, M.; Ryzhova, M.; Sharma, T.; Schrimpf, D.; Stichel, D.; Capper, D.; et al. Integrated molecular characterization of IDH-mutant glioblastomas. Neuropathol. Appl. Neurobiol. 2019, 45, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Appay, R.; Dehais, C.; Maurage, C.A.; Alentorn, A.; Carpentier, C.; Colin, C.; Ducray, F.; Escande, F.; Idbaih, A.; Kamoun, A.; et al. CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse malignant IDH-mutant gliomas. Neuro Oncol. 2019, 21, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Reis, G.F.; Pekmezci, M.; Hansen, H.M.; Rice, T.; Marshall, R.E.; Molinaro, A.M.; Phillips, J.J.; Vogel, H.; Wiencke, J.K.; Wrensch, M.R.; et al. CDKN2A loss is associated with shortened overall survival in lower-grade (World Health Organization Grades II-III) astrocytomas. J. Neuropathol. Exp. Neurol. 2015, 74, 442–452. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell. 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Hentschel, B.; Wick, W.; Capper, D.; Felsberg, J.; Simon, M.; Westphal, M.; Schackert, G.; Meyermann, R.; Pietsch, T.; et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: Implications for classification of gliomas. Acta Neuropathol. 2010, 120, 707–718. [Google Scholar] [CrossRef]
- Paz, M.F.; Yaya-Tur, R.; Rojas-Marcos, I.; Reynes, G.; Pollan, M.; Aguirre-Cruz, L.; Garcia-Lopez, J.L.; Piquer, J.; Safont, M.J.; Balana, C.; et al. CpG island hypermethylation of the DNA repair enzyme methyltransferase predicts response to temozolomide in primary gliomas. Clin. Cancer Res. 2004, 10, 4933–4938. [Google Scholar] [CrossRef]
- Gerson, S.L. MGMT: Its role in cancer aetiology and cancer therapeutics. Nat. Rev. Cancer 2004, 4, 296–307. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Chai, R.; Li, G.; Liu, Y.; Zhang, K.; Zhao, Z.; Wu, F.; Chang, Y.; Pang, B.; Li, J.; Li, Y.; et al. Predictive value of MGMT promoter methylation on the survival of TMZ treated IDH-mutant glioblastoma. Cancer Biol. Med. 2021, 18, 272–282. [Google Scholar] [CrossRef]
- Jin, K.; Zhang, S.Y.; Li, L.W.; Zou, Y.F.; Wu, B.; Xia, L.; Sun, C.X. Prognosis of Oligodendroglioma Patients Stratified by Age: A SEER Population-Based Analysis. Int. J. Gen. Med. 2021, 14, 9523–9536. [Google Scholar] [CrossRef] [PubMed]
- Sherman, J.H.; Prevedello, D.M.; Shah, L.; Raghavan, P.; Pouratian, N.; Starke, R.M.; Lopes, M.B.; Shaffrey, M.E.; Schiff, D. MR imaging characteristics of oligodendroglial tumors with assessment of 1p/19q deletion status. Acta Neurochir. (Wien) 2010, 152, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, K.; Snuderl, M. Molecular Pathology of Gliomas. Surg. Pathol. Clin. 2021, 14, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Bou Zerdan, M.; Assi, H.I. Oligodendroglioma: A Review of Management and Pathways. Front. Mol. Neurosci. 2021, 14, 722396. [Google Scholar] [CrossRef]
- Smits, M. Imaging of oligodendroglioma. Br. J. Radiol. 2016, 89, 20150857. [Google Scholar] [CrossRef]
- McNamara, C.; Mankad, K.; Thust, S.; Dixon, L.; Limback-Stanic, C.; D’Arco, F.; Jacques, T.S.; Lobel, U. 2021 WHO classification of tumours of the central nervous system: A review for the neuroradiologist. Neuroradiology 2022, 64, 1919–1950. [Google Scholar] [CrossRef]
- Bouvier-Labit, C.; Liprandi, A.; Monti, G.; Pellissier, J.F.; Figarella-Branger, D. CD44H is expressed by cells of the oligodendrocyte lineage and by oligodendrogliomas in humans. J. Neurooncol. 2002, 60, 127–134. [Google Scholar] [CrossRef]
- Lindberg, N.; Kastemar, M.; Olofsson, T.; Smits, A.; Uhrbom, L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene 2009, 28, 2266–2275. [Google Scholar] [CrossRef]
- Dai, C.; Celestino, J.C.; Okada, Y.; Louis, D.N.; Fuller, G.N.; Holland, E.C. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001, 15, 1913–1925. [Google Scholar] [CrossRef]
- Tirosh, I.; Venteicher, A.S.; Hebert, C.; Escalante, L.E.; Patel, A.P.; Yizhak, K.; Fisher, J.M.; Rodman, C.; Mount, C.; Filbin, M.G.; et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 2016, 539, 309–313. [Google Scholar] [CrossRef]
- Griffin, C.A.; Burger, P.; Morsberger, L.; Yonescu, R.; Swierczynski, S.; Weingart, J.D.; Murphy, K.M. Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J. Neuropathol. Exp. Neurol. 2006, 65, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.B.; Blair, H.; Ballman, K.V.; Giannini, C.; Arusell, R.M.; Law, M.; Flynn, H.; Passe, S.; Felten, S.; Brown, P.D.; et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006, 66, 9852–9861. [Google Scholar] [CrossRef] [PubMed]
- Arita, H.; Narita, Y.; Fukushima, S.; Tateishi, K.; Matsushita, Y.; Yoshida, A.; Miyakita, Y.; Ohno, M.; Collins, V.P.; Kawahara, N.; et al. Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol. 2013, 126, 267–276. [Google Scholar] [CrossRef]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef]
- Bettegowda, C.; Agrawal, N.; Jiao, Y.; Sausen, M.; Wood, L.D.; Hruban, R.H.; Rodriguez, F.J.; Cahill, D.P.; McLendon, R.; Riggins, G.; et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 2011, 333, 1453–1455. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.; Butterfield, Y.S.; Morozova, O.; Chittaranjan, S.; Blough, M.D.; An, J.; Birol, I.; Chesnelong, C.; Chiu, R.; Chuah, E.; et al. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J. Pathol. 2012, 226, 7–16. [Google Scholar] [CrossRef]
- Wesseling, P.; van den Bent, M.; Perry, A. Oligodendroglioma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 809–827. [Google Scholar] [CrossRef]
- Arvold, N.D.; Lee, E.Q.; Mehta, M.P.; Margolin, K.; Alexander, B.M.; Lin, N.U.; Anders, C.K.; Soffietti, R.; Camidge, D.R.; Vogelbaum, M.A.; et al. Updates in the management of brain metastases. Neuro Oncol. 2016, 18, 1043–1065. [Google Scholar] [CrossRef]
- Wiestler, B.; Capper, D.; Sill, M.; Jones, D.T.; Hovestadt, V.; Sturm, D.; Koelsche, C.; Bertoni, A.; Schweizer, L.; Korshunov, A.; et al. Integrated DNA methylation and copy-number profiling identify three clinically and biologically relevant groups of anaplastic glioma. Acta Neuropathol. 2014, 128, 561–571. [Google Scholar] [CrossRef]
- Wiestler, B.; Capper, D.; Hovestadt, V.; Sill, M.; Jones, D.T.; Hartmann, C.; Felsberg, J.; Platten, M.; Feiden, W.; Keyvani, K.; et al. Assessing CpG island methylator phenotype, 1p/19q codeletion, and MGMT promoter methylation from epigenome-wide data in the biomarker cohort of the NOA-04 trial. Neuro Oncol. 2014, 16, 1630–1638. [Google Scholar] [CrossRef]
- Figarella-Branger, D.; Mokhtari, K.; Dehais, C.; Carpentier, C.; Colin, C.; Jouvet, A.; Uro-Coste, E.; Forest, F.; Maurage, C.A.; Vignaud, J.M.; et al. Mitotic index, microvascular proliferation, and necrosis define 3 pathological subgroups of prognostic relevance among 1p/19q co-deleted anaplastic oligodendrogliomas. Neuro Oncol. 2016, 18, 888–890. [Google Scholar] [CrossRef] [PubMed]
- Giannini, C.; Scheithauer, B.W.; Weaver, A.L.; Burger, P.C.; Kros, J.M.; Mork, S.; Graeber, M.B.; Bauserman, S.; Buckner, J.C.; Burton, J.; et al. Oligodendrogliomas: Reproducibility and prognostic value of histologic diagnosis and grading. J. Neuropathol. Exp. Neurol. 2001, 60, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Tesileanu, C.M.S.; Dirven, L.; Wijnenga, M.M.J.; Koekkoek, J.A.F.; Vincent, A.; Dubbink, H.J.; Atmodimedjo, P.N.; Kros, J.M.; van Duinen, S.G.; Smits, M.; et al. Survival of diffuse astrocytic glioma, IDH1/2 wildtype, with molecular features of glioblastoma, WHO grade IV: A confirmation of the cIMPACT-NOW criteria. Neuro Oncol. 2020, 22, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaih, A.; Reardon, D.A.; Cloughesy, T.; Sumrall, A.; Baehring, J.; van den Bent, M.; Bahr, O.; et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro Oncol. 2023, 25, 123–134. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cote, D.J.; Ascha, M.; Kruchko, C.; Barnholtz-Sloan, J.S. Adult Glioma Incidence and Survival by Race or Ethnicity in the United States From 2000 to 2014. JAMA Oncol. 2018, 4, 1254–1262. [Google Scholar] [CrossRef]
- Dono, A.; Wang, E.; Lopez-Rivera, V.; Ramesh, A.V.; Tandon, N.; Ballester, L.Y.; Esquenazi, Y. Molecular characteristics and clinical features of multifocal glioblastoma. J. Neurooncol. 2020, 148, 389–397. [Google Scholar] [CrossRef]
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef]
- Aprile, I.; Giorgi, C.; Guiducci, A.; Conti, G.; Ottaviano, I.; Ottaviano, P. Characterization of glioblastoma by contrast-enhanced flair sequences. Neuroradiol. J. 2008, 21, 196–203. [Google Scholar] [CrossRef]
- Canoll, P.; Goldman, J.E. The interface between glial progenitors and gliomas. Acta Neuropathol. 2008, 116, 465–477. [Google Scholar] [CrossRef]
- Alcantara Llaguno, S.; Chen, J.; Kwon, C.H.; Jackson, E.L.; Li, Y.; Burns, D.K.; Alvarez-Buylla, A.; Parada, L.F. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009, 15, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Bjorland, L.S.; Daehli Kurz, K.; Fluge, O.; Gilje, B.; Mahesparan, R.; Saetran, H.; Ushakova, A.; Farbu, E. Butterfly glioblastoma: Clinical characteristics, treatment strategies and outcomes in a population-based cohort. Neurooncol. Adv. 2022, 4, vdac102. [Google Scholar] [CrossRef]
- Abou-El-Ardat, K.; Seifert, M.; Becker, K.; Eisenreich, S.; Lehmann, M.; Hackmann, K.; Rump, A.; Meijer, G.; Carvalho, B.; Temme, A.; et al. Comprehensive molecular characterization of multifocal glioblastoma proves its monoclonal origin and reveals novel insights into clonal evolution and heterogeneity of glioblastomas. Neuro Oncol. 2017, 19, 546–557. [Google Scholar] [CrossRef]
- Kikuchi, Z.; Shibahara, I.; Yamaki, T.; Yoshioka, E.; Shofuda, T.; Ohe, R.; Matsuda, K.I.; Saito, R.; Kanamori, M.; Kanemura, Y.; et al. TERT promoter mutation associated with multifocal phenotype and poor prognosis in patients with IDH wild-type glioblastoma. Neurooncol. Adv. 2020, 2, vdaa114. [Google Scholar] [CrossRef]
- Kong, D.S.; Song, S.Y.; Kim, D.H.; Joo, K.M.; Yoo, J.S.; Koh, J.S.; Dong, S.M.; Suh, Y.L.; Lee, J.I.; Park, K.; et al. Prognostic significance of c-Met expression in glioblastomas. Cancer 2009, 115, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Ferris, S.P.; Hofmann, J.W.; Solomon, D.A.; Perry, A. Characterization of gliomas: From morphology to molecules. Virchows Arch. 2017, 471, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.; Aldape, K.D.; George, D.H.; Burger, P.C. Small cell astrocytoma: An aggressive variant that is clinicopathologically and genetically distinct from anaplastic oligodendroglioma. Cancer 2004, 101, 2318–2326. [Google Scholar] [CrossRef]
- Meis, J.M.; Ho, K.L.; Nelson, J.S. Gliosarcoma: A histologic and immunohistochemical reaffirmation. Mod. Pathol. 1990, 3, 19–24. [Google Scholar]
- Poyuran, R.; Chandrasekharan, K.; Easwer, H.V.; Narasimhaiah, D. Glioblastoma with primitive neuronal component: An immunohistochemical study and review of literature. J. Clin. Neurosci. 2021, 93, 130–136. [Google Scholar] [CrossRef]
- Rodriguez, F.J.; Scheithauer, B.W.; Giannini, C.; Bryant, S.C.; Jenkins, R.B. Epithelial and pseudoepithelial differentiation in glioblastoma and gliosarcoma: A comparative morphologic and molecular genetic study. Cancer 2008, 113, 2779–2789. [Google Scholar] [CrossRef]
- Larkin, C.J.; Jennings, L.J.; Heimberger, A.B.; Horbinski, C. Next-Generation Sequencing of a Glioblastoma with True Epithelial Differentiation. J. Neuropathol. Exp. Neurol. 2022, 81, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Rickert, C.H.; Riemenschneider, M.J.; Schachenmayr, W.; Richter, H.P.; Bockhorn, J.; Reifenberger, G.; Paulus, W. Glioblastoma with adipocyte-like tumor cell differentiation—Histological and molecular features of a rare differentiation pattern. Brain Pathol. 2009, 19, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Mork, S.J.; Rubinstein, L.J.; Kepes, J.J.; Perentes, E.; Uphoff, D.F. Patterns of epithelial metaplasia in malignant gliomas. II. Squamous differentiation of epithelial-like formations in gliosarcomas and glioblastomas. J. Neuropathol. Exp. Neurol. 1988, 47, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Zhu, X.; Wang, Y.; Liu, B.; Yang, X.; Wang, Q.; Du, J.; Ma, Y.; Lin, L.; Fu, P.; et al. Clinicopathological, Immunohistochemical and Molecular Genetic Study on Epithelioid Glioblastoma: A Series of Fifteen Cases with Literature Review. Onco Targets Ther. 2020, 13, 3943–3952. [Google Scholar] [CrossRef] [PubMed]
- Geddes, J.F.; Thom, M.; Robinson, S.F.; Revesz, T. Granular cell change in astrocytic tumors. Am. J. Surg. Pathol. 1996, 20, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Verdugo, E.; Puerto, I.; Medina, M.A. An update on the molecular biology of glioblastoma, with clinical implications and progress in its treatment. Cancer Commun. (Lond.) 2022, 42, 1083–1111. [Google Scholar] [CrossRef]
- Zhang, Y.; Dube, C.; Gibert, M., Jr.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef]
- Peraud, A.; Watanabe, K.; Plate, K.H.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. p53 mutations versus EGF receptor expression in giant cell glioblastomas. J. Neuropathol. Exp. Neurol. 1997, 56, 1236–1241. [Google Scholar] [CrossRef]
- Kleinschmidt-DeMasters, B.K.; Aisner, D.L.; Birks, D.K.; Foreman, N.K. Epithelioid GBMs show a high percentage of BRAF V600E mutation. Am. J. Surg. Pathol. 2013, 37, 685–698. [Google Scholar] [CrossRef]
- Lee, M.; Kang, S.Y.; Suh, Y.L. Genetic Alterations of Epidermal Growth Factor Receptor in Glioblastoma: The Usefulness of Immunohistochemistry. Appl. Immunohistochem. Mol. Morphol. 2019, 27, 589–598. [Google Scholar] [CrossRef]
- Hasselblatt, M.; Jaber, M.; Reuss, D.; Grauer, O.; Bibo, A.; Terwey, S.; Schick, U.; Ebel, H.; Niederstadt, T.; Stummer, W.; et al. Diffuse Astrocytoma, IDH-Wildtype: A Dissolving Diagnosis. J. Neuropathol. Exp. Neurol. 2018, 77, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Weber, R.G.; Willscher, E.; Riehmer, V.; Hentschel, B.; Kreuz, M.; Felsberg, J.; Beyer, U.; Loffler-Wirth, H.; Kaulich, K.; et al. Molecular classification of diffuse cerebral WHO grade II/III gliomas using genome- and transcriptome-wide profiling improves stratification of prognostically distinct patient groups. Acta Neuropathol. 2015, 129, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Aibaidula, A.; Chan, A.K.; Shi, Z.; Li, Y.; Zhang, R.; Yang, R.; Li, K.K.; Chung, N.Y.; Yao, Y.; Zhou, L.; et al. Adult IDH wild-type lower-grade gliomas should be further stratified. Neuro Oncol. 2017, 19, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Nakamura, H.; Suzuki, H.; Matsuo, K.; Kataoka, K.; Shimamura, T.; Motomura, K.; Ohka, F.; Shiina, S.; Yamamoto, T.; et al. Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro Oncol. 2018, 20, 66–77. [Google Scholar] [CrossRef]
- Stichel, D.; Ebrahimi, A.; Reuss, D.; Schrimpf, D.; Ono, T.; Shirahata, M.; Reifenberger, G.; Weller, M.; Hanggi, D.; Wick, W.; et al. Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol. 2018, 136, 793–803. [Google Scholar] [CrossRef]
- Fujimoto, K.; Arita, H.; Satomi, K.; Yamasaki, K.; Matsushita, Y.; Nakamura, T.; Miyakita, Y.; Umehara, T.; Kobayashi, K.; Tamura, K.; et al. TERT promoter mutation status is necessary and sufficient to diagnose IDH-wildtype diffuse astrocytic glioma with molecular features of glioblastoma. Acta Neuropathol. 2021, 142, 323–338. [Google Scholar] [CrossRef]
- Jeong, D.E.; Woo, S.R.; Nam, H.; Nam, D.H.; Lee, J.H.; Joo, K.M. Preclinical and clinical implications of TERT promoter mutation in glioblastoma multiforme. Oncol. Lett. 2017, 14, 8213–8219. [Google Scholar] [CrossRef]
- Diplas, B.H.; He, X.; Brosnan-Cashman, J.A.; Liu, H.; Chen, L.H.; Wang, Z.; Moure, C.J.; Killela, P.J.; Loriaux, D.B.; Lipp, E.S.; et al. The genomic landscape of TERT promoter wildtype-IDH wildtype glioblastoma. Nat. Commun. 2018, 9, 2087. [Google Scholar] [CrossRef]
- Karschnia, P.; Young, J.S.; Dono, A.; Hani, L.; Juenger, S.T.; Sciortino, T.; Bruno, F.; Teske, N.; Morshed, R.A.; Haddad, A.F.; et al. TERT promotor status does not add prognostic information in IDH-wildtype glioblastomas fulfilling other diagnostic WHO criteria: A report of the RANO resect group. Neurooncol. Adv. 2022, 4, vdac158. [Google Scholar] [CrossRef]
- Binabaj, M.M.; Bahrami, A.; ShahidSales, S.; Joodi, M.; Joudi Mashhad, M.; Hassanian, S.M.; Anvari, K.; Avan, A. The prognostic value of MGMT promoter methylation in glioblastoma: A meta-analysis of clinical trials. J. Cell. Physiol. 2018, 233, 378–386. [Google Scholar] [CrossRef]
- Rivera, A.L.; Pelloski, C.E.; Gilbert, M.R.; Colman, H.; De La Cruz, C.; Sulman, E.P.; Bekele, B.N.; Aldape, K.D. MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neuro Oncol. 2010, 12, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigenic Target | Signal | Diagnostic Implications | Caveats |
|---|---|---|---|
| IDH1 p.R132H | Cytoplasmic POSITIVITY in tumour expressing the mutant IDH1 R132H epitope | The tumour harbours the most common IDH mutation stratifying it into the IDH-mutant astrocytoma vs. IDH-mutant oligodendroglioma diagnostic pathway. | A small percentage of IDH mutant gliomas are immunonegative secondary to non-R132H mutations in IDH1 or mutations affecting IDH2 requiring a secondary genetic assay for verification. |
| ATRX | Nuclear POSITIVITY in tumour cells is associated with no genomic alteration of ATRX | In the context of an IDH mutant glioma, the presence of ATRX nuclear expression is associated with oligodendroglioma but requires confirmatory genetic testing for 1p19q-codeletion (LOH PCR, FISH, copy number microarray, etc.). This is also associated with TERT mutation. The presence of nuclear ATRX expression is also seen in a majority of IDH-wildtype glioblastomas, while loss of nuclear staining is associated with IDH-mutant astrocytoma | Can be technically challenging to interpret. Best performed in conjunction with internal positive controls such as endothelial cells. |
| H3 p.K28M (K27M) | Nuclear POSITIVITY in tumour cells expressing the mutant H3 p.K28M (K27M) epitope | This mutant epitope is found in DMG and defines this entity. However, there are other less common alterations that are also associated with this diagnosis. | Can be technically challenging to interpret and DMGs harbouring less common disease-defining alterations affecting codon 27 (e.g., H3 p.K28I [K27I]), EZHIP over-expression, or EGFR mutation are not identified with this antibody. Note this antibody should be used in conjunction with the H3 p.K28me3 (K27me3) antibody to increase accuracy. This alteration is not specific to DMG and can also be relevant in the workup of ependymomas. |
| H3 p.K28me3 (K27me3) | Nuclear POSITIVITY in tumour cells is associated with the preservation of the tri-methylated mark of H3 K27, which is anti-correlated with mutations of H3 K27. | LOSS of H3 K27me3 nuclear reactivity supports the POSITIVE IHC finding of H3 K27M mutation. It also provides supporting evidence of a DMG in cases with non-H3 K27M drivers. | Interpretation is best performed in conjunction with H3 p.K28M (K27M) IHC. |
| H3 p.G35R (G34R) | Nuclear POSITIVITY in tumour cells expressing the mutant H3 G34R epitope | This mutant epitope is found in DHG and defines this entity. However, the less common alteration, H3 p.G35V (G34V), is also associated with this diagnosis. | Can be technically challenging to interpret and false negative immunoreactivity in H3 G34R mutant DHG cases has been described. Additionally, the less common H3 G34V mutant cases are not identified with this antibody. |
| BRAF p.V600E | Cytoplasmic POSITIVITY in tumours with BRAF p.V600E mutation. | Supports the diagnosis of an epithelioid glioblastoma, ganglioglioma, and other low-grade gliomas but interpretation is context dependent. | The p.V600E mutation is found in multiple types of brain tumours and its interpretation has to be made in an integrative manner. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, K.C.; Ma, C.; Yip, S. From Theory to Practice: Implementing the WHO 2021 Classification of Adult Diffuse Gliomas in Neuropathology Diagnosis. Brain Sci. 2023, 13, 817. https://doi.org/10.3390/brainsci13050817
Martin KC, Ma C, Yip S. From Theory to Practice: Implementing the WHO 2021 Classification of Adult Diffuse Gliomas in Neuropathology Diagnosis. Brain Sciences. 2023; 13(5):817. https://doi.org/10.3390/brainsci13050817
Chicago/Turabian StyleMartin, Karina Chornenka, Crystal Ma, and Stephen Yip. 2023. "From Theory to Practice: Implementing the WHO 2021 Classification of Adult Diffuse Gliomas in Neuropathology Diagnosis" Brain Sciences 13, no. 5: 817. https://doi.org/10.3390/brainsci13050817
APA StyleMartin, K. C., Ma, C., & Yip, S. (2023). From Theory to Practice: Implementing the WHO 2021 Classification of Adult Diffuse Gliomas in Neuropathology Diagnosis. Brain Sciences, 13(5), 817. https://doi.org/10.3390/brainsci13050817