
Reelin Signaling in Neurodevelopmental Disorders and Neurodegenerative Diseases


{kind=link}
{kind=link}
Abstract
1. Introduction
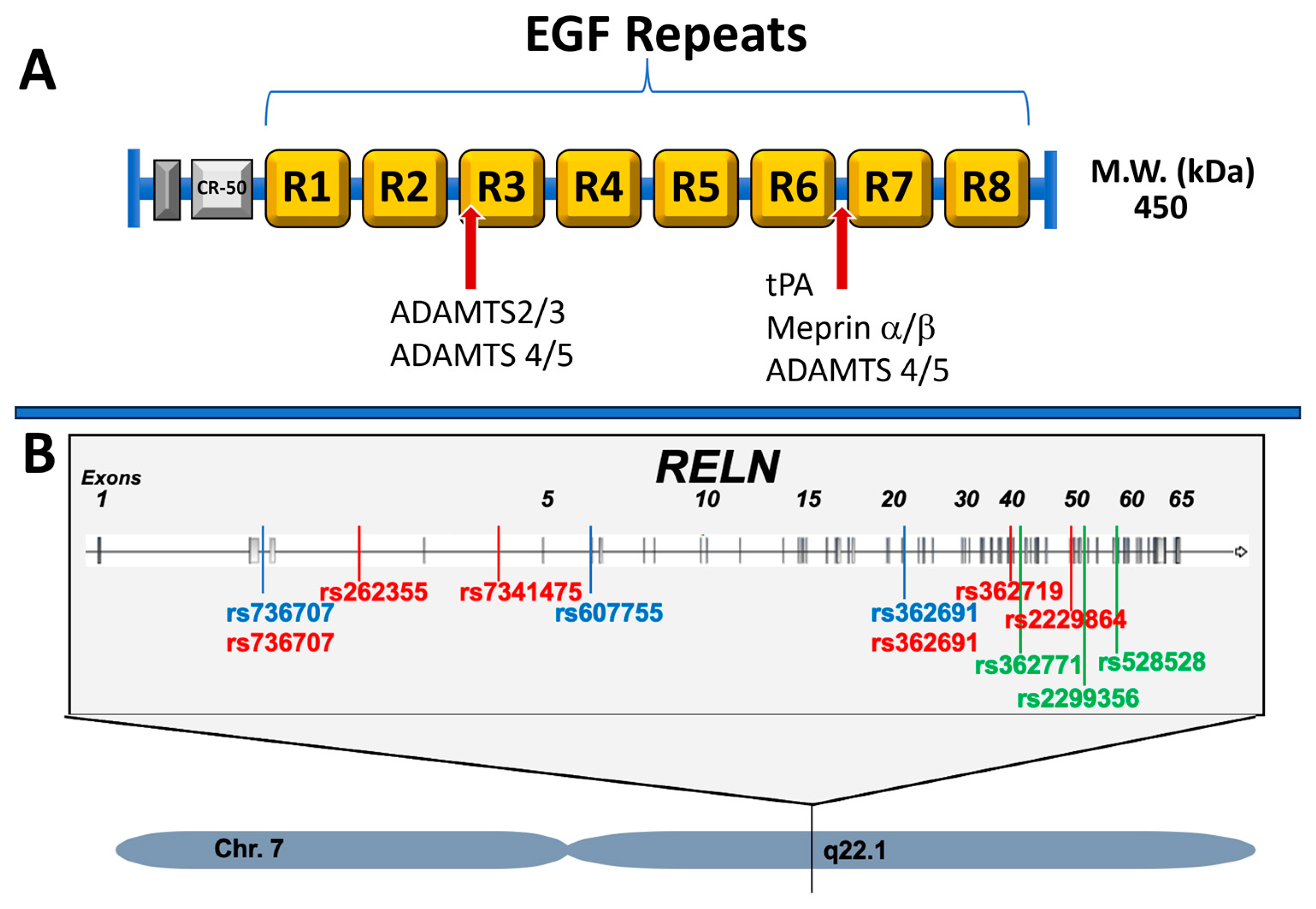
1.1. Reelin Protein
1.2. Reelin Signaling
2. Reelin in Aging and Disease
2.1. Aging
2.2. Reelin in Ataxias
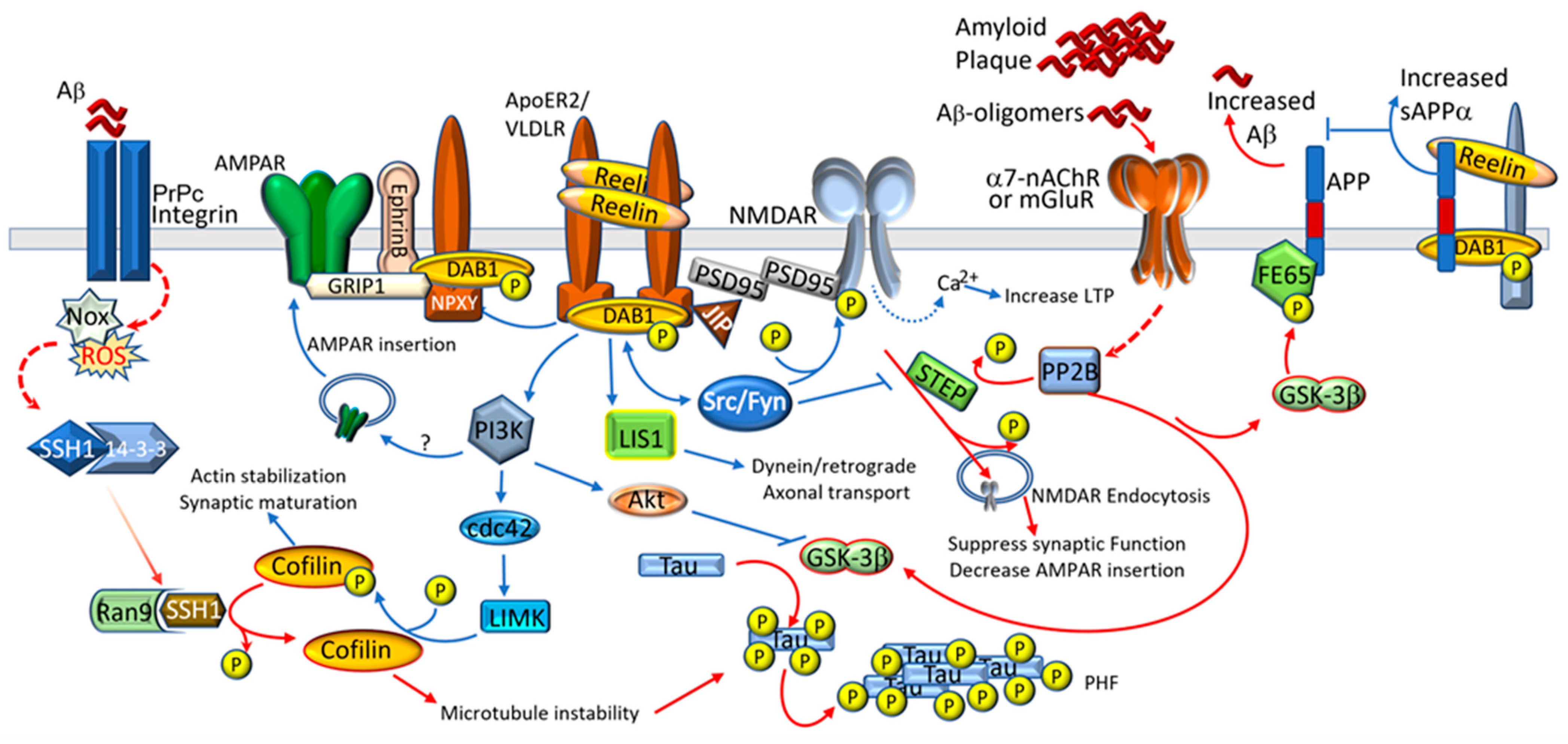
2.3. Alzheimer’s Disease
2.4. Reelin in Schizophrenia
2.5. Autism Spectrum Disorders
2.6. Traumatic Brain Injury
3. Potential Therapeutic Interventions
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Falconer, D.S. Two new mutants, ‘trembler’ and ‘reeler’, with neurological actions in the house mouse (Mus musculus L.). J. Genet. 1951, 50, 192–201. [Google Scholar] [CrossRef]
- Hamburgh, M. Analysis of the Postnatal Developmental Effects of “Reeler,” a Neurological Mutation in Mice. A Study in Developmental Genetics. Dev. Biol. 1963, 8, 165–185. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, G.; Miao, G.G.; Chen, S.C.; Soares, H.D.; Morgan, J.I.; Curran, T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 1995, 374, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Royaux, I.; Lambert de Rouvroit, C.; D’Arcangelo, G.; Demirov, D.; Goffinet, A.M. Genomic organization of the mouse reelin gene. Genomics 1997, 46, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Jossin, Y.; Ignatova, N.; Hiesberger, T.; Herz, J.; Lambert de Rouvroit, C.; Goffinet, A.M. The central fragment of Reelin, generated by proteolytic processing in vivo, is critical to its function during cortical plate development. J. Neurosci. 2004, 24, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Koie, M.; Okumura, K.; Hisanaga, A.; Kamei, T.; Sasaki, K.; Deng, M.; Baba, A.; Kohno, T.; Hattori, M. Cleavage within Reelin repeat 3 regulates the duration and range of the signaling activity of Reelin protein. J. Biol. Chem. 2014, 289, 12922–12930. [Google Scholar] [CrossRef] [PubMed]
- Krstic, D.; Rodriguez, M.; Knuesel, I. Regulated proteolytic processing of Reelin through interplay of tissue plasminogen activator (tPA), ADAMTS-4, ADAMTS-5, and their modulators. PLoS ONE 2012, 7, e47793. [Google Scholar] [CrossRef] [PubMed]
- Trotter, J.H.; Lussier, A.L.; Psilos, K.E.; Mahoney, H.L.; Sponaugle, A.E.; Hoe, H.S.; Rebeck, G.W.; Weeber, E.J. Extracellular proteolysis of reelin by tissue plasminogen activator following synaptic potentiation. Neuroscience 2014, 274, 299–307. [Google Scholar] [CrossRef]
- Sato, Y.; Kobayashi, D.; Kohno, T.; Kidani, Y.; Prox, J.; Becker-Pauly, C.; Hattori, M. Determination of cleavage site of Reelin between its sixth and seventh repeat and contribution of meprin metalloproteases to the cleavage. J. Biochem. 2016, 159, 305–312. [Google Scholar] [CrossRef][Green Version]
- Yamakage, Y.; Kato, M.; Hongo, A.; Ogino, H.; Ishii, K.; Ishizuka, T.; Kamei, T.; Tsuiji, H.; Miyamoto, T.; Oishi, H.; et al. A disintegrin and metalloproteinase with thrombospondin motifs 2 cleaves and inactivates Reelin in the postnatal cerebral cortex and hippocampus, but not in the cerebellum. Mol. Cell Neurosci. 2019, 100, 103401. [Google Scholar] [CrossRef]
- Ogino, H.; Hisanaga, A.; Kohno, T.; Kondo, Y.; Okumura, K.; Kamei, T.; Sato, T.; Asahara, H.; Tsuiji, H.; Fukata, M.; et al. Secreted Metalloproteinase ADAMTS-3 Inactivates Reelin. J. Neurosci. 2017, 37, 3181–3191. [Google Scholar] [CrossRef] [PubMed]
- Quattrocchi, C.C.; Wannenes, F.; Persico, A.M.; Ciafre, S.A.; D’Arcangelo, G.; Farace, M.G.; Keller, F. Reelin is a serine protease of the extracellular matrix. J. Biol. Chem. 2002, 277, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Hattori, M. Re-evaluation of protease activity of reelin. Biol. Pharm. Bull. 2010, 33, 1047–1049. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Kohno, T. Regulation of Reelin functions by specific proteolytic processing in the brain. J. Biochem. 2021, 169, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Pesold, C.; Impagnatiello, F.; Pisu, M.G.; Uzunov, D.P.; Costa, E.; Guidotti, A.; Caruncho, H.J. Reelin is preferentially expressed in neurons synthesizing gamma-aminobutyric acid in cortex and hippocampus of adult rats. Proc. Natl. Acad. Sci. USA 1998, 95, 3221–3226. [Google Scholar] [CrossRef] [PubMed]
- Stranahan, A.M.; Erion, J.R.; Wosiski-Kuhn, M. Reelin signaling in development, maintenance, and plasticity of neural networks. Ageing Res. Rev. 2013, 12, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Herz, J.; Chen, Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat. Rev. Neurosci. 2006, 7, 850–859. [Google Scholar] [CrossRef]
- Hirota, Y.; Kubo, K.; Katayama, K.; Honda, T.; Fujino, T.; Yamamoto, T.T.; Nakajima, K. Reelin receptors ApoER2 and VLDLR are expressed in distinct spatiotemporal patterns in developing mouse cerebral cortex. J. Comp. Neurol. 2015, 523, 463–478. [Google Scholar] [CrossRef]
- Jossin, Y. Reelin Functions, Mechanisms of Action and Signaling Pathways During Brain Development and Maturation. Biomolecules 2020, 10, 964. [Google Scholar] [CrossRef]
- Jossin, Y.; Goffinet, A.M. Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Mol. Cell Biol. 2007, 27, 7113–7124. [Google Scholar] [CrossRef]
- Ogawa, B.; Wang, L.; Ohishi, T.; Taniai, E.; Akane, H.; Suzuki, K.; Mitsumori, K.; Shibutani, M. Reversible aberration of neurogenesis targeting late-stage progenitor cells in the hippocampal dentate gyrus of rat offspring after maternal exposure to acrylamide. Arch. Toxicol. 2012, 86, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Frotscher, M. Role for Reelin in stabilizing cortical architecture. Trends Neurosci. 2010, 33, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Alcantara, S.; Ruiz, M.; D’Arcangelo, G.; Ezan, F.; de Lecea, L.; Curran, T.; Sotelo, C.; Soriano, E. Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J. Neurosci. 1998, 18, 7779–7799. [Google Scholar] [CrossRef] [PubMed]
- Hiesberger, T.; Trommsdorff, M.; Howell, B.W.; Goffinet, A.; Mumby, M.C.; Cooper, J.A.; Herz, J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 1999, 24, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Beffert, U.; Morfini, G.; Bock, H.H.; Reyna, H.; Brady, S.T.; Herz, J. Reelin-mediated signaling locally regulates protein kinase B/Akt and glycogen synthase kinase 3beta. J. Biol. Chem. 2002, 277, 49958–49964. [Google Scholar] [CrossRef] [PubMed]
- Trommsdorff, M.; Gotthardt, M.; Hiesberger, T.; Shelton, J.; Stockinger, W.; Nimpf, J.; Hammer, R.E.; Richardson, J.A.; Herz, J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 1999, 97, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Weeber, E.J.; Beffert, U.; Jones, C.; Christian, J.M.; Forster, E.; Sweatt, J.D.; Herz, J. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J. Biol. Chem. 2002, 277, 39944–39952. [Google Scholar] [CrossRef] [PubMed]
- Tissir, F.; Lambert de Rouvroit, C.; Goffinet, A.M. The role of reelin in the development and evolution of the cerebral cortex. Braz. J. Med. Biol. Res. 2002, 35, 1473–1484. [Google Scholar] [CrossRef]
- Jacquelin, C.; Lalonde, R.; Jantzen-Ossola, C.; Strazielle, C. Neurobehavioral performances and brain regional metabolism in Dab1(scm) (scrambler) mutant mice. Behav. Brain Res. 2013, 252, 92–100. [Google Scholar] [CrossRef]
- Howell, B.W.; Hawkes, R.; Soriano, P.; Cooper, J.A. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature 1997, 389, 733–737. [Google Scholar] [CrossRef]
- Sheldon, M.; Rice, D.S.; D’Arcangelo, G.; Yoneshima, H.; Nakajima, K.; Mikoshiba, K.; Howell, B.W.; Cooper, J.A.; Goldowitz, D.; Curran, T. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature 1997, 389, 730–733. [Google Scholar] [CrossRef]
- Alexander, A.; Herz, J.; Calvier, L. Reelin through the years: From brain development to inflammation. Cell Rep. 2023, 42, 112669. [Google Scholar] [CrossRef] [PubMed]
- Strasser, V.; Fasching, D.; Hauser, C.; Mayer, H.; Bock, H.H.; Hiesberger, T.; Herz, J.; Weeber, E.J.; Sweatt, J.D.; Pramatarova, A.; et al. Receptor clustering is involved in Reelin signaling. Mol. Cell Biol. 2004, 24, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, G.; Homayouni, R.; Keshvara, L.; Rice, D.S.; Sheldon, M.; Curran, T. Reelin is a ligand for lipoprotein receptors. Neuron 1999, 24, 471–479. [Google Scholar] [CrossRef]
- Howell, B.W.; Herrick, T.M.; Cooper, J.A. Reelin-induced tyrosine [corrected] phosphorylation of disabled 1 during neuronal positioning. Genes. Dev. 1999, 13, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Howell, B.W.; Herrick, T.M.; Hildebrand, J.D.; Zhang, Y.; Cooper, J.A. Dab1 tyrosine phosphorylation sites relay positional signals during mouse brain development. Curr. Biol. 2000, 10, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Jossin, Y.; Ogawa, M.; Metin, C.; Tissir, F.; Goffinet, A.M. Inhibition of SRC family kinases and non-classical protein kinases C induce a reeler-like malformation of cortical plate development. J. Neurosci. 2003, 23, 9953–9959. [Google Scholar] [CrossRef] [PubMed]
- Ballif, B.A.; Arnaud, L.; Cooper, J.A. Tyrosine phosphorylation of Disabled-1 is essential for Reelin-stimulated activation of Akt and Src family kinases. Mol. Brain Res. 2003, 117, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, L.; Ballif, B.A.; Forster, E.; Cooper, J.A. Fyn tyrosine kinase is a critical regulator of disabled-1 during brain development. Curr. Biol. 2003, 13, 9–17. [Google Scholar] [CrossRef]
- Bock, H.H.; Jossin, Y.; Liu, P.; Forster, E.; May, P.; Goffinet, A.M.; Herz, J. Phosphatidylinositol 3-kinase interacts with the adaptor protein Dab1 in response to Reelin signaling and is required for normal cortical lamination. J. Biol. Chem. 2003, 278, 38772–38779. [Google Scholar] [CrossRef]
- Lee, G.H.; Chhangawala, Z.; von Daake, S.; Savas, J.N.; Yates, J.R., 3rd; Comoletti, D.; D’Arcangelo, G. Reelin induces Erk1/2 signaling in cortical neurons through a non-canonical pathway. J. Biol. Chem. 2014, 289, 20307–20317. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Setsu, T.; Okuyama-Yamamoto, A.; Terashima, T. Histological study in the brain of the reelin/Dab1-compound mutant mouse. Anat. Sci. Int. 2009, 84, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, N.; Lee, Y.D.; Morishima, A.; Terashima, T.; Kikkawa, S.; Tohyama, M.; Sakanaka, M.; Tanaka, J.; Maeda, N.; Vitek, M.P.; et al. Apolipoprotein E and Reelin ligands modulate tau phosphorylation through an apolipoprotein E receptor/disabled-1/glycogen synthase kinase-3beta cascade. FASEB J. 2003, 17, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, T.; Ogawa, M.; Veeranna; Hirasawa, M.; Longenecker, G.; Ishiguro, K.; Pant, H.C.; Brady, R.O.; Kulkarni, A.B.; Mikoshiba, K. Synergistic contributions of cyclin-dependant kinase 5/p35 and Reelin/Dab1 to the positioning of cortical neurons in the developing mouse brain. Proc. Natl. Acad. Sci. USA 2001, 98, 2764–2769. [Google Scholar] [CrossRef] [PubMed]
- Yoneshima, H.; Nagata, E.; Matsumoto, M.; Yamada, M.; Nakajima, K.; Miyata, T.; Ogawa, M.; Mikoshiba, K. A novel neurological mutant mouse, yotari, which exhibits reeler-like phenotype but expresses CR-50 antigen/reelin. Neurosci. Res. 1997, 29, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Assadi, A.H.; Zhang, G.; Beffert, U.; McNeil, R.S.; Renfro, A.L.; Niu, S.; Quattrocchi, C.C.; Antalffy, B.A.; Sheldon, M.; Armstrong, D.D.; et al. Interaction of reelin signaling and Lis1 in brain development. Nat. Genet. 2003, 35, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Frotscher, M. How does Reelin signaling regulate the neuronal cytoskeleton during migration? Neurogenesis 2016, 3, e1242455. [Google Scholar] [CrossRef]
- Pandey, J.P.; Shi, L.; Brebion, R.A.; Smith, D.S. LIS1 and NDEL1 Regulate Axonal Trafficking of Mitochondria in Mature Neurons. Front. Mol. Neurosci. 2022, 15, 841047. [Google Scholar] [CrossRef]
- Kohno, S.; Kohno, T.; Nakano, Y.; Suzuki, K.; Ishii, M.; Tagami, H.; Baba, A.; Hattori, M. Mechanism and significance of specific proteolytic cleavage of Reelin. Biochem. Biophys. Res. Commun. 2009, 380, 93–97. [Google Scholar] [CrossRef]
- Nakano, Y.; Kohno, T.; Hibi, T.; Kohno, S.; Baba, A.; Mikoshiba, K.; Nakajima, K.; Hattori, M. The extremely conserved C-terminal region of Reelin is not necessary for secretion but is required for efficient activation of downstream signaling. J. Biol. Chem. 2007, 282, 20544–20552. [Google Scholar] [CrossRef]
- Ogino, H.; Nakajima, T.; Hirota, Y.; Toriuchi, K.; Aoyama, M.; Nakajima, K.; Hattori, M. The Secreted Glycoprotein Reelin Suppresses the Proliferation and Regulates the Distribution of Oligodendrocyte Progenitor Cells in the Embryonic Neocortex. J. Neurosci. 2020, 40, 7625–7636. [Google Scholar] [CrossRef] [PubMed]
- Okugawa, E.; Ogino, H.; Shigenobu, T.; Yamakage, Y.; Tsuiji, H.; Oishi, H.; Kohno, T.; Hattori, M. Physiological significance of proteolytic processing of Reelin revealed by cleavage-resistant Reelin knock-in mice. Sci. Rep. 2020, 10, 4471. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.; Tripathi, P.P.; Mihalas, A.B.; Hevner, R.F.; Beier, D.R. C-Terminal Region Truncation of RELN Disrupts an Interaction with VLDLR, Causing Abnormal Development of the Cerebral Cortex and Hippocampus. J. Neurosci. 2017, 37, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Honda, T.; Kubo, K.; Nakano, Y.; Tsuchiya, A.; Murakami, T.; Banno, H.; Nakajima, K.; Hattori, M. Importance of Reelin C-terminal region in the development and maintenance of the postnatal cerebral cortex and its regulation by specific proteolysis. J. Neurosci. 2015, 35, 4776–4787. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.; Stottmann, R.W.; Furley, A.J.; Beier, D.R. A forward genetic screen in mice identifies mutants with abnormal cortical patterning. Cereb. Cortex 2015, 25, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Morrill, N.K.; Moerman-Herzog, A.M.; Barger, S.W.; Joly-Amado, A.; Peters, M.; Soueidan, H.; Diemler, C.; Prabhudeva, S.; Weeber, E.J.; et al. Central repeat fragment of reelin leads to active reelin intracellular signaling and rescues cognitive deficits in a mouse model of reelin deficiency. Cell Signal 2023, 109, 110763. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Rusiana, I.; Trotter, J.; Zhao, L.; Donaldson, E.; Pak, D.T.; Babus, L.W.; Peters, M.; Banko, J.L.; Chavis, P.; et al. Reelin supplementation enhances cognitive ability, synaptic plasticity, and dendritic spine density. Learn. Mem. 2011, 18, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Beffert, U.; Ertunc, M.; Tang, T.S.; Kavalali, E.T.; Bezprozvanny, I.; Herz, J. Reelin modulates NMDA receptor activity in cortical neurons. J. Neurosci. 2005, 25, 8209–8216. [Google Scholar] [CrossRef]
- Pfennig, S.; Foss, F.; Bissen, D.; Harde, E.; Treeck, J.C.; Segarra, M.; Acker-Palmer, A. GRIP1 Binds to ApoER2 and EphrinB2 to Induce Activity-Dependent AMPA Receptor Insertion at the Synapse. Cell Rep. 2017, 21, 84–96. [Google Scholar] [CrossRef]
- Senkov, O.; Andjus, P.; Radenovic, L.; Soriano, E.; Dityatev, A. Neural ECM molecules in synaptic plasticity, learning, and memory. Prog. Brain Res. 2014, 214, 53–80. [Google Scholar] [CrossRef]
- Qiu, S.; Weeber, E.J. Reelin signaling facilitates maturation of CA1 glutamatergic synapses. J. Neurophysiol. 2007, 97, 2312–2321. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Zhao, L.F.; Korwek, K.M.; Weeber, E.J. Differential reelin-induced enhancement of NMDA and AMPA receptor activity in the adult hippocampus. J. Neurosci. 2006, 26, 12943–12955. [Google Scholar] [CrossRef] [PubMed]
- Pujadas, L.; Gruart, A.; Bosch, C.; Delgado, L.; Teixeira, C.M.; Rossi, D.; de Lecea, L.; Martinez, A.; Delgado-Garcia, J.M.; Soriano, E. Reelin regulates postnatal neurogenesis and enhances spine hypertrophy and long-term potentiation. J. Neurosci. 2010, 30, 4636–4649. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Korwek, K.M.; Pratt-Davis, A.R.; Peters, M.; Bergman, M.Y.; Weeber, E.J. Cognitive disruption and altered hippocampus synaptic function in Reelin haploinsufficient mice. Neurobiol. Learn. Mem. 2006, 85, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Zhao, L.; Trotter, J.H.; Rusiana, I.; Peters, M.M.; Li, Q.; Donaldson, E.; Banko, J.L.; Keenoy, K.E.; Rebeck, G.W.; et al. Reelin supplementation recovers sensorimotor gating, synaptic plasticity and associative learning deficits in the heterozygous reeler mouse. J. Psychopharmacol. 2013, 27, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Yabut, O.; D’Arcangelo, G. The Reelin signaling pathway promotes dendritic spine development in hippocampal neurons. J. Neurosci. 2008, 28, 10339–10348. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Renfro, A.; Quattrocchi, C.C.; Sheldon, M.; D’Arcangelo, G. Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron 2004, 41, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Cuchillo-Ibanez, I.; Balmaceda, V.; Mata-Balaguer, T.; Lopez-Font, I.; Saez-Valero, J. Reelin in Alzheimer’s Disease, Increased Levels but Impaired Signaling: When More is Less. J. Alzheimers Dis. 2016, 52, 403–416. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Snow, A.V.; Stary, J.M.; Araghi-Niknam, M.; Reutiman, T.J.; Lee, S.; Brooks, A.I.; Pearce, D.A. Reelin signaling is impaired in autism. Biol. Psychiatry 2005, 57, 777–787. [Google Scholar] [CrossRef]
- Folsom, T.D.; Fatemi, S.H. The involvement of Reelin in neurodevelopmental disorders. Neuropharmacology 2013, 68, 122–135. [Google Scholar] [CrossRef]
- Ishii, K.; Kubo, K.I.; Nakajima, K. Reelin and Neuropsychiatric Disorders. Front. Cell. Neurosci. 2016, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.E.; Shugart, Y.Y.; Huang, D.T.; Shahwan, S.A.; Grant, P.E.; Hourihane, J.O.; Martin, N.D.; Walsh, C.A. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet. 2000, 26, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Knuesel, I.; Nyffeler, M.; Mormede, C.; Muhia, M.; Meyer, U.; Pietropaolo, S.; Yee, B.K.; Pryce, C.R.; LaFerla, F.M.; Marighetto, A.; et al. Age-related accumulation of Reelin in amyloid-like deposits. Neurobiol. Aging 2009, 30, 697–716. [Google Scholar] [CrossRef] [PubMed]
- Stranahan, A.M.; Haberman, R.P.; Gallagher, M. Cognitive decline is associated with reduced reelin expression in the entorhinal cortex of aged rats. Cereb. Cortex 2011, 21, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Perez, E.J.; Roberts, J.A.; Roberts, M.T.; Rapp, P.R. Reelin in the Years: Decline in the number of reelin immunoreactive neurons in layer II of the entorhinal cortex in aged monkeys with memory impairment. Neurobiol. Aging 2020, 87, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Doehner, J.; Madhusudan, A.; Konietzko, U.; Fritschy, J.M.; Knuesel, I. Co-localization of Reelin and proteolytic AbetaPP fragments in hippocampal plaques in aged wild-type mice. J. Alzheimers Dis. 2010, 19, 1339–1357. [Google Scholar] [CrossRef] [PubMed]
- Madhusudan, A.; Sidler, C.; Knuesel, I. Accumulation of reelin-positive plaques is accompanied by a decline in basal forebrain projection neurons during normal aging. Eur. J. Neurosci. 2009, 30, 1064–1076. [Google Scholar] [CrossRef]
- Teive, H.A.G.; Ashizawa, T. Primary and secondary ataxias. Curr. Opin. Neurol. 2015, 28, 413–422. [Google Scholar] [CrossRef]
- Castagna, C.; Merighi, A.; Lossi, L. Cell death and neurodegeneration in the postnatal development of cerebellar vermis in normal and Reeler mice. Ann. Anat. 2016, 207, 76–90. [Google Scholar] [CrossRef]
- Terashima, T.; Inoue, K.; Inoue, Y.; Mikoshiba, K.; Tsukada, Y. Observations on Golgi epithelial cells and granule cells in the cerebellum of the reeler mutant mouse. Brain Res. 1985, 350, 103–112. [Google Scholar] [CrossRef]
- Magdaleno, S.; Keshvara, L.; Curran, T. Rescue of ataxia and preplate splitting by ectopic expression of Reelin in reeler mice. Neuron 2002, 33, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Kohno, T.; Hattori, M. Postnatal injection of Reelin protein into the cerebellum ameliorates the motor functions in reeler mouse. Neurosci. Res. 2023, 194, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Canet-Pons, J.; Schubert, R.; Duecker, R.P.; Schrewe, R.; Wolke, S.; Kieslich, M.; Schnolzer, M.; Chiocchetti, A.; Auburger, G.; Zielen, S.; et al. Ataxia telangiectasia alters the ApoB and reelin pathway. Neurogenetics 2018, 19, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Herzog, K.-H.; Chong, M.J.; Kapsetaki, M.; Morgan, J.I.; McKinnon, P.J. Requirement for Atm in Ionizing Radiation-Induced Cell Death in the Developing Central Nervous System. Science 1998, 280, 1089–1091. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; Yau, W.Y.; O’Connor, E.; Houlden, H. Spinocerebellar ataxia: An update. J. Neurol. 2019, 266, 533–544. [Google Scholar] [CrossRef]
- McCullough, S.D.; Xu, X.; Dent, S.Y.; Bekiranov, S.; Roeder, R.G.; Grant, P.A. Reelin is a target of polyglutamine expanded ataxin-7 in human spinocerebellar ataxia type 7 (SCA7) astrocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 21319–21324. [Google Scholar] [CrossRef]
- Corral-Juan, M.; Serrano-Munuera, C.; Rábano, A.; Cota-González, D.; Segarra-Roca, A.; Ispierto, L.; Cano-Orgaz, A.T.; Adarmes, A.D.; Méndez-Del-Barrio, C.; Jesús, S.; et al. Clinical, genetic and neuropathological characterization of spinocerebellar ataxia type 37. Brain 2018, 141, 1981–1997. [Google Scholar] [CrossRef]
- Seixas, A.I.; Loureiro, J.R.; Costa, C.; Ordóñez-Ugalde, A.; Marcelino, H.; Oliveira, C.L.; Loureiro, J.L.; Dhingra, A.; Brandão, E.; Cruz, V.T.; et al. A Pentanucleotide ATTTC Repeat Insertion in the Non-coding Region of DAB1, Mapping to SCA37, Causes Spinocerebellar Ataxia. Am. J. Hum. Genet. 2017, 101, 87–103. [Google Scholar] [CrossRef]
- Loureiro, J.R.; Oliveira, C.L.; Mota, C.; Castro, A.F.; Costa, C.; Loureiro, J.L.; Coutinho, P.; Martins, S.; Sequeiros, J.; Silveira, I. Mutational mechanism for DAB1 (ATTTC)(n) insertion in SCA37: ATTTT repeat lengthening and nucleotide substitution. Hum. Mutat. 2019, 40, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Okamoto, S.; Lipton, S.A.; Xu, H. Oligomeric Abeta-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav. Brain Res. 2008, 192, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Bracher-Smith, M.; Leonenko, G.; Baker, E.; Crawford, K.; Graham, A.C.; Salih, D.A.; Howell, B.W.; Hardy, J.; Escott-Price, V. Whole genome analysis in APOE4 homozygotes identifies the DAB1-RELN pathway in Alzheimer’s disease pathogenesis. Neurobiol. Aging 2022, 119, 67–76. [Google Scholar] [CrossRef]
- Hoe, H.S.; Lee, K.J.; Carney, R.S.; Lee, J.; Markova, A.; Lee, J.Y.; Howell, B.W.; Hyman, B.T.; Pak, D.T.; Bu, G.; et al. Interaction of reelin with amyloid precursor protein promotes neurite outgrowth. J. Neurosci. 2009, 29, 7459–7473. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.; Massaro, C.M.; Palop, J.J.; Thwin, M.T.; Yu, G.Q.; Bien-Ly, N.; Bender, A.; Mucke, L. Reelin depletion in the entorhinal cortex of human amyloid precursor protein transgenic mice and humans with Alzheimer’s disease. J. Neurosci. 2007, 27, 2727–2733. [Google Scholar] [CrossRef] [PubMed]
- Herring, A.; Donath, A.; Steiner, K.M.; Widera, M.P.; Hamzehian, S.; Kanakis, D.; Kolble, K.; ElAli, A.; Hermann, D.M.; Paulus, W.; et al. Reelin depletion is an early phenomenon of Alzheimer’s pathology. J. Alzheimers Dis. 2012, 30, 963–979. [Google Scholar] [CrossRef]
- Kocherhans, S.; Madhusudan, A.; Doehner, J.; Breu, K.S.; Nitsch, R.M.; Fritschy, J.M.; Knuesel, I. Reduced Reelin expression accelerates amyloid-beta plaque formation and tau pathology in transgenic Alzheimer’s disease mice. J. Neurosci. 2010, 30, 9228–9240. [Google Scholar] [CrossRef]
- Yu, N.N.; Tan, M.S.; Yu, J.T.; Xie, A.M.; Tan, L. The Role of Reelin Signaling in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 5692–5700. [Google Scholar] [CrossRef]
- Botella-Lopez, A.; Burgaya, F.; Gavin, R.; Garcia-Ayllon, M.S.; Gomez-Tortosa, E.; Pena-Casanova, J.; Urena, J.M.; Del Rio, J.A.; Blesa, R.; Soriano, E.; et al. Reelin expression and glycosylation patterns are altered in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5573–5578. [Google Scholar] [CrossRef]
- Lidon, L.; Urrea, L.; Llorens, F.; Gil, V.; Alvarez, I.; Diez-Fairen, M.; Aguilar, M.; Pastor, P.; Zerr, I.; Alcolea, D.; et al. Disease-Specific Changes in Reelin Protein and mRNA in Neurodegenerative Diseases. Cells 2020, 9, 1252. [Google Scholar] [CrossRef] [PubMed]
- Saez-Valero, J.; Costell, M.; Sjogren, M.; Andreasen, N.; Blennow, K.; Luque, J.M. Altered levels of cerebrospinal fluid reelin in frontotemporal dementia and Alzheimer’s disease. J. Neurosci. Res. 2003, 72, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Botella-Lopez, A.; Cuchillo-Ibanez, I.; Cotrufo, T.; Mok, S.S.; Li, Q.X.; Barquero, M.S.; Dierssen, M.; Soriano, E.; Saez-Valero, J. Beta-amyloid controls altered Reelin expression and processing in Alzheimer’s disease. Neurobiol. Dis. 2010, 37, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Cuchillo-Ibanez, I.; Mata-Balaguer, T.; Balmaceda, V.; Arranz, J.J.; Nimpf, J.; Saez-Valero, J. The beta-amyloid peptide compromises Reelin signaling in Alzheimer’s disease. Sci. Rep. 2016, 6, 31646. [Google Scholar] [CrossRef] [PubMed]
- Pujadas, L.; Rossi, D.; Andres, R.; Teixeira, C.M.; Serra-Vidal, B.; Parcerisas, A.; Maldonado, R.; Giralt, E.; Carulla, N.; Soriano, E. Reelin delays amyloid-beta fibril formation and rescues cognitive deficits in a model of Alzheimer’s disease. Nat. Commun. 2014, 5, 3443. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, R.; Zhu, D.; Wang, Q.; Li, Z.; Cheng, L.; Ren, J.; Guo, Y.; Chai, H.; Wang, M.; et al. Effect of the Reelin-Dab1 signaling pathway on the abnormal metabolism of Abeta protein induced by aluminum. Toxicol. Ind. Health 2023, 39, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.E.; Zamora, D.; Horowitz, M.; Jahanipour, J.; Keyes, G.; Li, X.; Murray, H.C.; Curtis, M.A.; Faull, R.M.; Sedlock, A.; et al. ApoER2-Dab1 disruption as the origin of pTau-related neurodegeneration in sporadic Alzheimer’s disease. Res. Sq. 2023, preprint. [Google Scholar] [CrossRef]
- Lane-Donovan, C.; Philips, G.T.; Wasser, C.R.; Durakoglugil, M.S.; Masiulis, I.; Upadhaya, A.; Pohlkamp, T.; Coskun, C.; Kotti, T.; Steller, L.; et al. Reelin protects against amyloid beta toxicity in vivo. Sci. Signal 2015, 8, ra67. [Google Scholar] [CrossRef]
- Han, S.; Miller, J.E.; Byun, S.; Kim, D.; Risacher, S.L.; Saykin, A.J.; Lee, Y.; Nho, K. Identification of exon skipping events associated with Alzheimer’s disease in the human hippocampus. BMC Med. Genom. 2019, 12, 51–61. [Google Scholar] [CrossRef]
- Bufill, E.; Roura-Poch, P.; Sala-Matavera, I.; Anton, S.; Lleo, A.; Sanchez-Saudinos, B.; Tomas-Abadal, L.; Puig, T.; Abos, J.; Bernades, S.; et al. Reelin signaling pathway genotypes and Alzheimer disease in a Spanish population. Alzheimer Dis. Assoc. Disord. 2015, 29, 169–172. [Google Scholar] [CrossRef]
- Lopera, F.; Marino, C.; Chandrahas, A.S.; O’Hare, M.; Villalba-Moreno, N.D.; Aguillon, D.; Baena, A.; Sanchez, J.S.; Vila-Castelar, C.; Ramirez Gomez, L.; et al. Resilience to autosomal dominant Alzheimer’s disease in a Reelin-COLBOS heterozygous man. Nat. Med. 2023, 29, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Brich, J.; Shie, F.S.; Howell, B.W.; Li, R.; Tus, K.; Wakeland, E.K.; Jin, L.W.; Mumby, M.; Churchill, G.; Herz, J.; et al. Genetic modulation of tau phosphorylation in the mouse. J. Neurosci. 2003, 23, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Cuchillo-Ibanez, I.; Balmaceda, V.; Botella-Lopez, A.; Rabano, A.; Avila, J.; Saez-Valero, J. Beta-amyloid impairs reelin signaling. PLoS ONE 2013, 8, e72297. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lang, M. New insight into protein glycosylation in the development of Alzheimer’s disease. Cell Death Discov. 2023, 9, 314. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Gruart, A.; Contreras-Murillo, G.; Muhaisen, A.; Avila, J.; Delgado-Garcia, J.M.; Pujadas, L.; Soriano, E. Reelin reverts biochemical, physiological and cognitive alterations in mouse models of Tauopathy. Prog. Neurobiol. 2020, 186, 101743. [Google Scholar] [CrossRef]
- Marckx, A.T.; Fritschle, K.E.; Calvier, L.; Herz, J. Reelin changes hippocampal learning in aging and Alzheimer’s disease. Behav. Brain Res. 2021, 414, 113482. [Google Scholar] [CrossRef] [PubMed]
- Dineley, K.T.; Westerman, M.; Bui, D.; Bell, K.; Ashe, K.H.; Sweatt, J.D. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer’s disease. J. Neurosci. 2001, 21, 4125–4133. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.; Lacor, P.N.; Velasco, P.T.; Xu, J.; Contractor, A.; Klein, W.L.; Triller, A. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron 2010, 66, 739–754. [Google Scholar] [CrossRef]
- Lauren, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef]
- Kurup, P.; Zhang, Y.; Xu, J.; Venkitaramani, D.V.; Haroutunian, V.; Greengard, P.; Nairn, A.C.; Lombroso, P.J. Abeta-mediated NMDA receptor endocytosis in Alzheimer’s disease involves ubiquitination of the tyrosine phosphatase STEP61. J. Neurosci. 2010, 30, 5948–5957. [Google Scholar] [CrossRef] [PubMed]
- Pelkey, K.A.; Askalan, R.; Paul, S.; Kalia, L.V.; Nguyen, T.H.; Pitcher, G.M.; Salter, M.W.; Lombroso, P.J. Tyrosine phosphatase STEP is a tonic brake on induction of long-term potentiation. Neuron 2002, 34, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Liu, J.; Lombroso, P.J. Striatal enriched phosphatase 61 dephosphorylates Fyn at phosphotyrosine 420. J. Biol. Chem. 2002, 277, 24274–24279. [Google Scholar] [CrossRef] [PubMed]
- Kamceva, M.; Benedict, J.; Nairn, A.C.; Lombroso, P.J. Role of Striatal-Enriched Tyrosine Phosphatase in Neuronal Function. Neural Plast. 2016, 2016, 8136925. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, S.P.; Paul, S.; Nairn, A.C.; Lombroso, P.J. Synaptic plasticity: One STEP at a time. Trends Neurosci. 2006, 29, 452–458. [Google Scholar] [CrossRef]
- Durakoglugil, M.S.; Chen, Y.; White, C.L.; Kavalali, E.T.; Herz, J. Reelin signaling antagonizes beta-amyloid at the synapse. Proc. Natl. Acad. Sci. USA 2009, 106, 15938–15943. [Google Scholar] [CrossRef] [PubMed]
- Durakoglugil, M.S.; Wasser, C.R.; Wong, C.H.; Pohlkamp, T.; Xian, X.; Lane-Donovan, C.; Fritschle, K.; Naestle, L.; Herz, J. Reelin Regulates Neuronal Excitability through Striatal-Enriched Protein Tyrosine Phosphatase (STEP61) and Calcium Permeable AMPARs in an NMDAR-Dependent Manner. J. Neurosci. 2021, 41, 7340–7349. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chatterjee, M.; Baguley, T.D.; Brouillette, J.; Kurup, P.; Ghosh, D.; Kanyo, J.; Zhang, Y.; Seyb, K.; Ononenyi, C.; et al. Inhibitor of the tyrosine phosphatase STEP reverses cognitive deficits in a mouse model of Alzheimer’s disease. PLoS Biol. 2014, 12, e1001923. [Google Scholar] [CrossRef]
- Jo, J.; Whitcomb, D.J.; Olsen, K.M.; Kerrigan, T.L.; Lo, S.C.; Bru-Mercier, G.; Dickinson, B.; Scullion, S.; Sheng, M.; Collingridge, G.; et al. Abeta(1-42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3beta. Nat. Neurosci. 2011, 14, 545–547. [Google Scholar] [CrossRef]
- Zhu, L.Q.; Wang, S.H.; Liu, D.; Yin, Y.Y.; Tian, Q.; Wang, X.C.; Wang, Q.; Chen, J.G.; Wang, J.Z. Activation of glycogen synthase kinase-3 inhibits long-term potentiation with synapse-associated impairments. J. Neurosci. 2007, 27, 12211–12220. [Google Scholar] [CrossRef]
- Gu, Z.; Liu, W.; Yan, Z. {beta}-Amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J. Biol. Chem. 2009, 284, 10639–10649. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Roselli, F.; Tirard, M.; Lu, J.; Hutzler, P.; Lamberti, P.; Livrea, P.; Morabito, M.; Almeida, O.F. Soluble beta-amyloid1-40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J. Neurosci. 2005, 25, 11061–11070. [Google Scholar] [CrossRef]
- Almeida, C.G.; Tampellini, D.; Takahashi, R.H.; Greengard, P.; Lin, M.T.; Snyder, E.M.; Gouras, G.K. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 2005, 20, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Banks, W.A.; Butterfield, D.A. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: Insights into their potential roles for loss of synapses and memory, accumulation of Abeta, and neurodegeneration in a prodromal stage of Alzheimer’s disease. J. Neurosci. Res. 2010, 88, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.P.; Koutsilieri, E.; Bartl, J.; Neuen-Jacob, E.; Arzberger, T.; Zander, N.; Ravid, R.; Roggendorf, W.; Riederer, P.; Grunblatt, E. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J. Alzheimers Dis. 2007, 11, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Jeong, Y.; Chang, Y.C. Extracellular matrix protein reelin regulate dendritic spine density through CaMKIIbeta. Neurosci. Lett. 2015, 599, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Hoe, H.S.; Rebeck, G.W. Regulated proteolysis of APP and ApoE receptors. Mol. Neurobiol. 2008, 37, 64–72. [Google Scholar] [CrossRef]
- Lane-Donovan, C.; Herz, J. The ApoE receptors Vldlr and Apoer2 in central nervous system function and disease. J. Lipid Res. 2017, 58, 1036–1043. [Google Scholar] [CrossRef]
- Hoe, H.S.; Magill, L.A.; Guenette, S.; Fu, Z.; Vicini, S.; Rebeck, G.W. FE65 interaction with the ApoE receptor ApoEr2. J. Biol. Chem. 2006, 281, 24521–24530. [Google Scholar] [CrossRef]
- Hoe, H.S.; Tran, T.S.; Matsuoka, Y.; Howell, B.W.; Rebeck, G.W. DAB1 and Reelin effects on amyloid precursor protein and ApoE receptor 2 trafficking and processing. J. Biol. Chem. 2006, 281, 35176–35185. [Google Scholar] [CrossRef] [PubMed]
- Hoe, H.S.; Minami, S.S.; Makarova, A.; Lee, J.; Hyman, B.T.; Matsuoka, Y.; Rebeck, G.W. Fyn modulation of Dab1 effects on amyloid precursor protein and ApoE receptor 2 processing. J. Biol. Chem. 2008, 283, 6288–6299. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Cooley, K.; Chung, C.H.; Dashti, N.; Tang, J. Apolipoprotein receptor 2 and X11 alpha/beta mediate apolipoprotein E-induced endocytosis of amyloid-beta precursor protein and beta-secretase, leading to amyloid-beta production. J. Neurosci. 2007, 27, 4052–4060. [Google Scholar] [CrossRef] [PubMed]
- Dumanis, S.B.; Chamberlain, K.A.; Jin Sohn, Y.; Jin Lee, Y.; Guenette, S.Y.; Suzuki, T.; Mathews, P.M.; Pak, D.; Rebeck, G.W.; Suh, Y.H.; et al. FE65 as a link between VLDLR and APP to regulate their trafficking and processing. Mol. Neurodegener. 2012, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Park, J.H.; Trotter, J.H.; Maher, J.N.; Keenoy, K.E.; Jang, Y.M.; Lee, Y.; Kim, J.I.; Weeber, E.J.; Hoe, H.S. Reelin and APP Cooperatively Modulate Dendritic Spine Formation In Vitro and In Vivo. Exp. Neurobiol. 2023, 32, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Chow, W.N.V.; Lau, K.F. Phosphorylation of FE65 at threonine 579 by GSK3beta stimulates amyloid precursor protein processing. Sci. Rep. 2017, 7, 12456. [Google Scholar] [CrossRef] [PubMed]
- Minami, S.S.; Sung, Y.M.; Dumanis, S.B.; Chi, S.H.; Burns, M.P.; Ann, E.J.; Suzuki, T.; Turner, R.S.; Park, H.S.; Pak, D.T.; et al. The cytoplasmic adaptor protein X11alpha and extracellular matrix protein Reelin regulate ApoE receptor 2 trafficking and cell movement. FASEB J. 2010, 24, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Caroni, P.; Chowdhury, A.; Lahr, M. Synapse rearrangements upon learning: From divergent-sparse connectivity to dedicated sub-circuits. Trends Neurosci. 2014, 37, 604–614. [Google Scholar] [CrossRef]
- Mizuno, K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal 2013, 25, 457–469. [Google Scholar] [CrossRef]
- Bosch, C.; Masachs, N.; Exposito-Alonso, D.; Martinez, A.; Teixeira, C.M.; Fernaud, I.; Pujadas, L.; Ulloa, F.; Comella, J.X.; DeFelipe, J.; et al. Reelin Regulates the Maturation of Dendritic Spines, Synaptogenesis and Glial Ensheathment of Newborn Granule Cells. Cereb. Cortex 2016, 26, 4282–4298. [Google Scholar] [CrossRef]
- Woo, J.A.; Liu, T.; Fang, C.C.; Cazzaro, S.; Kee, T.; LePochat, P.; Yrigoin, K.; Penn, C.; Zhao, X.; Wang, X.; et al. Activated cofilin exacerbates tau pathology by impairing tau-mediated microtubule dynamics. Commun. Biol. 2019, 2, 112. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.A.; Boggess, T.; Uhlar, C.; Wang, X.; Khan, H.; Cappos, G.; Joly-Amado, A.; De Narvaez, E.; Majid, S.; Minamide, L.S.; et al. RanBP9 at the intersection between cofilin and Abeta pathologies: Rescue of neurodegenerative changes by RanBP9 reduction. Cell Death Dis. 2015, 6, 1676. [Google Scholar] [CrossRef] [PubMed]
- Kho, S.H.; Yee, J.Y.; Puang, S.J.; Han, L.; Chiang, C.; Rapisarda, A.; Goh, W.W.B.; Lee, J.; Sng, J.C.G. DNA methylation levels of RELN promoter region in ultra-high risk, first episode and chronic schizophrenia cohorts of schizophrenia. Schizophrenia 2022, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhou, D.; Yan, T.; Chen, W.; Xie, H.; Xiong, Y. Association between CpG island DNA methylation in the promoter region of RELN and positive and negative types of schizophrenia. J. Int. Med. Res. 2022, 50, 03000605221100345. [Google Scholar] [CrossRef] [PubMed]
- Nawa, Y.; Kimura, H.; Mori, D.; Kato, H.; Toyama, M.; Furuta, S.; Yu, Y.; Ishizuka, K.; Kushima, I.; Aleksic, B.; et al. Rare single-nucleotide DAB1 variants and their contribution to Schizophrenia and autism spectrum disorder susceptibility. Hum. Genome Var. 2020, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Sobue, A.; Kushima, I.; Nagai, T.; Shan, W.; Kohno, T.; Aleksic, B.; Aoyama, Y.; Mori, D.; Arioka, Y.; Kawano, N.; et al. Genetic and animal model analyses reveal the pathogenic role of a novel deletion of RELN in schizophrenia. Sci. Rep. 2018, 8, 13046. [Google Scholar] [CrossRef]
- Ping, J.; Zhang, J.; Wan, J.; Banerjee, A.; Huang, C.; Yu, J.; Jiang, T.; Du, B. Correlation of Four Single Nucleotide Polymorphisms of the RELN Gene With Schizophrenia. East. Asian Arch. Psychiatry 2021, 31, 112–118. [Google Scholar] [CrossRef]
- Sozuguzel, M.D.; Sazci, A.; Yildiz, M. Female gender specific association of the Reelin (RELN) gene rs7341475 variant with schizophrenia. Mol. Biol. Rep. 2019, 46, 3411–3416. [Google Scholar] [CrossRef]
- Kuang, W.J.; Sun, R.F.; Zhu, Y.S.; Li, S.B. A new single-nucleotide mutation (rs362719) of the reelin (RELN) gene associated with schizophrenia in female Chinese Han. Genet. Mol.Res. GMR 2011, 10, 1650–1658. [Google Scholar] [CrossRef]
- Zhou, Z.; Hu, Z.; Zhang, L.; Hu, Z.; Liu, H.; Liu, Z.; Du, J.; Zhao, J.; Zhou, L.; Xia, K.; et al. Identification of RELN variation p.Thr3192Ser in a Chinese family with schizophrenia. Sci. Rep. 2016, 6, 24327. [Google Scholar] [CrossRef]
- Marzan, S.; Aziz, M.A.; Islam, M.S. Association Between REELIN Gene Polymorphisms (rs7341475 and rs262355) and Risk of Schizophrenia: An Updated Meta-analysis. J. Mol. Neurosci. 2021, 71, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Shoji, H.; Kohno, T.; Miyakawa, T.; Hattori, M. Mice that lack the C-terminal region of Reelin exhibit behavioral abnormalities related to neuropsychiatric disorders. Sci. Rep. 2016, 6, 28636. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Bao, Y.; Xue, Y.; Sun, Y.; Hu, D.; Meng, S.; Lu, L.; Shi, J. Meta-analyses of RELN variants in neuropsychiatric disorders. Behav. Brain Res. 2017, 332, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Kushima, I.; Aleksic, B.; Nakatochi, M.; Shimamura, T.; Shiino, T.; Yoshimi, A.; Kimura, H.; Takasaki, Y.; Wang, C.; Xing, J.; et al. High-resolution copy number variation analysis of schizophrenia in Japan. Mol. Psychiatry 2017, 22, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Yu, H.; Li, J.; Wang, L.; Li, L.; Chang, H.; Zhang, D.; Yue, W.; Li, M. Further evidence for the association between LRP8 and schizophrenia. Schizophr. Res. 2020, 215, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Nie, F.; Zhang, Q.; Ma, J.; Wang, P.; Gu, R.; Han, J.; Zhang, R. Schizophrenia risk candidate EGR3 is a novel transcriptional regulator of RELN and regulates neurite outgrowth via the Reelin signal pathway in vitro. J. Neurochem. 2021, 157, 1745–1758. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Kroll, J.L.; Stary, J.M. Altered levels of Reelin and its isoforms in schizophrenia and mood disorders. Neuroreport 2001, 12, 3209–3215. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Earle, J.A.; McMenomy, T. Reduction in Reelin immunoreactivity in hippocampus of subjects with schizophrenia, bipolar disorder and major depression. Mol. Psychiatry 2000, 5, 654–663, 571. [Google Scholar] [CrossRef]
- Guidotti, A.; Auta, J.; Davis, J.M.; Di-Giorgi-Gerevini, V.; Dwivedi, Y.; Grayson, D.R.; Impagnatiello, F.; Pandey, G.; Pesold, C.; Sharma, R.; et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: A postmortem brain study. Arch. Gen. Psychiatry 2000, 57, 1061–1069. [Google Scholar] [CrossRef]
- Yin, J.; Lu, Y.; Yu, S.; Dai, Z.; Zhang, F.; Yuan, J. Exploring the mRNA expression level of RELN in peripheral blood of schizophrenia patients before and after antipsychotic treatment. Hereditas 2020, 157, 43. [Google Scholar] [CrossRef]
- Bai, W.; Niu, Y.; Yu, X.; Yi, J.; Zhen, Q.; Kou, C. Decreased serum levels of reelin in patients with schizophrenia. Asian J. Psychiatr. 2020, 49, 101995. [Google Scholar] [CrossRef] [PubMed]
- Nabil Fikri, R.M.; Norlelawati, A.T.; Nour El-Huda, A.R.; Hanisah, M.N.; Kartini, A.; Norsidah, K.; Nor Zamzila, A. Reelin (RELN) DNA methylation in the peripheral blood of schizophrenia. J. Psychiatr. Res. 2017, 88, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Sawahata, M.; Mori, D.; Arioka, Y.; Kubo, H.; Kushima, I.; Kitagawa, K.; Sobue, A.; Shishido, E.; Sekiguchi, M.; Kodama, A.; et al. Generation and analysis of novel Reln-deleted mouse model corresponding to exonic Reln deletion in schizophrenia. Psychiatry Clin. Neurosci. 2020, 74, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Dong, G.; Wulaer, B.; Sawahata, M.; Mizoguchi, H.; Mori, D.; Ozaki, N.; Nabeshima, T.; Nagai, T.; Yamada, K. Mice with exonic RELN deletion identified from a patient with schizophrenia have impaired visual discrimination learning and reversal learning in touchscreen operant tasks. Behav. Brain Res. 2022, 416, 113569. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Hidalgo, A.C.; Martin-Cuevas, C.; Crespo-Facorro, B.; Garrido-Torres, N. Reelin Alterations, Behavioral Phenotypes, and Brain Anomalies in Schizophrenia: A Systematic Review of Insights From Rodent Models. Front. Neuroanat. 2022, 16, 844737. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Shoji, H.; Ogata, M.; Kagawa, Y.; Owada, Y.; Miyakawa, T.; Sakimura, K.; Terashima, T.; Katsuyama, Y. Dorsal Forebrain-Specific Deficiency of Reelin-Dab1 Signal Causes Behavioral Abnormalities Related to Psychiatric Disorders. Cereb. Cortex 2017, 27, 3485–3501. [Google Scholar] [CrossRef] [PubMed]
- Marrone, M.C.; Marinelli, S.; Biamonte, F.; Keller, F.; Sgobio, C.A.; Ammassari-Teule, M.; Bernardi, G.; Mercuri, N.B. Altered cortico-striatal synaptic plasticity and related behavioural impairments in reeler mice. Eur. J. Neurosci. 2006, 24, 2061–2070. [Google Scholar] [CrossRef] [PubMed]
- Ammassari-Teule, M.; Sgobio, C.; Biamonte, F.; Marrone, C.; Mercuri, N.B.; Keller, F. Reelin haploinsufficiency reduces the density of PV+ neurons in circumscribed regions of the striatum and selectively alters striatal-based behaviors. Psychopharmacology 2009, 204, 511–521. [Google Scholar] [CrossRef]
- Teixeira, C.M.; Martin, E.D.; Sahun, I.; Masachs, N.; Pujadas, L.; Corvelo, A.; Bosch, C.; Rossi, D.; Martinez, A.; Maldonado, R.; et al. Overexpression of Reelin prevents the manifestation of behavioral phenotypes related to schizophrenia and bipolar disorder. Neuropsychopharmacology 2011, 36, 2395–2405. [Google Scholar] [CrossRef]
- Pardo, M.; Gregorio, S.; Montalban, E.; Pujadas, L.; Elias-Tersa, A.; Masachs, N.; Vilchez-Acosta, A.; Parent, A.; Auladell, C.; Girault, J.A.; et al. Adult-specific Reelin expression alters striatal neuronal organization: Implications for neuropsychiatric disorders. Front. Cell. Neurosci. 2023, 17, 1143319. [Google Scholar] [CrossRef]
- Ibi, D.; Nakasai, G.; Koide, N.; Sawahata, M.; Kohno, T.; Takaba, R.; Nagai, T.; Hattori, M.; Nabeshima, T.; Yamada, K.; et al. Reelin Supplementation Into the Hippocampus Rescues Abnormal Behavior in a Mouse Model of Neurodevelopmental Disorders. Front. Cell. Neurosci. 2020, 14, 285. [Google Scholar] [CrossRef]
- Iafrati, J.; Malvache, A.; Gonzalez Campo, C.; Orejarena, M.J.; Lassalle, O.; Bouamrane, L.; Chavis, P. Multivariate synaptic and behavioral profiling reveals new developmental endophenotypes in the prefrontal cortex. Sci. Rep. 2016, 6, 35504. [Google Scholar] [CrossRef] [PubMed]
- Ali, Z.A.; Yasseen, A.A.; McAllister, K.A.; Al-Dujailli, A.; Al-Karaqully, A.J.; Jumaah, A.S. SNP-PCR genotyping links alterations in the GABAA receptor (GABRG3: rs208129) and RELN (rs73670) genes to autism spectrum disorder among peadiatric Iraqi Arabs. Mol. Biol. Rep. 2022, 49, 6019–6028. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Zhu, Y.; Wang, T.; Zhang, X.; Zhang, K.; Zhang, Z. Genetic risk factors for autism-spectrum disorders: A systematic review based on systematic reviews and meta-analysis. J. Neural. Transm. 2021, 128, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Garcia, I.; Chamorro, A.J.; Ternavasio-de la Vega, H.G.; Carbonell, C.; Marcos, M.; Miron-Canelo, J.A. Association of Allelic Variants of the Reelin Gene with Autistic Spectrum Disorder: A Systematic Review and Meta-Analysis of Candidate Gene Association Studies. Int. J. Environ. Res. Public. Health 2020, 17, 8010. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Sanchez, S.M.; Magdalon, J.; Griesi-Oliveira, K.; Yamamoto, G.L.; Santacruz-Perez, C.; Fogo, M.; Passos-Bueno, M.R.; Sertie, A.L. Rare RELN variants affect Reelin-DAB1 signal transduction in autism spectrum disorder. Hum. Mutat. 2018, 39, 1372–1383. [Google Scholar] [CrossRef] [PubMed]
- Teles, E.S.A.L.; Glaser, T.; Griesi-Oliveira, K.; Correa-Velloso, J.; Wang, J.Y.T.; da Silva Campos, G.; Ulrich, H.; Balan, A.; Zarrei, M.; Higginbotham, E.J.; et al. Rare CACNA1H and RELN variants interact through mTORC1 pathway in oligogenic autism spectrum disorder. Transl. Psychiatry 2022, 12, 234. [Google Scholar] [CrossRef] [PubMed]
- Cuchillo-Ibanez, I.; Andreo-Lillo, P.; Pastor-Ferrandiz, L.; Carratala-Marco, F.; Saez-Valero, J. Elevated Plasma Reelin Levels in Children With Autism. Front. Psychiatry 2020, 11, 242. [Google Scholar] [CrossRef] [PubMed]
- Steffenburg, S.; Gillberg, C.L.; Steffenburg, U.; Kyllerman, M. Autism in Angelman syndrome: A population-based study. Pediatr. Neurol. 1996, 14, 131–136. [Google Scholar] [CrossRef]
- Peters, S.U.; Horowitz, L.; Barbieri-Welge, R.; Taylor, J.L.; Hundley, R.J. Longitudinal follow-up of autism spectrum features and sensory behaviors in Angelman syndrome by deletion class. J. Child. Psychol. Psychiatry 2012, 53, 152–159. [Google Scholar] [CrossRef]
- Williams, C.A.; Beaudet, A.L.; Clayton-Smith, J.; Knoll, J.H.; Kyllerman, M.; Laan, L.A.; Magenis, R.E.; Moncla, A.; Schinzel, A.A.; Summers, J.A.; et al. Angelman syndrome 2005: Updated consensus for diagnostic criteria. Am. J. Med. Genet. A 2006, 140, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.H.; Wauki, K.; Liu, Q.; Bressler, J.; Pan, Y.; Kashork, C.D.; Shaffer, L.G.; Beaudet, A.L. Genomic analysis of the chromosome 15q11-q13 Prader-Willi syndrome region and characterization of transcripts for GOLGA8E and WHCD1L1 from the proximal breakpoint region. BMC Genom. 2008, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- van Woerden, G.M.; Harris, K.D.; Hojjati, M.R.; Gustin, R.M.; Qiu, S.; de Avila Freire, R.; Jiang, Y.H.; Elgersma, Y.; Weeber, E.J. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat. Neurosci. 2007, 10, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Hethorn, W.R.; Ciarlone, S.L.; Filonova, I.; Rogers, J.T.; Aguirre, D.; Ramirez, R.A.; Grieco, J.C.; Peters, M.M.; Gulick, D.; Anderson, A.E.; et al. Reelin supplementation recovers synaptic plasticity and cognitive deficits in a mouse model for Angelman syndrome. Eur. J. Neurosci. 2015, 41, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, D.R.; Jenkins, T.M.; Addington, C.P.; Stabenfeldt, S.E.; Lifshitz, J. Extracellular matrix proteins are time-dependent and regional-specific markers in experimental diffuse brain injury. Brain Behav. 2020, 10, e01767. [Google Scholar] [CrossRef] [PubMed]
- Dal Pozzo, V.; Crowell, B.; Briski, N.; Crockett, D.P.; D’Arcangelo, G. Reduced Reelin Expression in the Hippocampus after Traumatic Brain Injury. Biomolecules 2020, 10, 975. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Font, I.; Lennol, M.P.; Iborra-Lazaro, G.; Zetterberg, H.; Blennow, K.; Saez-Valero, J. Altered Balance of Reelin Proteolytic Fragments in the Cerebrospinal Fluid of Alzheimer’s Disease Patients. Int. J. Mol. Sci. 2022, 23, 7522. [Google Scholar] [CrossRef] [PubMed]
- Morrill, N.K.; Li, Q.; Joly-Amado, A.; Weeber, E.J.; Nash, K.R. A novel Reelin construct, R36, recovered behavioural deficits in the heterozygous reeler mouse. Eur. J. Neurosci. 2023, 57, 1657–1670. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joly-Amado, A.; Kulkarni, N.; Nash, K.R. Reelin Signaling in Neurodevelopmental Disorders and Neurodegenerative Diseases. Brain Sci. 2023, 13, 1479. https://doi.org/10.3390/brainsci13101479
Joly-Amado A, Kulkarni N, Nash KR. Reelin Signaling in Neurodevelopmental Disorders and Neurodegenerative Diseases. Brain Sciences. 2023; 13(10):1479. https://doi.org/10.3390/brainsci13101479
Chicago/Turabian StyleJoly-Amado, Aurelie, Neel Kulkarni, and Kevin R. Nash. 2023. "Reelin Signaling in Neurodevelopmental Disorders and Neurodegenerative Diseases" Brain Sciences 13, no. 10: 1479. https://doi.org/10.3390/brainsci13101479
APA StyleJoly-Amado, A., Kulkarni, N., & Nash, K. R. (2023). Reelin Signaling in Neurodevelopmental Disorders and Neurodegenerative Diseases. Brain Sciences, 13(10), 1479. https://doi.org/10.3390/brainsci13101479