

Clinico-Radiological Phenotype of UBTF c.628G>A Pathogenic Variant-Related Neurodegeneration in Childhood: A Case Report and Literature Review



Abstract
:1. Introduction
2. Materials and Methods
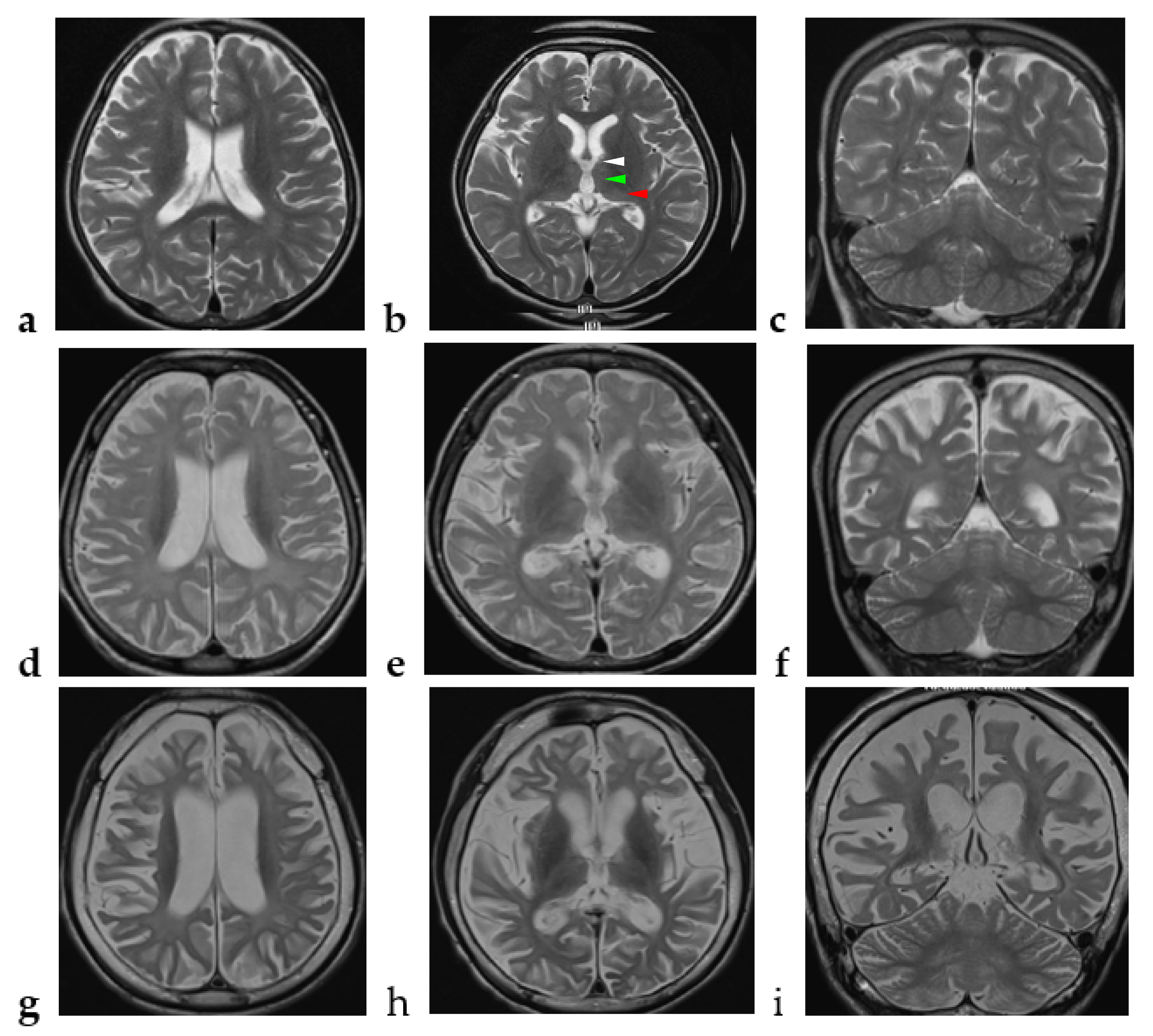
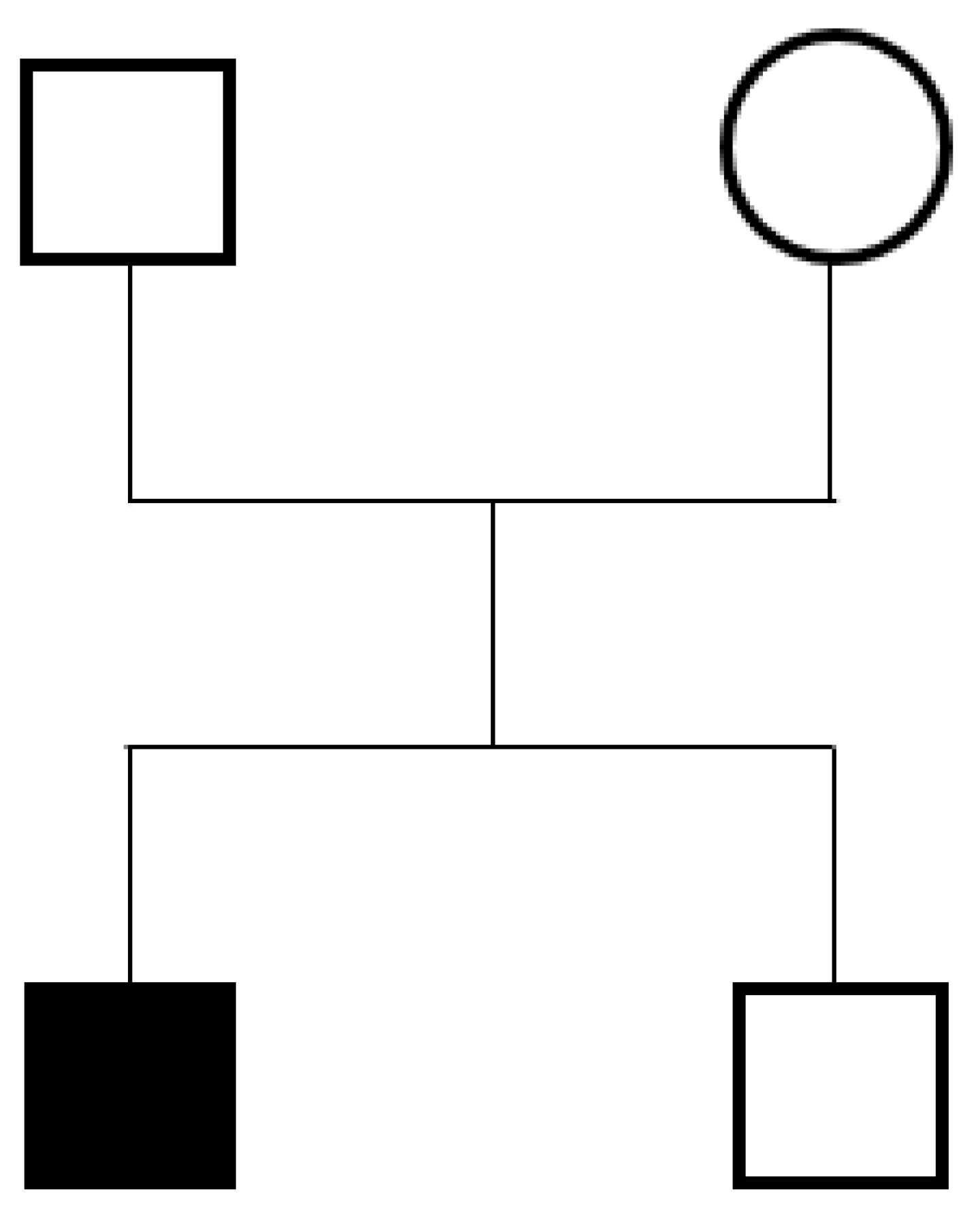
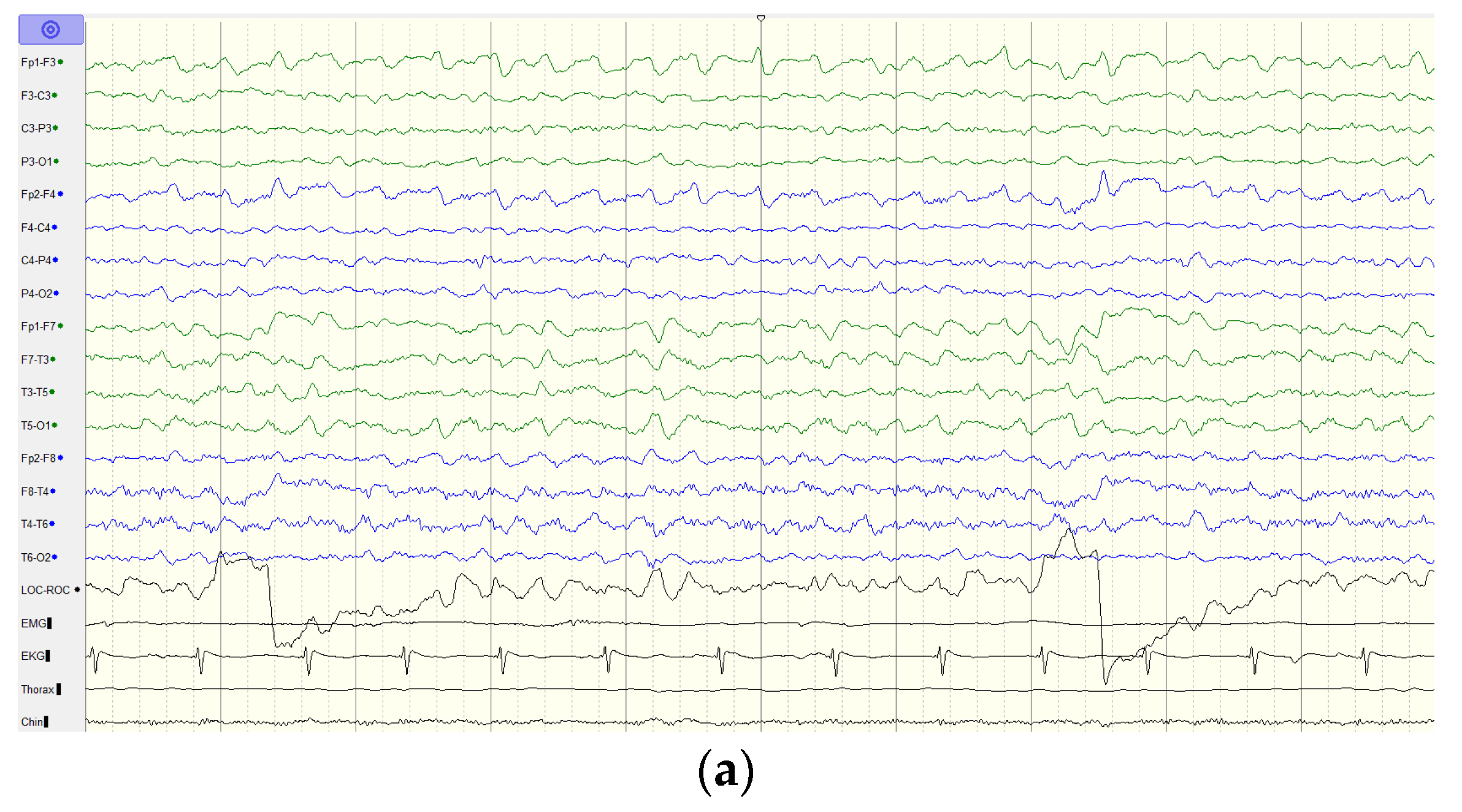
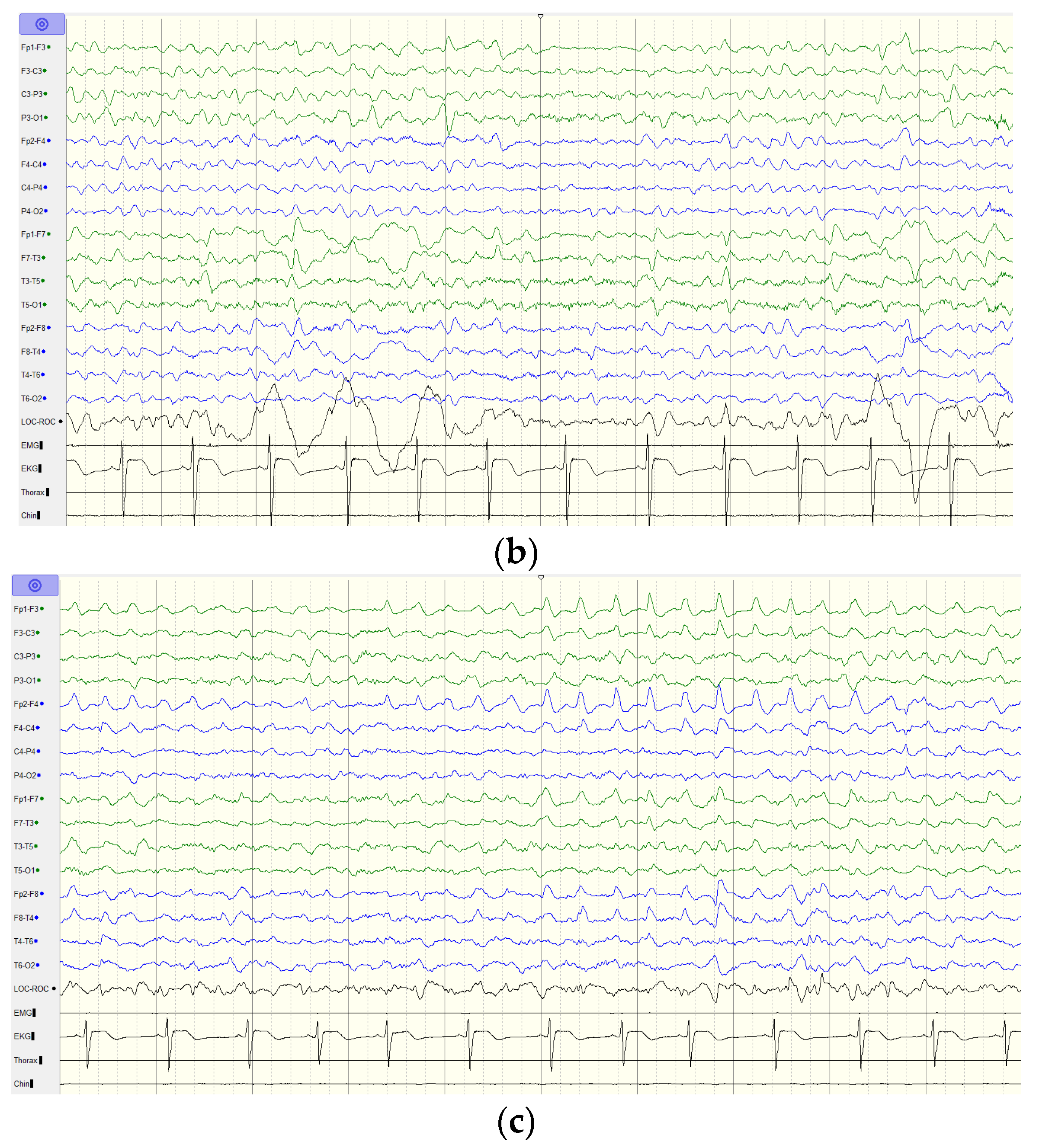
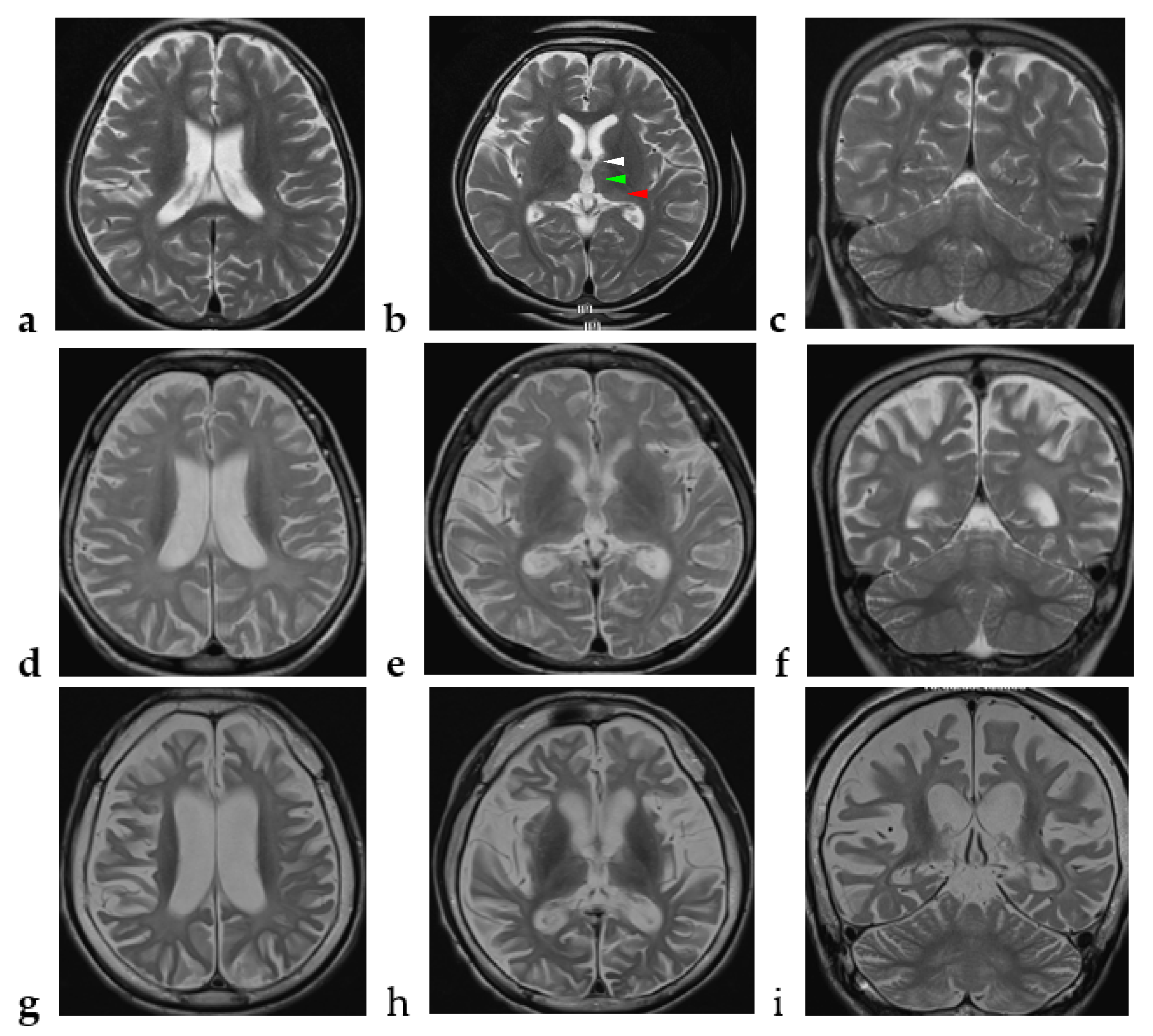
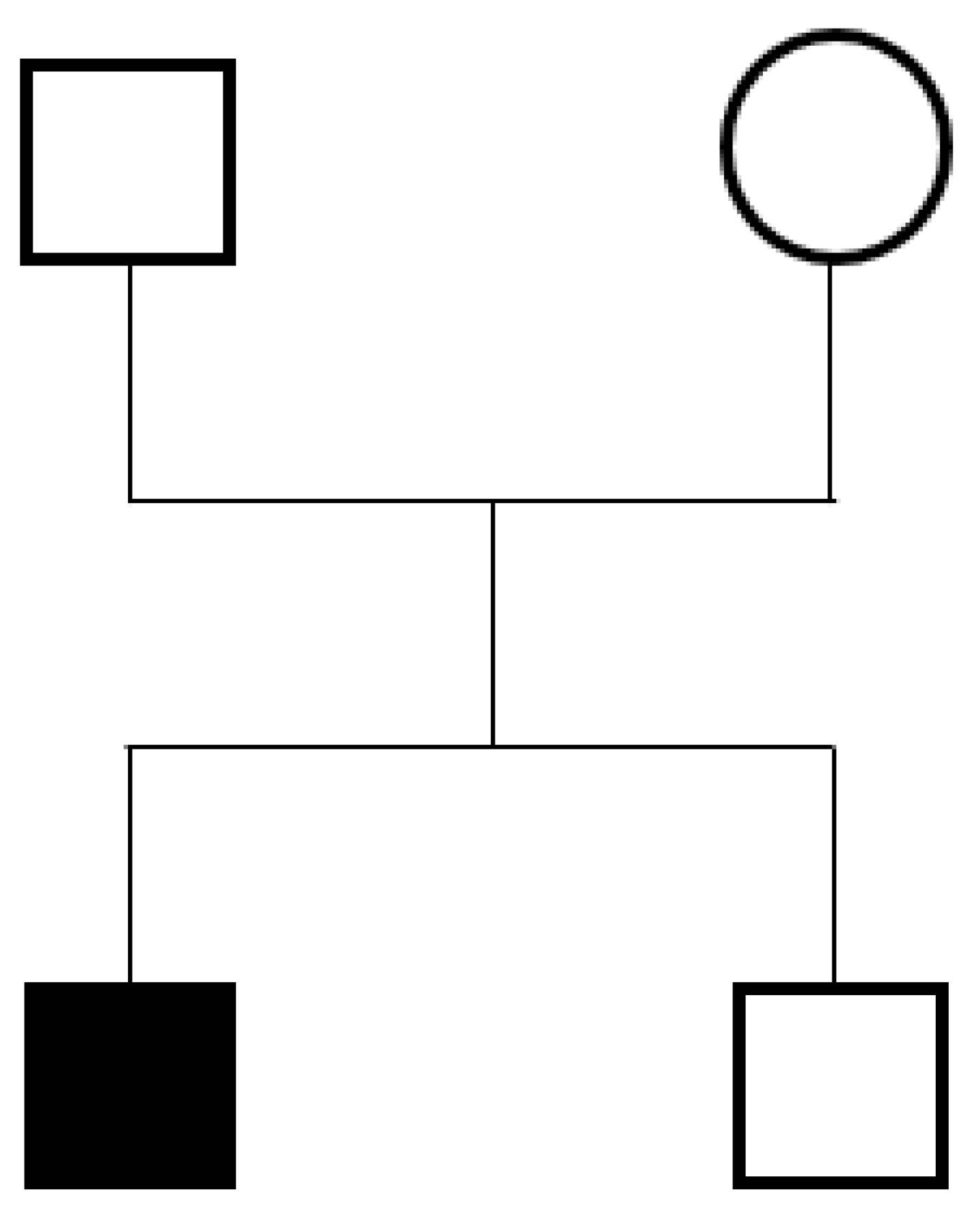
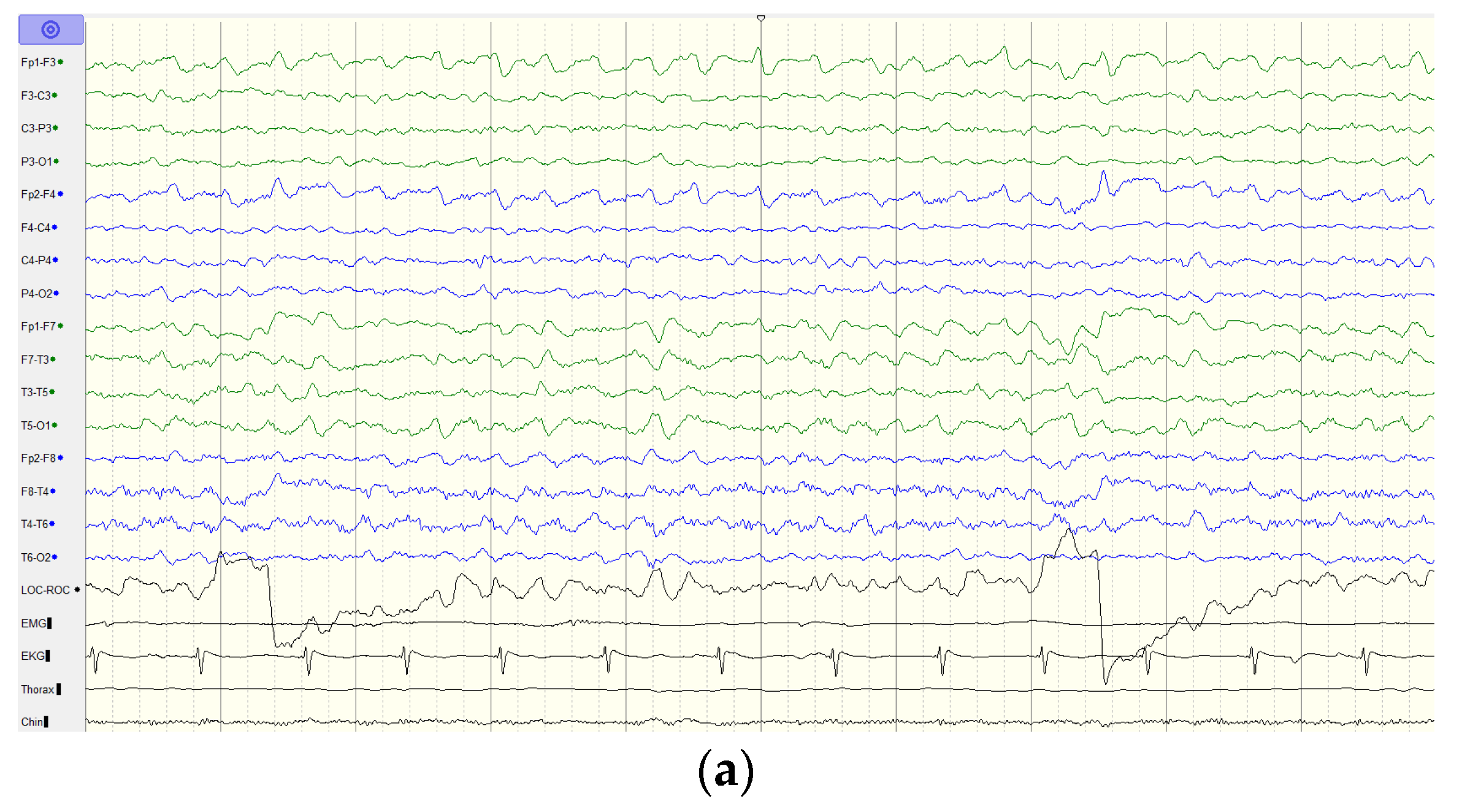
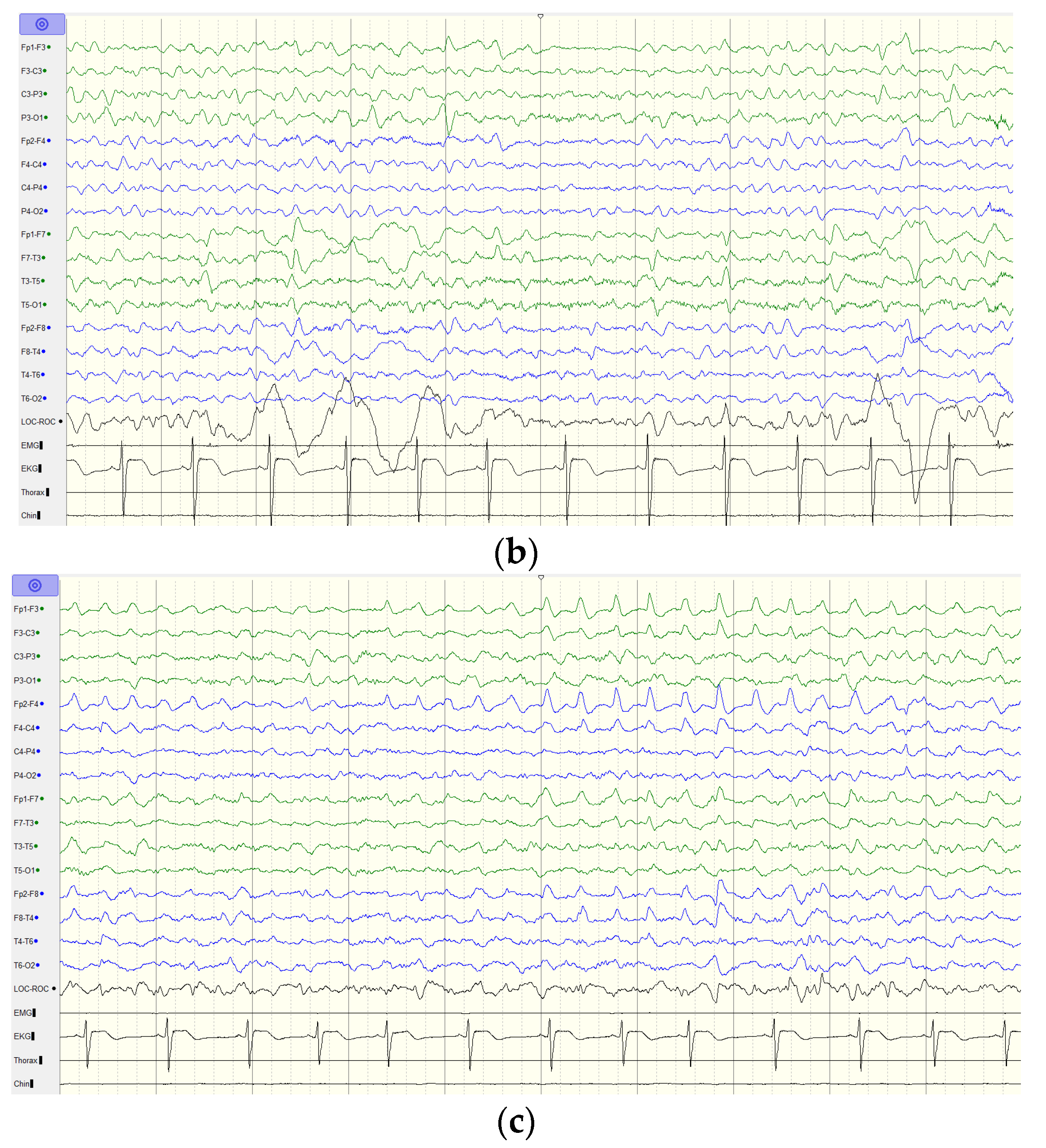
2.1. Case Report
2.2. Literature Review
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Mastrangelo, M. Clinical approach to neurodegenerative disorders in childhood: An updated overview. Acta Neurol. Belg. 2019, 119, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Pierre, G. Neurodegenerative disorders and metabolic disease. Arch. Dis. Child. 2013, 98, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Edvardson, S.; Nicolae, C.M.; Agrawal, P.B.; Mignot, C.; Payne, K.; Prasad, A.N.; Prasad, C.; Sadler, L.; Nava, C.; Mullen, T.E.; et al. Heterozygous de novo UBTF gain-of-function variant is associated with neurodegeneration in childhood. Am. J. Hum. Genet. 2017, 101, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Toro, C.; Hori, R.T.; Malicdan, M.C.V.; Tifft, C.J.; Goldstein, A.; Gahl, W.A.; Adams, D.R.; Fauni, H.B.; Wolfe, L.A.; Xiao, J.; et al. A recurrent de no missense mutation in UBTF causes developmental regression. Hum. Mol. Genet. 2018, 27, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Sedláčková, L.; Laššuthová, P.; Štěrbová, K.; Haberlová, J.; Vyhnálková, E.; Neupauerová, J.; Staněk, D.; Šedivá, M.; Kršek, P.; Seeman, P. UBTF mutation causes complex phenotype of neurodegeneration and severe epilepsy in childhood. Neuropediatrics 2019, 50, 57–60. [Google Scholar] [CrossRef]
- Bastos, F.; Quinodoz, M.; Addor, M.C.; Royer-Bertrand, B.; Fodstad, H.; Rivolta, C.; Poloni, C.; Superti-Furga, A.; Roulet Perez, E.; Lebon, S. Childhood neurodegeneration associated with a specific UBTF variant: A new case report and review of the literature. BMC Neurol. 2020, 20, 17. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, C.; Kawarai, T.; Setoyama, C.; Orlacchio, A.; Imanura, H. Recurrent de novo missense variant E210K in UBTF causes juvenile dystonia-parkinsonism. Neurol. Sci. 2021, 42, 1217–1219. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.B.; Smirniotopoulos, J.G.; Rushing, E.J.; Goldstein, S.J. Bilateral thalamic lesions. AJR Am. J. Roentgenol. 2009, 192, W53–W62. [Google Scholar] [CrossRef] [PubMed]
- Zuccoli, G.; Yannes, M.P.; Nardone, R.; Bailey, A.; Goldstein, A. Bilateral symmetrical basal ganglia and thalamic lesions in children: An update. Neuroradiology 2015, 57, 973–989. [Google Scholar] [CrossRef]
- Paprocka, J.; Machnikowska-Sokołowska, M.; Gruszczyńska, K.; Emich-Widera, E. Neuroimaging of basal ganglia in neurometabolic diseases in children. Brain Sci. 2020, 10, 849. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UBTF c.628 G>A (p.Glu210Lys) Pathogenic Variant-Related Neurodegeneration in Childhood | Total Number, N = 15 |
|---|---|
| Gender, male: female | 6:9 |
| Clinical manifestation | |
| Developmental delay noted at ≤2 years, n (%) | 10 (67) |
| Age at onset of neuroregression, years, median (range) | 3 (2–7) |
| Initial motor regression, n (%) | 6 (40) |
| Initial speech regression, n (%) | 2 (13) |
| Initial motor and speech regression, n (%) | 7 (47) |
| Microcephaly | 8 (53) |
| Age at onset of epilepsy, years, median (range) | 10.5 (5–15) |
| Neurologic examination | |
| Spasticity, n (%) | 14 (93) |
| Dystonia, n (%) | 11 (73) |
| Chorea, n (%) | 3 (20) |
| Parkinsonism, n (%) | 3 (20) |
| Ataxia, n (%) | 10 (67) |
| Brain magnetic resonance imaging | |
| Supratentorial cerebral atrophy | 15 (100) |
| Cerebellar atrophy | 11 (73) |
| Diffuse white matter T2 hyperintensity | 12 (80) |
| Thalamus involvement | 2 (13) |
| Clinical outcome | |
| Profound intellectual disability | 15 (100) |
| Non-verbal | 13 (87) |
| Non-ambulatory | 12 (80) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chi, C.-S.; Lee, H.-F.; Tsai, C.-R. Clinico-Radiological Phenotype of UBTF c.628G>A Pathogenic Variant-Related Neurodegeneration in Childhood: A Case Report and Literature Review. Brain Sci. 2022, 12, 1262. https://doi.org/10.3390/brainsci12091262
Chi C-S, Lee H-F, Tsai C-R. Clinico-Radiological Phenotype of UBTF c.628G>A Pathogenic Variant-Related Neurodegeneration in Childhood: A Case Report and Literature Review. Brain Sciences. 2022; 12(9):1262. https://doi.org/10.3390/brainsci12091262
Chicago/Turabian StyleChi, Ching-Shiang, Hsiu-Fen Lee, and Chi-Ren Tsai. 2022. "Clinico-Radiological Phenotype of UBTF c.628G>A Pathogenic Variant-Related Neurodegeneration in Childhood: A Case Report and Literature Review" Brain Sciences 12, no. 9: 1262. https://doi.org/10.3390/brainsci12091262
APA StyleChi, C.-S., Lee, H.-F., & Tsai, C.-R. (2022). Clinico-Radiological Phenotype of UBTF c.628G>A Pathogenic Variant-Related Neurodegeneration in Childhood: A Case Report and Literature Review. Brain Sciences, 12(9), 1262. https://doi.org/10.3390/brainsci12091262