
Acid-Sensing Ion Channel 1a Contributes to the Prefrontal Cortex Ischemia-Enhanced Neuronal Activities in the Amygdala



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. ET-1-Induced Brain Ischemia Model and Chemical Infusion
2.3. Brain Slice Preparation and Patch–Clamp Recording of Amygdala Pyramidal Neurons
2.4. Tail Suspension and Forced Swimming Tests
2.5. Statistical Analysis
3. Results
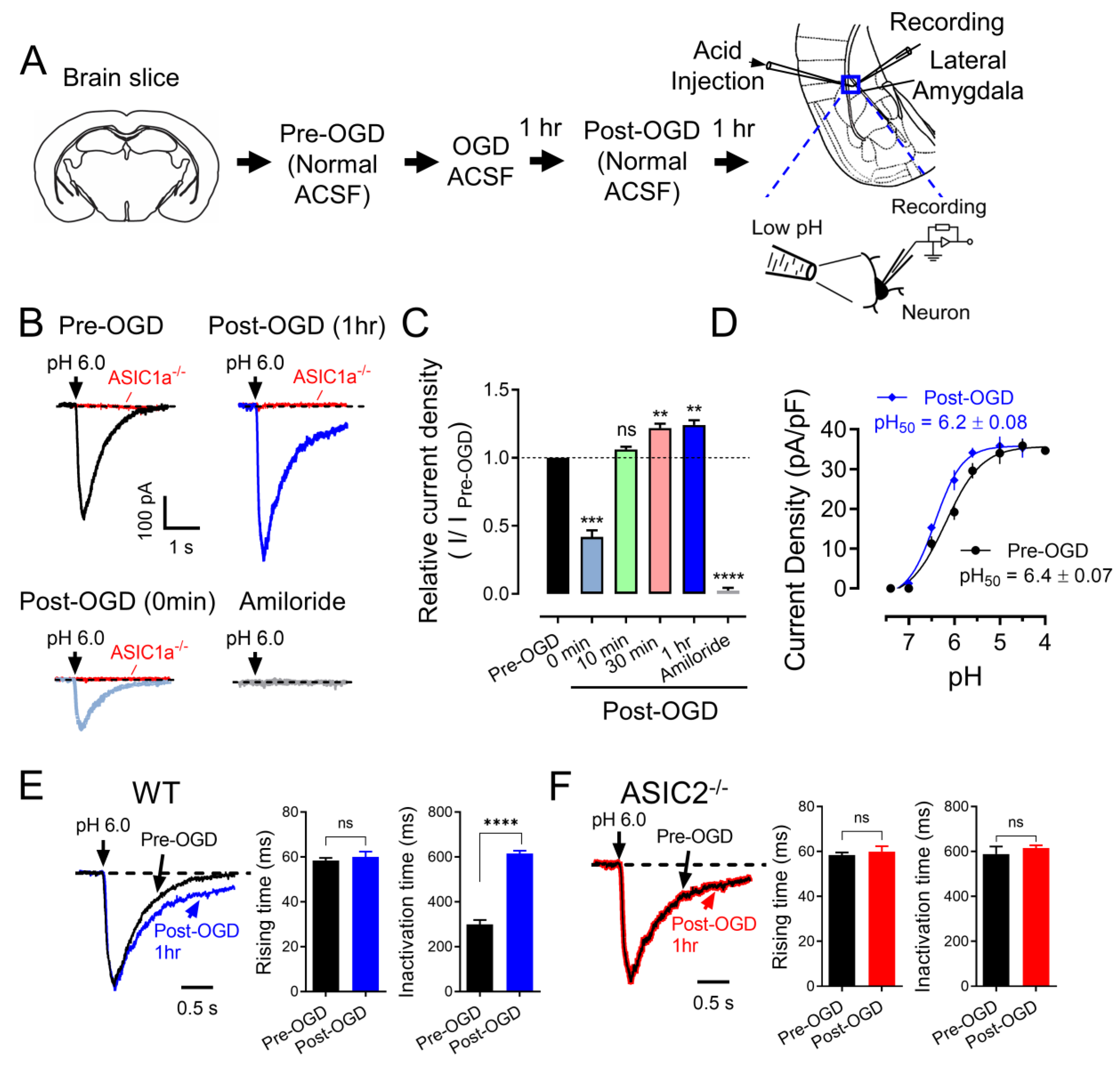
3.1. OGD (In Vitro Ischemia) Enhances ASIC Currents in the Amygdala
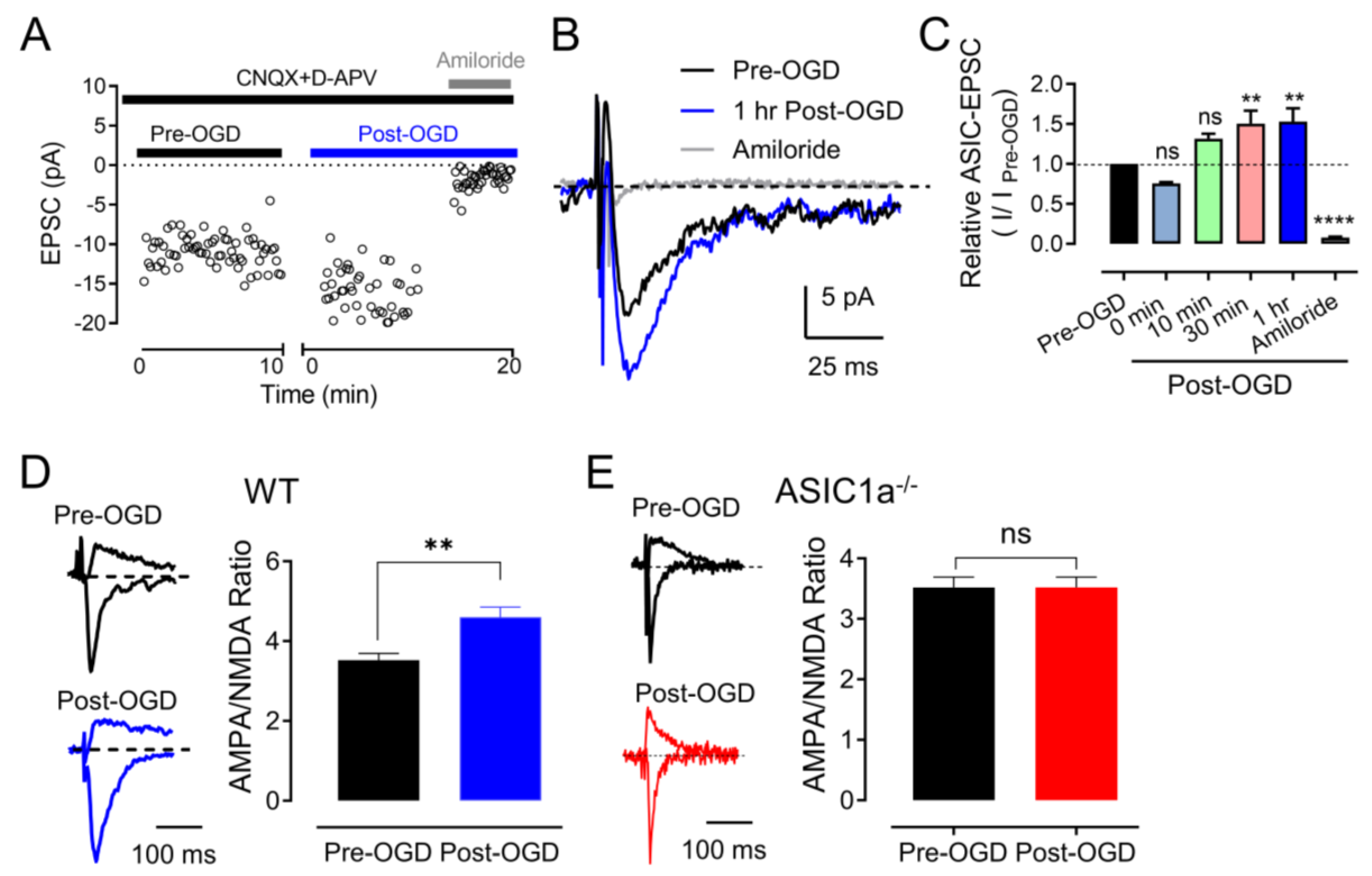
3.2. OGD (In Vitro Ischemia) Enhances ASIC1a–Dependent Synaptic Transmission in the Amygdala
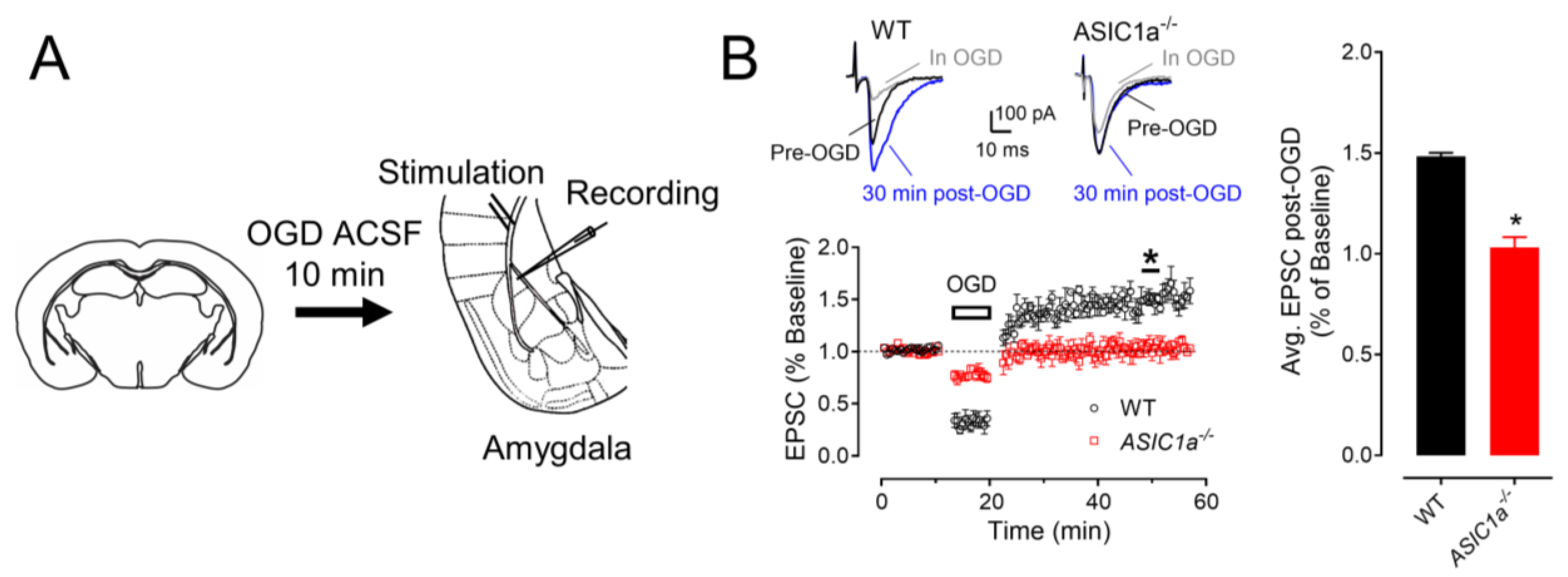
3.3. ASIC1a Contributes to OGD–Induced LTP
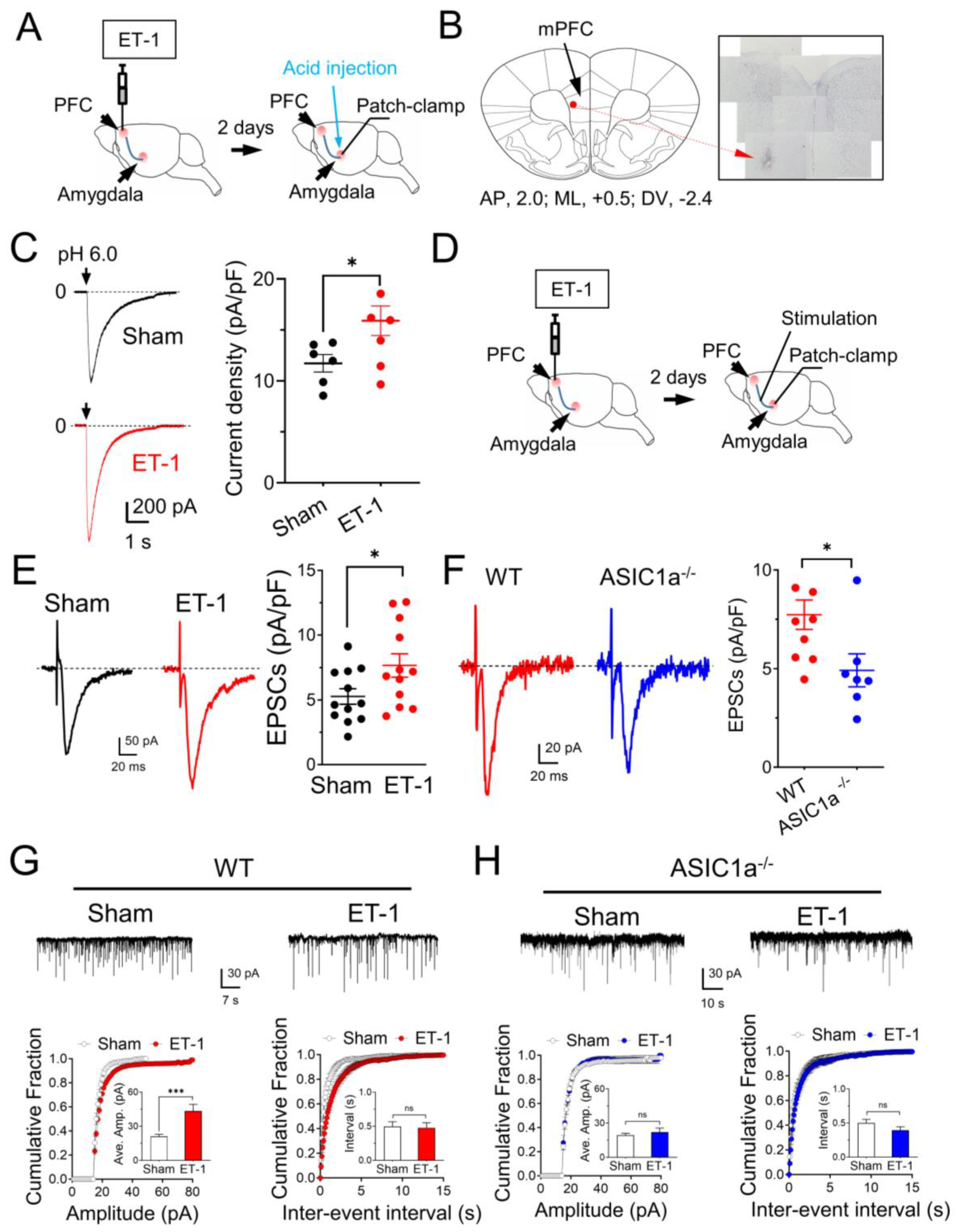
3.4. In Vivo Ischemia Enhances ASIC Currents and Neuronal Activities in the Amygdala
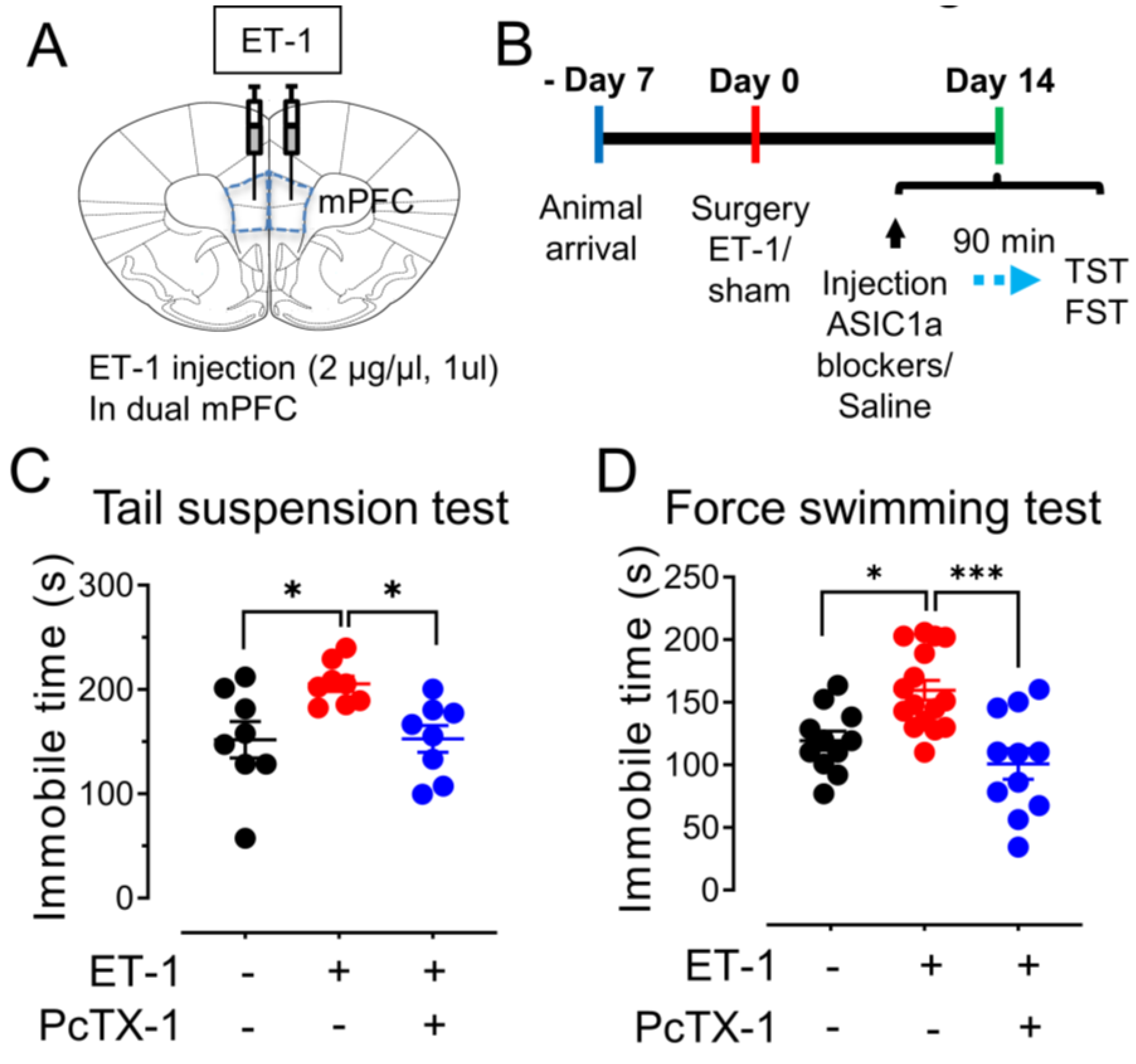
3.5. The mPFC Ischemia–Induced Depression–like Behaviors Are ASICs–Dependent
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hackett, M.L.; Kohler, S.; O’Brien, J.T.; Mead, G.E. Neuropsychiatric outcomes of stroke. Lancet Neurol. 2014, 13, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.H.; Corbett, D. Plasticity during stroke recovery: From synapse to behaviour. Nat. Rev. Neurosci. 2009, 10, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Xerri, C.; Zennou-Azogui, Y.; Sadlaoud, K.; Sauvajon, D. Interplay between intra- and interhemispheric remodeling of neural networks as a substrate of functional recovery after stroke: Adaptive versus maladaptive reorganization. Neuroscience 2014, 283, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Naghavi, F.S.; Koffman, E.E.; Lin, B.; Du, J. Post-stroke neuronal circuits and mental illnesses. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 1–11. [Google Scholar] [PubMed]
- Janak, P.H.; Tye, K.M. From circuits to behaviour in the amygdala. Nature 2015, 517, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Forkert, N.D.; Zavaglia, M.; Hilgetag, C.C.; Golsari, A.; Siemonsen, S.; Fiehler, J.; Pedraza, S.; Puig, J.; Cho, T.H.; et al. Influence of stroke infarct location on functional outcome measured by the modified rankin scale. Stroke 2014, 45, 1695–1702. [Google Scholar] [CrossRef] [PubMed]
- Rehncrona, S. Brain acidosis. Ann. Emerg. Med. 1985, 14, 770–776. [Google Scholar] [CrossRef]
- Siesjo, B.K.; Katsura, K.; Kristian, T. Acidosis-related damage. Adv. Neurol. 1996, 71, 209–233. [Google Scholar]
- Nedergaard, M.; Kraig, R.P.; Tanabe, J.; Pulsinelli, W.A. Dynamics of interstitial and intracellular pH in evolving brain infarct. Am. J. Physiol. 1991, 260, R581–R588. [Google Scholar] [CrossRef]
- Siesjo, B.K. Acidosis and ischemic brain damage. Neurochem. Pathol. 1988, 9, 31–88. [Google Scholar] [CrossRef]
- Choi, D.W. Calcium: Still center-stage in hypoxic-ischemic neuronal death. Trends Neurosci. 1995, 18, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Reznikov, L.R.; Price, M.P.; Zha, X.M.; Lu, Y.; Moninger, T.O.; Wemmie, J.A.; Welsh, M.J. Protons are a neurotransmitter that regulates synaptic plasticity in the lateral amygdala. Proc. Natl. Acad. Sci. USA 2014, 111, 8961–8966. [Google Scholar] [CrossRef] [PubMed]
- Coryell, M.W.; Wunsch, A.M.; Haenfler, J.M.; Allen, J.E.; Schnizler, M.; Ziemann, A.E.; Cook, M.N.; Dunning, J.P.; Price, M.P.; Rainier, J.D.; et al. Acid-sensing ion channel-1a in the amygdala, a novel therapeutic target in depression-related behavior. J. Neurosci. 2009, 29, 5381–5388. [Google Scholar] [CrossRef] [PubMed]
- Wemmie, J.A.; Coryell, M.W.; Askwith, C.C.; Lamani, E.; Leonard, A.S.; Sigmund, C.D.; Welsh, M.J. Overexpression of acid-sensing ion channel 1a in transgenic mice increases acquired fear-related behavior. Proc. Natl. Acad. Sci. USA 2004, 101, 3621–3626. [Google Scholar] [CrossRef] [PubMed]
- Coryell, M.W.; Ziemann, A.E.; Westmoreland, P.J.; Haenfler, J.M.; Kurjakovic, Z.; Zha, X.M.; Price, M.; Schnizler, M.K.; Wemmie, J.A. Targeting ASIC1a reduces innate fear and alters neuronal activity in the fear circuit. Biol. Psychiatry 2007, 62, 1140–1148. [Google Scholar] [CrossRef]
- Waldmann, R.; Champigny, G.; Bassilana, F.; Heurteaux, C.; Lazdunski, M. A proton-gated cation channel involved in acid-sensing. Nature 1997, 386, 173–177. [Google Scholar] [CrossRef]
- Waldmann, R.; Lazdunski, M. H(+)-gated cation channels: Neuronal acid sensors in the NaC/DEG family of ion channels. Curr. Opin. Neurobiol. 1998, 8, 418–424. [Google Scholar] [CrossRef]
- Xiong, Z.G.; Zhu, X.M.; Chu, X.P.; Minami, M.; Hey, J.; Wei, W.L.; MacDonald, J.F.; Wemmie, J.A.; Price, M.P.; Welsh, M.J.; et al. Neuroprotection in ischemia: Blocking calcium-permeable acid-sensing ion channels. Cell 2004, 118, 687–698. [Google Scholar] [CrossRef]
- Chesler, M. Regulation and modulation of pH in the brain. Physiol. Rev. 2003, 83, 1183–1221. [Google Scholar] [CrossRef]
- Takmakov, P.; Zachek, M.K.; Keithley, R.B.; Bucher, E.S.; McCarty, G.S.; Wightman, R.M. Characterization of local pH changes in brain using fast-scan cyclic voltammetry with carbon microelectrodes. Anal. Chem. 2010, 82, 9892–9900. [Google Scholar] [CrossRef]
- Kai Kaila, B.R.R. pH and Brain Function, 1st ed.; Wiley-Liss: New York City, NY, USA, 1998; p. 688. [Google Scholar]
- Du, J.; Price, M.P.; Taugher, R.J.; Grigsby, D.; Ash, J.J.; Stark, A.C.; Hossain Saad, M.Z.; Singh, K.; Mandal, J.; Wemmie, J.A.; et al. Transient acidosis while retrieving a fear-related memory enhances its lability. eLife 2017, 6, e22564. [Google Scholar] [CrossRef] [PubMed]
- Bielajew, C.; Konkle, A.T.; Kentner, A.C.; Baker, S.L.; Stewart, A.; Hutchins, A.A.; Santa-Maria Barbagallo, L.; Fouriezos, G. Strain and gender specific effects in the forced swim test: Effects of previous stress exposure. Stress 2003, 6, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Fujisaki, C.; Utsuyama, M.; Kuroda, Y.; Watanabe, A.; Seidler, H.; Watanabe, S.; Kitagawa, M.; Hirokawa, K. An immnosuppressive drug, cyclosporine-A acts like anti-depressant for rats under unpredictable chronic stress. J. Med. Dent. Sci. 2003, 50, 93–100. [Google Scholar] [PubMed]
- Yankelevitch-Yahav, R.; Franko, M.; Huly, A.; Doron, R. The forced swim test as a model of depressive-like behavior. J. Vis. Exp. 2015, 97, e52587. [Google Scholar] [CrossRef]
- Askwith, C.C.; Wemmie, J.A.; Price, M.P.; Rokhlina, T.; Welsh, M.J. Acid-sensing ion channel 2 (ASIC2) modulates ASIC1 H+-activated currents in hippocampal neurons. J. Biol. Chem. 2004, 279, 18296–18305. [Google Scholar] [CrossRef]
- Rao, V.R.; Finkbeiner, S. NMDA and AMPA receptors: Old channels, new tricks. Trends Neurosci. 2007, 30, 284–291. [Google Scholar] [CrossRef]
- Calabresi, P.; Saulle, E.; Centonze, D.; Pisani, A.; Marfia, G.A.; Bernardi, G. Post-ischaemic long-term synaptic potentiation in the striatum: A putative mechanism for cell type-specific vulnerability. Brain 2002, 125, 844–860. [Google Scholar] [CrossRef]
- Horie, N.; Maag, A.L.; Hamilton, S.A.; Shichinohe, H.; Bliss, T.M.; Steinberg, G.K. Mouse model of focal cerebral ischemia using endothelin-1. J. Neurosci. Methods 2008, 173, 286–290. [Google Scholar] [CrossRef]
- Chan, H.H.; Cooperrider, J.L.; Park, H.J.; Wathen, C.A.; Gale, J.T.; Baker, K.B.; Machado, A.G. Crossed Cerebellar Atrophy of the Lateral Cerebellar Nucleus in an Endothelin-1-Induced, Rodent Model of Ischemic Stroke. Front. Aging Neurosci. 2017, 9, 10. [Google Scholar] [CrossRef]
- Xiong, Z.G.; Pignataro, G.; Li, M.; Chang, S.Y.; Simon, R.P. Acid-sensing ion channels (ASICs) as pharmacological targets for neurodegenerative diseases. Curr. Opin. Pharmacol. 2008, 8, 25–32. [Google Scholar] [CrossRef]
- Quintana, P.; Soto, D.; Poirot, O.; Zonouzi, M.; Kellenberger, S.; Muller, D.; Chrast, R.; Cull-Candy, S.G. Acid-sensing ion channel 1a drives AMPA receptor plasticity following ischaemia and acidosis in hippocampal CA1 neurons. J. Physiol. 2015, 593, 4373–4386. [Google Scholar] [CrossRef] [PubMed]
- Castagne, V.; Moser, P.; Porsolt, R.D. Behavioral Assessment of Antidepressant Activity in Rodents. In Methods of Behavior Analysis in Neuroscience, 2nd ed.; Buccafusco, J.J., Ed.; Frontiers in Neuroscience: Boca Raton, FL, USA, 2009. [Google Scholar]
- Hofmeijer, J.; van Putten, M.J. Ischemic cerebral damage: An appraisal of synaptic failure. Stroke 2012, 43, 607–615. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, G.; Ge, Q.; Jin, Z.; Du, J. Acid-Sensing Ion Channel 1a Contributes to the Prefrontal Cortex Ischemia-Enhanced Neuronal Activities in the Amygdala. Brain Sci. 2023, 13, 1684. https://doi.org/10.3390/brainsci13121684
Park G, Ge Q, Jin Z, Du J. Acid-Sensing Ion Channel 1a Contributes to the Prefrontal Cortex Ischemia-Enhanced Neuronal Activities in the Amygdala. Brain Sciences. 2023; 13(12):1684. https://doi.org/10.3390/brainsci13121684
Chicago/Turabian StylePark, Gyeongah, Qian Ge, Zhen Jin, and Jianyang Du. 2023. "Acid-Sensing Ion Channel 1a Contributes to the Prefrontal Cortex Ischemia-Enhanced Neuronal Activities in the Amygdala" Brain Sciences 13, no. 12: 1684. https://doi.org/10.3390/brainsci13121684
APA StylePark, G., Ge, Q., Jin, Z., & Du, J. (2023). Acid-Sensing Ion Channel 1a Contributes to the Prefrontal Cortex Ischemia-Enhanced Neuronal Activities in the Amygdala. Brain Sciences, 13(12), 1684. https://doi.org/10.3390/brainsci13121684