
Environmental Impact on the Epigenetic Mechanisms Underlying Parkinson’s Disease Pathogenesis: A Narrative Review

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Methods
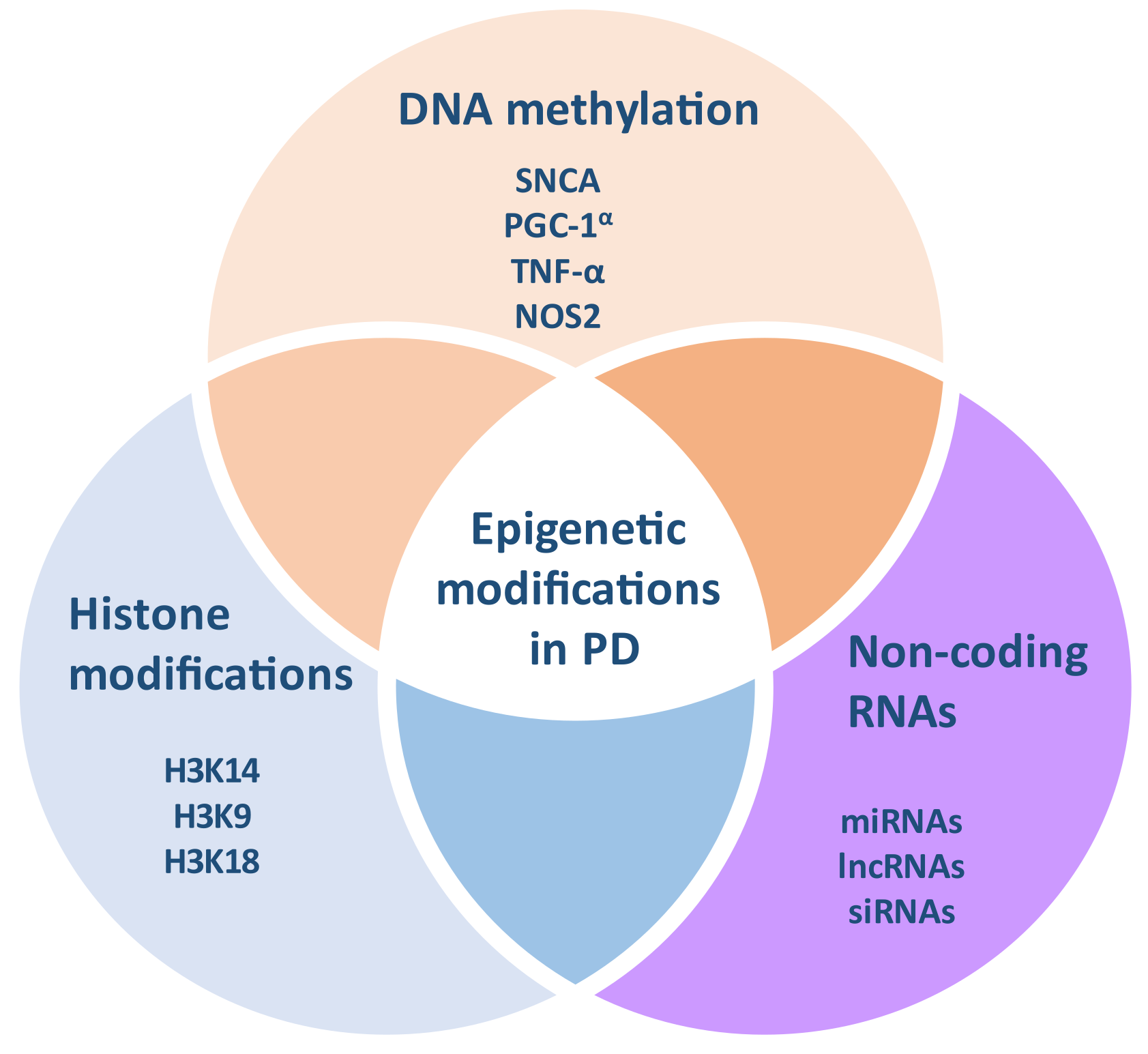
3. Epigenetic Modifications in PD
3.1. DNA Methylation in PD
3.2. Histone Acetylation and Other Modifications in PD
3.3. MiRNAs and Other Non-Coding RNAs in PD
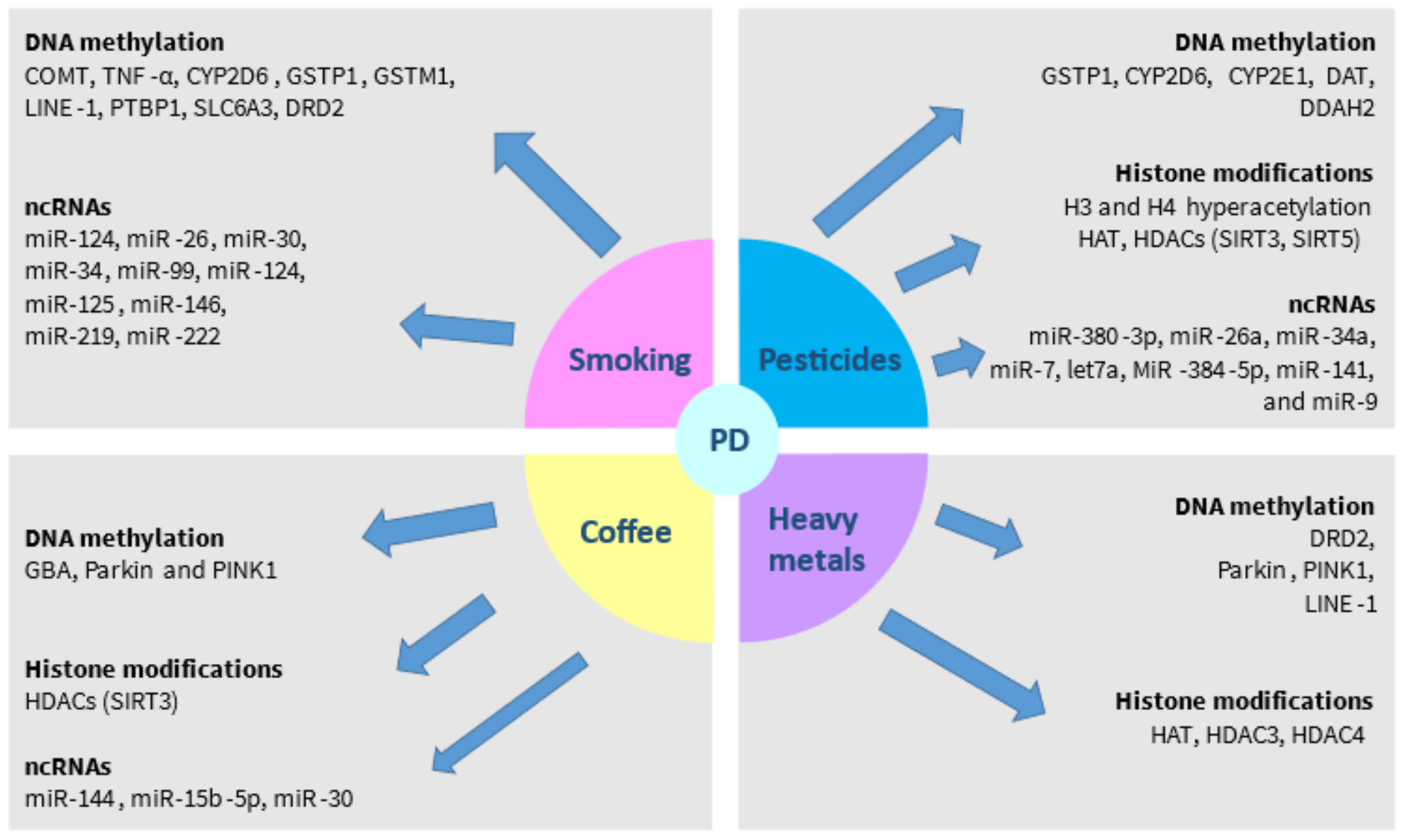
4. Environmental Impact on Epigenetic Modifications in PD
4.1. Smoking
4.2. Exposure to Pesticides and Insecticides
4.3. Coffee Consumption
4.4. Exposure to Heavy Metals
5. Potential Implications for Diagnosis and Therapy
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PD | Parkinson’s disease |
| SNpc | Substantia nigra pars compacta |
| GWAS | Genome wide association studies |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| miRNAs | microRNAs |
| CNS | Central nervous system |
| DNMTs | DNA methyltransferases |
| MBD | Methyl CpG-binding domain |
| CpGs | CpG dinucleotides |
| DAT | Dopamine transporter |
| HATs | Histone acetyltransferases |
| HDACs | Histone deacetylases |
| Nurr1 | Nuclear receptor related 1 protein |
| lncRNAs | Long non-coding RNAs |
| DMRs | Differentially methylated regions |
| LINE-1 | Long interspersed nucleotide element 1 |
| PTBP1 | Polypyrimidine Tract Binding Protein 1 |
| GLP-1 | Glucagon-like peptide 1 |
| CYP | Cytochrome P450 |
References
- de Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Marino, B.L.B.; de Souza, L.R.; Sousa, K.P.A.; Ferreira, J.V.; Padilha, E.C.; da Silva, C.; Taft, C.A.; Hage-Melim, L.I.S. Parkinson’s Disease: A Review from Pathophysiology to Treatment. Mini Rev. Med. Chem. 2020, 20, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Badanjak, K.; Fixemer, S.; Smajic, S.; Skupin, A.; Grunewald, A. The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4676. [Google Scholar] [CrossRef] [PubMed]
- Marogianni, C.; Sokratous, M.; Dardiotis, E.; Hadjigeorgiou, G.M.; Bogdanos, D.; Xiromerisiou, G. Neurodegeneration and Inflammation-An Interesting Interplay in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8421. [Google Scholar] [CrossRef]
- Gruchot, J.; Kremer, D.; Kury, P. Neural Cell Responses Upon Exposure to Human Endogenous Retroviruses. Front. Genet. 2019, 10, 655. [Google Scholar] [CrossRef]
- Kury, P.; Nath, A.; Creange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.P.; Perron, H. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379–394. [Google Scholar] [CrossRef]
- Sun, F.; Salinas, A.G.; Filser, S.; Blumenstock, S.; Medina-Luque, J.; Herms, J.; Sgobio, C. Impact of alpha-synuclein spreading on the nigrostriatal dopaminergic pathway depends on the onset of the pathology. Brain Pathol. 2021, e13036. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.Y.; Ho, P.W.; Liu, H.F.; Leung, C.T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.L. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl. Neurodegener. 2019, 8, 23. [Google Scholar] [CrossRef]
- Marques, S.C.; Oliveira, C.R.; Pereira, C.M.; Outeiro, T.F. Epigenetics in neurodegeneration: A new layer of complexity. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 348–355. [Google Scholar] [CrossRef]
- Migliore, L.; Coppede, F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat. Res. 2009, 667, 82–97. [Google Scholar] [CrossRef]
- Singh, M.; Khan, A.J.; Shah, P.P.; Shukla, R.; Khanna, V.K.; Parmar, D. Polymorphism in environment responsive genes and association with Parkinson disease. Mol. Cell. Biochem. 2008, 312, 131–138. [Google Scholar] [CrossRef]
- Dunn, A.R.; O’Connell, K.M.S.; Kaczorowski, C.C. Gene-by-environment interactions in Alzheimer’s disease and Parkinson’s disease. Neurosci. Biobehav. Rev. 2019, 103, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Bellou, V.; Belbasis, L.; Tzoulaki, I.; Evangelou, E.; Ioannidis, J.P. Environmental risk factors and Parkinson’s disease: An umbrella review of meta-analyses. Parkinsonism Relat. Disord. 2016, 23, 1–9. [Google Scholar] [CrossRef]
- Coppede, F. Genetics and epigenetics of Parkinson’s disease. Sci. World J. 2012, 2012, 489830. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Jankovic, J.; Wu, Y.C. Epigenetic mechanisms in Parkinson’s disease. J. Neurol. Sci. 2015, 349, 3–9. [Google Scholar] [CrossRef]
- Pavlou, M.A.S.; Outeiro, T.F. Epigenetics in Parkinson’s Disease. Adv. Exp. Med. Biol. 2017, 978, 363–390. [Google Scholar] [CrossRef] [PubMed]
- van Heesbeen, H.J.; Smidt, M.P. Entanglement of Genetics and Epigenetics in Parkinson’s Disease. Front. Neurosci. 2019, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- Karakaidos, P.; Karagiannis, D.; Rampias, T. Resolving DNA Damage: Epigenetic Regulation of DNA Repair. Molecules 2020, 25, 2496. [Google Scholar] [CrossRef]
- Creighton, S.D.; Stefanelli, G.; Reda, A.; Zovkic, I.B. Epigenetic Mechanisms of Learning and Memory: Implications for Aging. Int. J. Mol. Sci. 2020, 21, 6918. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Gowher, H.; Jeltsch, A. Mammalian DNA methyltransferases: New discoveries and open questions. Biochem. Soc. Trans. 2018, 46, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Dumaop, W.; Galasko, D.; Desplats, P. Distinctive patterns of DNA methylation associated with Parkinson disease: Identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics 2013, 8, 1030–1038. [Google Scholar] [CrossRef]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. Alpha-synuclein sequesters Dnmt1 from the nucleus: A novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef] [PubMed]
- International Parkinson’s Disease Genomics, C.; Wellcome Trust Case Control, C. A two-stage meta-analysis identifies several new loci for Parkinson’s disease. PLoS Genet. 2011, 7, e1002142. [Google Scholar] [CrossRef]
- Rubino, A.; D’Addario, C.; Di Bartolomeo, M.; Michele Salamone, E.; Locuratolo, N.; Fattapposta, F.; Vanacore, N.; Pascale, E. DNA methylation of the 5’-UTR DAT 1 gene in Parkinson’s disease patients. Acta Neurol. Scand. 2020, 142, 275–280. [Google Scholar] [CrossRef]
- Pieper, H.C.; Evert, B.O.; Kaut, O.; Riederer, P.F.; Waha, A.; Wullner, U. Different methylation of the TNF-alpha promoter in cortex and substantia nigra: Implications for selective neuronal vulnerability. Neurobiol. Dis. 2008, 32, 521–527. [Google Scholar] [CrossRef]
- Kaut, O.; Schmitt, I.; Wullner, U. Genome-scale methylation analysis of Parkinson’s disease patients’ brains reveals DNA hypomethylation and increased mRNA expression of cytochrome P450 2E1. Neurogenetics 2012, 13, 87–91. [Google Scholar] [CrossRef]
- Shahabi, H.N.; Westberg, L.; Melke, J.; Hakansson, A.; Belin, A.C.; Sydow, O.; Olson, L.; Holmberg, B.; Nissbrandt, H. Cytochrome P450 2E1 gene polymorphisms/haplotypes and Parkinson’s disease in a Swedish population. J. Neural Transm. 2009, 116, 567–573. [Google Scholar] [CrossRef]
- Kakade, A.; Kumari, B.; Dholaniya, P.S. Feature selection using logistic regression in case-control DNA methylation data of Parkinson’s disease: A comparative study. J. Theor. Biol. 2018, 457, 14–18. [Google Scholar] [CrossRef]
- Jazvinscak Jembrek, M.; Orsolic, N.; Mandic, L.; Sadzak, A.; Segota, S. Anti-Oxidative, Anti-Inflammatory and Anti-Apoptotic Effects of Flavonols: Targeting Nrf2, NF-kappaB and p53 Pathways in Neurodegeneration. Antioxidants 2021, 10, 1628. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Shao, Y.; Zhao, Y.; Li, Q.; Li, R.; Xiao, H.; Zhang, F.; Zhang, Y.; Chang, X.; Zhang, Y.; et al. Endoplasmic reticulum stress-related neuroinflammation and neural stem cells decrease in mice exposure to paraquat. Sci. Rep. 2020, 10, 17757. [Google Scholar] [CrossRef] [PubMed]
- Coufal, N.G.; Garcia-Perez, J.L.; Peng, G.E.; Yeo, G.W.; Mu, Y.; Lovci, M.T.; Morell, M.; O’Shea, K.S.; Moran, J.V.; Gage, F.H. L1 retrotransposition in human neural progenitor cells. Nature 2009, 460, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Bartova, E.; Krejci, J.; Harnicarova, A.; Galiova, G.; Kozubek, S. Histone modifications and nuclear architecture: A review. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2008, 56, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Chrun, E.S.; Modolo, F.; Daniel, F.I. Histone modifications: A review about the presence of this epigenetic phenomenon in carcinogenesis. Pathol. Res. Pract. 2017, 213, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- van Heesbeen, H.J.; Mesman, S.; Veenvliet, J.V.; Smidt, M.P. Epigenetic mechanisms in the development and maintenance of dopaminergic neurons. Development 2013, 140, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Park, G.; Tan, J.; Garcia, G.; Kang, Y.; Salvesen, G.; Zhang, Z. Regulation of Histone Acetylation by Autophagy in Parkinson Disease. J. Biol. Chem. 2016, 291, 3531–3540. [Google Scholar] [CrossRef] [PubMed]
- Renani, P.G.; Taheri, F.; Rostami, D.; Farahani, N.; Abdolkarimi, H.; Abdollahi, E.; Taghizadeh, E.; Gheibi Hayat, S.M. Involvement of aberrant regulation of epigenetic mechanisms in the pathogenesis of Parkinson’s disease and epigenetic-based therapies. J. Cell. Physiol. 2019, 234, 19307–19319. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Zhu, K.; Chi, S.; Wang, C.; Xie, A. Emerging Role of Sirtuin 2 in Parkinson’s Disease. Front. Aging Neurosci. 2019, 11, 372. [Google Scholar] [CrossRef]
- Siddiqui, A.; Chinta, S.J.; Mallajosyula, J.K.; Rajagopolan, S.; Hanson, I.; Rane, A.; Melov, S.; Andersen, J.K. Selective binding of nuclear alpha-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: Implications for Parkinson’s disease. Free. Radic. Biol. Med. 2012, 53, 993–1003. [Google Scholar] [CrossRef]
- Hoss, A.G.; Labadorf, A.; Beach, T.G.; Latourelle, J.C.; Myers, R.H. microRNA Profiles in Parkinson’s Disease Prefrontal Cortex. Front. Aging Neurosci. 2016, 8, 36. [Google Scholar] [CrossRef]
- Nies, Y.H.; Mohamad Najib, N.H.; Lim, W.L.; Kamaruzzaman, M.A.; Yahaya, M.F.; Teoh, S.L. MicroRNA Dysregulation in Parkinson’s Disease: A Narrative Review. Front. Neurosci. 2021, 15, 660379. [Google Scholar] [CrossRef]
- Jankovic, J.; Chen, S.; Le, W.D. The role of Nurr1 in the development of dopaminergic neurons and Parkinson’s disease. Prog. Neurobiol. 2005, 77, 128–138. [Google Scholar] [CrossRef]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science 2007, 317, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Liu, G.; Jin, S.M.; Parisiadou, L.; Xie, C.; Yu, J.; Sun, L.; Ma, B.; Ding, J.; Vancraenenbroeck, R.; et al. MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum. Mol. Genet. 2013, 22, 608–620. [Google Scholar] [CrossRef]
- Wang, Z.H.; Zhang, J.L.; Duan, Y.L.; Zhang, Q.S.; Li, G.F.; Zheng, D.L. MicroRNA-214 participates in the neuroprotective effect of Resveratrol via inhibiting alpha-synuclein expression in MPTP-induced Parkinson’s disease mouse. Biomed. Pharmacother. Biomed. Pharmacother. 2015, 74, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Goh, S.Y.; Chao, Y.X.; Dheen, S.T.; Tan, E.K.; Tay, S.S. Role of MicroRNAs in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 5649. [Google Scholar] [CrossRef]
- Banerjee, A.; Waters, D.; Camacho, O.M.; Minet, E. Quantification of plasma microRNAs in a group of healthy smokers, ex-smokers and non-smokers and correlation to biomarkers of tobacco exposure. Biomark. Biochem. Indic. Expo. Response Susceptibility Chem. 2015, 20, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Aloizou, A.M.; Siokas, V.; Sapouni, E.M.; Sita, N.; Liampas, I.; Brotis, A.G.; Rakitskii, V.N.; Burykina, T.I.; Aschner, M.; Bogdanos, D.P.; et al. Parkinson’s disease and pesticides: Are microRNAs the missing link? Sci. Total Environ. 2020, 744, 140591. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, M.; Liang, J.; Wang, C.; Deng, Y.; Chen, Z. Epigenetic Regulation of Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4956. [Google Scholar] [CrossRef]
- Chuang, Y.-H.; Paul, K.C.; Bronstein, J.M.; Bordelon, Y.; Horvath, S.; Ritz, B. Parkinson’s disease is associated with DNA methylation levels in human blood and saliva. Genome Med. 2017, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Quik, M.; Bordia, T.; Zhang, D.; Perez, X.A. Nicotine and Nicotinic Receptor Drugs: Potential for Parkinson’s Disease and Drug-Induced Movement Disorders. Int. Rev. Neurobiol. 2015, 124, 247–271. [Google Scholar] [CrossRef]
- Lu, J.Y.D.; Su, P.; Barber, J.E.M.; Nash, J.E.; Le, A.D.; Liu, F.; Wong, A.H.C. The neuroprotective effect of nicotine in Parkinson’s disease models is associated with inhibiting PARP-1 and caspase-3 cleavage. PeerJ 2017, 5, e3933. [Google Scholar] [CrossRef]
- Mao, Y.; Huang, P.; Wang, Y.; Wang, M.; Li, M.D.; Yang, Z. Genome-wide methylation and expression analyses reveal the epigenetic landscape of immune-related diseases for tobacco smoking. Clin. Epigenet. 2021, 13, 215. [Google Scholar] [CrossRef]
- Ding, J.; Li, F.; Cong, Y.; Miao, J.; Wu, D.; Liu, B.; Wang, L. Trichostatin A inhibits skeletal muscle atrophy induced by cigarette smoke exposure in mice. Life Sci. 2019, 235, 116800. [Google Scholar] [CrossRef]
- Leus, N.G.; van den Bosch, T.; van der Wouden, P.E.; Krist, K.; Ourailidou, M.E.; Eleftheriadis, N.; Kistemaker, L.E.; Bos, S.; Gjaltema, R.A.; Mekonnen, S.A.; et al. HDAC1-3 inhibitor MS-275 enhances IL10 expression in RAW264.7 macrophages and reduces cigarette smoke-induced airway inflammation in mice. Sci. Rep. 2017, 7, 45047. [Google Scholar] [CrossRef]
- Pace, E.; Di Vincenzo, S.; Ferraro, M.; Siena, L.; Chiappara, G.; Dino, P.; Vitulo, P.; Bertani, A.; Saibene, F.; Lanata, L.; et al. Effects of Carbocysteine and Beclomethasone on Histone Acetylation/Deacetylation Processes in Cigarette Smoke Exposed Bronchial Epithelial Cells. J. Cell. Physiol. 2017, 232, 2851–2859. [Google Scholar] [CrossRef] [PubMed]
- Ganjali, M.; Kheirkhah, B.; Amini, K. Expression of miRNA-601 and PD-L1 among Iranian Patients with Lung Cancer and Their Relationship with Smoking and Mycoplasma Infection. Cell J. 2021, 23, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Kiyohara, C.; Miyake, Y.; Koyanagi, M.; Fujimoto, T.; Shirasawa, S.; Tanaka, K.; Fukushima, W.; Sasaki, S.; Tsuboi, Y.; Yamada, T.; et al. Genetic polymorphisms involved in dopaminergic neurotransmission and risk for Parkinson’s disease in a Japanese population. BMC Neurol. 2011, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- Mellick, G.D. CYP450, genetics and Parkinson’s disease: Gene x environment interactions hold the key. In Parkinson’s Disease and Related Disorders; Springer: Vienna, Austria, 2006; pp. 159–165. [Google Scholar] [CrossRef]
- Tiili, E.M.; Antikainen, M.S.; Mitiushkina, N.V.; Sukhovskaya, O.A.; Imyanitov, E.N.; Hirvonen, A.P. Effect of genotype and methylation of CYP2D6 on smoking behaviour. Pharm. Genom. 2015, 25, 531–540. [Google Scholar] [CrossRef]
- Kiyohara, C.; Miyake, Y.; Koyanagi, M.; Fujimoto, T.; Shirasawa, S.; Tanaka, K.; Fukushima, W.; Sasaki, S.; Tsuboi, Y.; Yamada, T.; et al. GST polymorphisms, interaction with smoking and pesticide use, and risk for Parkinson’s disease in a Japanese population. Parkinsonism Relat. Disord. 2010, 16, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Newman, B.; Dunne, M.P.; Silburn, P.A.; Mellick, G.D. Case-only study of interactions between genetic polymorphisms of GSTM1, P1, T1 and Z1 and smoking in Parkinson’s disease. Neurosci. Lett. 2004, 366, 326–331. [Google Scholar] [CrossRef] [PubMed]
- De Palma, G.; Dick, F.D.; Calzetti, S.; Scott, N.W.; Prescott, G.J.; Osborne, A.; Haites, N.; Mozzoni, P.; Negrotti, A.; Scaglioni, A.; et al. A case-control study of Parkinson’s disease and tobacco use: Gene-tobacco interactions. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, 912–919. [Google Scholar] [CrossRef]
- Korff, A.; Pfeiffer, B.; Smeyne, M.; Kocak, M.; Pfeiffer, R.F.; Smeyne, R.J. Alterations in glutathione S-transferase pi expression following exposure to MPP+ -induced oxidative stress in the blood of Parkinson’s disease patients. Parkinsonism Relat. Disord. 2011, 17, 765–768. [Google Scholar] [CrossRef]
- Hernandez, H.G.; Sandoval-Hernandez, A.G.; Garrido-Gil, P.; Labandeira-Garcia, J.L.; Zelaya, M.V.; Bayon, G.F.; Fernandez, A.F.; Fraga, M.F.; Arboleda, G.; Arboleda, H. Alzheimer’s disease DNA methylome of pyramidal layers in frontal cortex: Laser-assisted microdissection study. Epigenomics 2018, 10, 1365–1382. [Google Scholar] [CrossRef]
- Searles Nielsen, S.; Checkoway, H.; Butler, R.A.; Nelson, H.H.; Farin, F.M.; Longstreth, W.T., Jr.; Franklin, G.M.; Swanson, P.D.; Kelsey, K.T. LINE-1 DNA methylation, smoking and risk of Parkinson’s disease. J. Parkinson’s Dis. 2012, 2, 303–308. [Google Scholar] [CrossRef]
- Patchsung, M.; Boonla, C.; Amnattrakul, P.; Dissayabutra, T.; Mutirangura, A.; Tosukhowong, P. Long interspersed nuclear element-1 hypomethylation and oxidative stress: Correlation and bladder cancer diagnostic potential. PLoS ONE 2012, 7, e37009. [Google Scholar] [CrossRef]
- Rusiecki, J.A.; Baccarelli, A.; Bollati, V.; Tarantini, L.; Moore, L.E.; Bonefeld-Jorgensen, E.C. Global DNA hypomethylation is associated with high serum-persistent organic pollutants in Greenlandic Inuit. Environ. Health Perspect. 2008, 116, 1547–1552. [Google Scholar] [CrossRef] [PubMed]
- Lambrou, A.; Baccarelli, A.; Wright, R.O.; Weisskopf, M.; Bollati, V.; Amarasiriwardena, C.; Vokonas, P.; Schwartz, J. Arsenic exposure and DNA methylation among elderly men. Epidemiology 2012, 23, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.A.; Potashkin, J.A. Network-based metaanalysis identifies HNF4A and PTBP1 as longitudinally dynamic biomarkers for Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2015, 112, 2257–2262. [Google Scholar] [CrossRef] [PubMed]
- Knoch, K.P.; Meisterfeld, R.; Kersting, S.; Bergert, H.; Altkruger, A.; Wegbrod, C.; Jager, M.; Saeger, H.D.; Solimena, M. cAMP-dependent phosphorylation of PTB1 promotes the expression of insulin secretory granule proteins in beta cells. Cell Metab. 2006, 3, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Foltynie, T.; Aviles-Olmos, I. Exenatide as a potential treatment for patients with Parkinson’s disease: First steps into the clinic. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2014, 10, S38–S46. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, S.; Zhao, J.; Nie, S.; Sun, J.; Gao, X.; Lenahan, C.; Lin, Z.; Huang, Y.; Chen, G. Tobacco Smoking Increases Methylation of Polypyrimidine Tract Binding Protein 1 Promoter in Intracranial Aneurysms. Front. Aging Neurosci. 2021, 13, 688179. [Google Scholar] [CrossRef] [PubMed]
- Zhai, D.; Li, S.; Zhao, Y.; Lin, Z. SLC6A3 is a risk factor for Parkinson’s disease: A meta-analysis of sixteen years’ studies. Neurosci. Lett. 2014, 564, 99–104. [Google Scholar] [CrossRef]
- Tiili, E.M.; Mitiushkina, N.V.; Sukhovskaya, O.A.; Imyanitov, E.N.; Hirvonen, A.P. The effect of SLC6A3 variable number of tandem repeats and methylation levels on individual susceptibility to start tobacco smoking and on the ability of smokers to quit smoking. Pharm. Genom. 2020, 30, 117–123. [Google Scholar] [CrossRef]
- Ozaki, Y.; Yoshino, Y.; Yamazaki, K.; Ochi, S.; Iga, J.I.; Nagai, M.; Nomoto, M.; Ueno, S.I. DRD2 methylation to differentiate dementia with Lewy bodies from Parkinson’s disease. Acta Neurol. Scand. 2020, 141, 177–182. [Google Scholar] [CrossRef]
- Liu, Q.; Xu, Y.; Mao, Y.; Ma, Y.; Wang, M.; Han, H.; Cui, W.; Yuan, W.; Payne, T.J.; Xu, Y.; et al. Genetic and Epigenetic Analysis Revealing Variants in the NCAM1-TTC12-ANKK1-DRD2 Cluster Associated Significantly With Nicotine Dependence in Chinese Han Smokers. Nicotine Tob. Res. Off. J. Soc. Res. Nicotine Tob. 2020, 22, 1301–1309. [Google Scholar] [CrossRef]
- Qin, Z.; Wan, J.J.; Sun, Y.; Wu, T.; Wang, P.Y.; Du, P.; Su, D.F.; Yang, Y.; Liu, X. Nicotine protects against DSS colitis through regulating microRNA-124 and STAT3. J. Mol. Med. 2017, 95, 221–233. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Paudel, Y.N.; Piperi, C. miR-124 and Parkinson’s disease: A biomarker with therapeutic potential. Pharmacol. Res. 2019, 150, 104515. [Google Scholar] [CrossRef]
- Izzotti, A.; Calin, G.A.; Arrigo, P.; Steele, V.E.; Croce, C.M.; De Flora, S. Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 806–812. [Google Scholar] [CrossRef]
- Chambers-Richards, T.; Su, Y.; Chireh, B.; D’Arcy, C. Exposure to toxic occupations and their association with Parkinson’s disease: A systematic review with meta-analysis. Rev. Environ. Health 2021. [Google Scholar] [CrossRef]
- Cao, F.; Souders Ii, C.L.; Perez-Rodriguez, V.; Martyniuk, C.J. Elucidating Conserved Transcriptional Networks Underlying Pesticide Exposure and Parkinson’s Disease: A Focus on Chemicals of Epidemiological Relevance. Front. Genet. 2018, 9, 701. [Google Scholar] [CrossRef]
- Kanthasamy, A.G.; Kitazawa, M.; Yang, Y.; Anantharam, V.; Kanthasamy, A. Environmental neurotoxin dieldrin induces apoptosis via caspase-3-dependent proteolytic activation of protein kinase C delta (PKCdelta): Implications for neurodegeneration in Parkinson’s disease. Mol. Brain 2008, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef]
- Giambo, F.; Leone, G.M.; Gattuso, G.; Rizzo, R.; Cosentino, A.; Cina, D.; Teodoro, M.; Costa, C.; Tsatsakis, A.; Fenga, C.; et al. Genetic and Epigenetic Alterations Induced by Pesticide Exposure: Integrated Analysis of Gene Expression, microRNA Expression, and DNA Methylation Datasets. Int. J. Environ. Res. Public Health 2021, 18, 8697. [Google Scholar] [CrossRef]
- Sabarwal, A.; Kumar, K.; Singh, R.P. Hazardous effects of chemical pesticides on human health-Cancer and other associated disorders. Environ. Toxicol. Pharmacol. 2018, 63, 103–114. [Google Scholar] [CrossRef]
- Menegon, A.; Board, P.G.; Blackburn, A.C.; Mellick, G.D.; Le Couteur, D.G. Parkinson’s disease, pesticides, and glutathione transferase polymorphisms. Lancet 1998, 352, 1344–1346. [Google Scholar] [CrossRef]
- Wilk, J.B.; Tobin, J.E.; Suchowersky, O.; Shill, H.A.; Klein, C.; Wooten, G.F.; Lew, M.F.; Mark, M.H.; Guttman, M.; Watts, R.L.; et al. Herbicide exposure modifies GSTP1 haplotype association to Parkinson onset age: The GenePD Study. Neurology 2006, 67, 2206–2210. [Google Scholar] [CrossRef]
- Chen, J.; Liou, A.; Zhang, L.; Weng, Z.; Gao, Y.; Cao, G.; Zigmond, M.J.; Chen, J. GST P1, a novel downstream regulator of LRRK2, G2019S-induced neuronal cell death. Front. Biosci. 2012, 4, 2365–2377. [Google Scholar] [CrossRef]
- Rudyk, C.; Dwyer, Z.; Hayley, S.; membership, C. Leucine-rich repeat kinase-2 (LRRK2) modulates paraquat-induced inflammatory sickness and stress phenotype. J. Neuroinflamm. 2019, 16, 120. [Google Scholar] [CrossRef] [PubMed]
- Marder, K.; Wang, Y.; Alcalay, R.N.; Mejia-Santana, H.; Tang, M.X.; Lee, A.; Raymond, D.; Mirelman, A.; Saunders-Pullman, R.; Clark, L.; et al. Age-specific penetrance of LRRK2 G2019S in the Michael, J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology 2015, 85, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, A.; Levecque, C.; Clavel, J.; Vidal, J.S.; Richard, F.; Amouyel, P.; Alperovitch, A.; Chartier-Harlin, M.C.; Tzourio, C. CYP2D6 polymorphism, pesticide exposure, and Parkinson’s disease. Ann. Neurol. 2004, 55, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Yadav, A.; Singh, C. Autonomous regulation of inducible nitric oxide synthase and cytochrome P450 2E1-mediated oxidative stress in maneb- and paraquat-treated rat polymorphs. Pestic. Biochem. Physiol. 2021, 178, 104944. [Google Scholar] [CrossRef] [PubMed]
- Ritz, B.R.; Manthripragada, A.D.; Costello, S.; Lincoln, S.J.; Farrer, M.J.; Cockburn, M.; Bronstein, J. Dopamine transporter genetic variants and pesticides in Parkinson’s disease. Environ. Health Perspect. 2009, 117, 964–969. [Google Scholar] [CrossRef] [PubMed]
- Lind, L.; Penell, J.; Luttropp, K.; Nordfors, L.; Syvanen, A.C.; Axelsson, T.; Salihovic, S.; van Bavel, B.; Fall, T.; Ingelsson, E.; et al. Global DNA hypermethylation is associated with high serum levels of persistent organic pollutants in an elderly population. Environ. Int. 2013, 59, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Hancock, D.B.; Martin, E.R.; Vance, J.M.; Scott, W.K. Nitric oxide synthase genes and their interactions with environmental factors in Parkinson’s disease. Neurogenetics 2008, 9, 249–262. [Google Scholar] [CrossRef]
- Paul, K.C.; Sinsheimer, J.S.; Rhodes, S.L.; Cockburn, M.; Bronstein, J.; Ritz, B. Organophosphate Pesticide Exposures, Nitric Oxide Synthase Gene Variants, and Gene-Pesticide Interactions in a Case-Control Study of Parkinson’s Disease, California (USA). Environ. Health Perspect. 2016, 124, 570–577. [Google Scholar] [CrossRef]
- Fiedler, L. The DDAH/ADMA pathway is a critical regulator of NO signalling in vascular homeostasis. Cell Adhes. Migr. 2008, 2, 149–150. [Google Scholar] [CrossRef][Green Version]
- Duarte-Hospital, C.; Tete, A.; Brial, F.; Benoit, L.; Koual, M.; Tomkiewicz, C.; Kim, M.J.; Blanc, E.B.; Coumoul, X.; Bortoli, S. Mitochondrial Dysfunction as a Hallmark of Environmental Injury. Cells 2021, 11, 110. [Google Scholar] [CrossRef]
- Wang, R.; Sun, H.; Wang, G.; Ren, H. Imbalance of Lysine Acetylation Contributes to the Pathogenesis of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 7182. [Google Scholar] [CrossRef]
- Song, C.; Kanthasamy, A.; Anantharam, V.; Sun, F.; Kanthasamy, A.G. Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells: Relevance to epigenetic mechanisms of neurodegeneration. Mol. Pharmacol. 2010, 77, 621–632. [Google Scholar] [CrossRef]
- Song, C.; Kanthasamy, A.; Jin, H.; Anantharam, V.; Kanthasamy, A.G. Paraquat induces epigenetic changes by promoting histone acetylation in cell culture models of dopaminergic degeneration. Neurotoxicology 2011, 32, 586–595. [Google Scholar] [CrossRef]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; Di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 2003, 42, 8465–8471. [Google Scholar] [CrossRef]
- Feng, Y.; Liu, T.; Dong, S.Y.; Guo, Y.J.; Jankovic, J.; Xu, H.; Wu, Y.C. Rotenone affects p53 transcriptional activity and apoptosis via targeting SIRT1 and H3K9 acetylation in SH-SY5Y cells. J. Neurochem. 2015, 134, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Fang, J.; Liu, W. Superoxide dismutase coding of gene polymorphisms associated with susceptibility to Parkinson’s disease. J. Integr. Neurosci. 2019, 18, 299–303. [Google Scholar] [CrossRef]
- Farin, F.M.; Hitosis, Y.; Hallagan, S.E.; Kushleika, J.; Woods, J.S.; Janssen, P.S.; Smith-Weller, T.; Franklin, G.M.; Swanson, P.D.; Checkoway, H. Genetic polymorphisms of superoxide dismutase in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2001, 16, 705–707. [Google Scholar] [CrossRef]
- Zhang, X.; Ren, X.; Zhang, Q.; Li, Z.; Ma, S.; Bao, J.; Li, Z.; Bai, X.; Zheng, L.; Zhang, Z.; et al. PGC-1alpha/ERRalpha-Sirt3 Pathway Regulates DAergic Neuronal Death by Directly Deacetylating SOD2 and ATP Synthase beta. Antioxid. Redox Signal. 2016, 24, 312–328. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Peritore, C.; Ginsberg, J.; Shih, J.; Arun, S.; Donmez, G. Protective role of SIRT5 against motor deficit and dopaminergic degeneration in MPTP-induced mice model of Parkinson’s disease. Behav. Brain Res. 2015, 281, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ren, N.; Cai, Z.; Lin, Q.; Wang, Z.; Zhang, Q.; Wu, S.; Li, H. Paraquat and MPTP induce neurodegeneration and alteration in the expression profile of microRNAs: The role of transcription factor Nrf2. NPJ Parkinsons Dis. 2017, 3, 31. [Google Scholar] [CrossRef]
- Horst, C.H.; Schlemmer, F.; de Aguiar Montenegro, N.; Domingues, A.C.M.; Ferreira, G.G.; da Silva Ribeiro, C.Y.; de Andrade, R.R.; Del Bel Guimaraes, E.; Titze-de-Almeida, S.S.; Titze-de-Almeida, R. Signature of Aberrantly Expressed microRNAs in the Striatum of Rotenone-Induced Parkinsonian Rats. Neurochem. Res. 2018, 43, 2132–2140. [Google Scholar] [CrossRef]
- Rostamian Delavar, M.; Baghi, M.; Safaeinejad, Z.; Kiani-Esfahani, A.; Ghaedi, K.; Nasr-Esfahani, M.H. Differential expression of miR-34a, miR-141, and miR-9 in MPP+-treated differentiated PC12 cells as a model of Parkinson’s disease. Gene 2018, 662, 54–65. [Google Scholar] [CrossRef]
- Tao, H.; Liu, Y.; Hou, Y. miRNA3845p regulates the progression of Parkinson’s disease by targeting SIRT1 in mice and SHSY5Y cell. Int. J. Mol. Med. 2020, 45, 441–450. [Google Scholar] [CrossRef]
- Grossi, I.; Radeghieri, A.; Paolini, L.; Porrini, V.; Pilotto, A.; Padovani, A.; Marengoni, A.; Barbon, A.; Bellucci, A.; Pizzi, M.; et al. MicroRNA34a5p expression in the plasma and in its extracellular vesicle fractions in subjects with Parkinson’s disease: An exploratory study. Int. J. Mol. Med. 2021, 47, 533–546. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, X.; Lin, Y.; Zhang, Z.; Gao, Y.; Wu, I.X.Y. Non-Genetic Risk Factors for Parkinson’s Disease: An Overview of 46 Systematic Reviews. J. Parkinsons Dis. 2021, 11, 919–935. [Google Scholar] [CrossRef]
- Socala, K.; Szopa, A.; Serefko, A.; Poleszak, E.; Wlaz, P. Neuroprotective Effects of Coffee Bioactive Compounds: A Review. Int. J. Mol. Sci. 2020, 22, 107. [Google Scholar] [CrossRef] [PubMed]
- Domenighetti, C.; Sugier, P.E.; Sreelatha, A.A.K.; Schulte, C.; Grover, S.; Mohamed, O.; Portugal, B.; May, P.; Bobbili, D.R.; Radivojkov-Blagojevic, M.; et al. Mendelian Randomisation Study of Smoking, Alcohol, and Coffee Drinking in Relation to Parkinson’s Disease. J. Parkinsons Dis. 2021, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, Y.; Ye, T.; Yu, Q.; Yu, J.; Yuan, S.; Gao, X.; Wan, X.; Zhang, R.; Han, W.; et al. Effect of Coffee against MPTP-Induced Motor Deficits and Neurodegeneration in Mice Via Regulating Gut Microbiota. J. Agric. Food Chem. 2022, 70, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.H.; Quach, A.; Absher, D.; Assimes, T.; Horvath, S.; Ritz, B. Coffee consumption is associated with DNA methylation levels of human blood. Eur. J. Hum. Genet. EJHG 2017, 25, 608–616. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Bozi, M.; Simitsi, A.M.; Koros, C.; Antonelou, R.; Papagiannakis, N.; Maniati, M.; Poula, D.; Stamelou, M.; Vassilatis, D.K.; et al. The relationship between environmental factors and different Parkinson’s disease subtypes in Greece: Data analysis of the Hellenic Biobank of Parkinson’s disease. Parkinsonism Relat. Disord. 2019, 67, 105–112. [Google Scholar] [CrossRef]
- Singh, S.; Singh, K.; Gupta, S.P.; Patel, D.K.; Singh, V.K.; Singh, R.K.; Singh, M.P. Effect of caffeine on the expression of cytochrome P450 1A2, adenosine A2A receptor and dopamine transporter in control and 1-methyl 4-phenyl 1, 2, 3, 6-tetrahydropyridine treated mouse striatum. Brain Res. 2009, 1283, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Siokas, V.; Aloizou, A.M.; Tsouris, Z.; Liampas, I.; Liakos, P.; Calina, D.; Docea, A.O.; Tsatsakis, A.; Bogdanos, D.P.; Hadjigeorgiou, G.M.; et al. ADORA2A rs5760423 and CYP1A2 rs762551 Polymorphisms as Risk Factors for Parkinson’s Disease. J. Clin. Med. 2021, 10, 381. [Google Scholar] [CrossRef]
- Duan, W.J.; Liang, L.; Pan, M.H.; Lu, D.H.; Wang, T.M.; Li, S.B.; Zhong, H.B.; Yang, X.J.; Cheng, Y.; Liu, B.; et al. Theacrine, a purine alkaloid from kucha, protects against Parkinson’s disease through SIRT3 activation. Phytomed. Int. J. Phytother. Phytopharm. 2020, 77, 153281. [Google Scholar] [CrossRef]
- Romualdo, G.R.; Prata, G.B.; da Silva, T.C.; Evangelista, A.F.; Reis, R.M.; Vinken, M.; Moreno, F.S.; Cogliati, B.; Barbisan, L.F. The combination of coffee compounds attenuates early fibrosis-associated hepatocarcinogenesis in mice: Involvement of miRNA profile modulation. J. Nutr. Biochem. 2020, 85, 108479. [Google Scholar] [CrossRef]
- Nakayama, T.; Funakoshi-Tago, M.; Tamura, H. Coffee reduces KRAS expression in Caco-2 human colon carcinoma cells via regulation of miRNAs. Oncol. Lett. 2017, 14, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Kulshreshtha, D.; Ganguly, J.; Jog, M. Manganese and Movement Disorders: A Review. J. Mov. Disord. 2021, 14, 93–102. [Google Scholar] [CrossRef]
- Raj, K.; Kaur, P.; Gupta, G.D.; Singh, S. Metals associated neurodegeneration in Parkinson’s disease: Insight to physiological, pathological mechanisms and management. Neurosci. Lett. 2021, 753, 135873. [Google Scholar] [CrossRef]
- Dick, F.D.; De Palma, G.; Ahmadi, A.; Scott, N.W.; Prescott, G.J.; Bennett, J.; Semple, S.; Dick, S.; Counsell, C.; Mozzoni, P.; et al. Environmental risk factors for Parkinson’s disease and parkinsonism: The Geoparkinson study. Occup. Environ. Med. 2007, 64, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, M.M.; Jerrett, M. A study of the relationships between Parkinson’s disease and markers of traffic-derived and environmental manganese air pollution in two Canadian cities. Environ. Res. 2007, 104, 420–432. [Google Scholar] [CrossRef]
- Aschner, M.; Erikson, K.M.; Herrero Hernandez, E.; Tjalkens, R. Manganese and its role in Parkinson’s disease: From transport to neuropathology. Neuromol. Med. 2009, 11, 252–266. [Google Scholar] [CrossRef]
- Guo, Z.; Zhang, Z.; Wang, Q.; Zhang, J.; Wang, L.; Zhang, Q.; Li, H.; Wu, S. Manganese chloride induces histone acetylation changes in neuronal cells: Its role in manganese-induced damage. Neurotoxicology 2018, 65, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J., Jr.; Pajarillo, E.; Karki, P.; Kim, J.; Son, D.S.; Aschner, M.; Lee, E. Valproic acid attenuates manganese-induced reduction in expression of GLT-1 and GLAST with concomitant changes in murine dopaminergic neurotoxicity. Neurotoxicology 2018, 67, 112–120. [Google Scholar] [CrossRef]
- Tarale, P.; Sivanesan, S.; Daiwile, A.P.; Stoger, R.; Bafana, A.; Naoghare, P.K.; Parmar, D.; Chakrabarti, T.; Kannan, K. Global DNA methylation profiling of manganese-exposed human neuroblastoma SH-SY5Y cells reveals epigenetic alterations in Parkinson’s disease-associated genes. Arch. Toxicol. 2017, 91, 2629–2641. [Google Scholar] [CrossRef] [PubMed]
- Coon, S.; Stark, A.; Peterson, E.; Gloi, A.; Kortsha, G.; Pounds, J.; Chettle, D.; Gorell, J. Whole-body lifetime occupational lead exposure and risk of Parkinson’s disease. Environ. Health Perspect. 2006, 114, 1872–1876. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yang, X.; Xu, M.; Zhang, J.; Sun, N. Epigenetic marker (LINE-1 promoter) methylation level was associated with occupational lead exposure. Clin. Toxicol. 2013, 51, 225–229. [Google Scholar] [CrossRef]
- Pfaff, A.L.; Bubb, V.J.; Quinn, J.P.; Koks, S. An Increased Burden of Highly Active Retrotransposition Competent L1s Is Associated with Parkinson’s Disease Risk and Progression in the PPMI Cohort. Int. J. Mol. Sci. 2020, 21, 6562. [Google Scholar] [CrossRef]
- Lee, C.P.; Zhu, C.H.; Su, C.C. Increased prevalence of Parkinson’s disease in soils with high arsenic levels. Parkinsonism Relat. Disord. 2021, 88, 19–23. [Google Scholar] [CrossRef]
- Bailey, K.A.; Fry, R.C. Arsenic-Associated Changes to the Epigenome: What Are the Functional Consequences? Curr. Environ. Health Rep. 2014, 1, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Huo, M.; Zhang, J.; Huang, W.; Wang, Y. Interplay Among Metabolism, Epigenetic Modifications, and Gene Expression in Cancer. Front. Cell Dev. Biol. 2021, 9, 793428. [Google Scholar] [CrossRef]
- Martinez-Iglesias, O.; Naidoo, V.; Cacabelos, N.; Cacabelos, R. Epigenetic Biomarkers as Diagnostic Tools for Neurodegenerative Disorders. Int. J. Mol. Sci. 2021, 23, 13. [Google Scholar] [CrossRef]
- Margis, R.; Margis, R.; Rieder, C.R. Identification of blood microRNAs associated to Parkinsonis disease. J. Biotechnol. 2011, 152, 96–101. [Google Scholar] [CrossRef]
- Hao, H.; Shao, M.; An, J.; Chen, C.; Feng, X.; Xie, S.; Gu, Z.; Chan, P.; Chinese Parkinson Study, G. Association of Catechol-O-Methyltransferase and monoamine oxidase B gene polymorphisms with motor complications in parkinson’s disease in a Chinese population. Parkinsonism Relat. Disord. 2014, 20, 1041–1045. [Google Scholar] [CrossRef]
- Michalowska, M.; Chalimoniuk, M.; Jowko, E.; Przybylska, I.; Langfort, J.; Toczylowska, B.; Krygowska-Wajs, A.; Fiszer, U. Gene polymorphisms and motor levodopa-induced complications in Parkinson’s disease. Brain Behav. 2020, 10, e01537. [Google Scholar] [CrossRef] [PubMed]
- Wills, A.M.; Eberly, S.; Tennis, M.; Lang, A.E.; Messing, S.; Togasaki, D.; Tanner, C.M.; Kamp, C.; Chen, J.F.; Oakes, D.; et al. Caffeine consumption and risk of dyskinesia in CALM-PD. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Fardi, M.; Solali, S.; Farshdousti Hagh, M. Epigenetic mechanisms as a new approach in cancer treatment: An updated review. Genes Dis. 2018, 5, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Li, R.; Yang, Z.F.; Wang, Y.Z.; Gong, X.L.; Wang, X.M. A DNA methyltransferase inhibitor, 5-aza-2’-deoxycytidine, exacerbates neurotoxicity and upregulates Parkinson’s disease-related genes in dopaminergic neurons. CNS Neurosci. Ther. 2013, 19, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Abascal, J.; Chiaino, E.; Frosini, M.; Davey, G.P.; Valoti, M. beta-Naphthoflavone and Ethanol Reverse Mitochondrial Dysfunction in A Parkinsonian Model of Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 3955. [Google Scholar] [CrossRef]
- Corrigendum. J. Pineal Res. 2019, 66, e12529. [CrossRef]
- Cacabelos, R.; Carrera, I.; Martinez, O.; Alejo, R.; Fernandez-Novoa, L.; Cacabelos, P.; Corzo, L.; Rodriguez, S.; Alcaraz, M.; Nebril, L.; et al. Atremorine in Parkinson’s disease: From dopaminergic neuroprotection to pharmacogenomics. Med. Res. Rev. 2021, 41, 2841–2886. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Li, W.W.; Lu, C.Z. Histone decacetylase inhibitors prevent mitochondrial fragmentation and elicit early neuroprotection against MPP+. CNS Neurosci. Ther. 2014, 20, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Wu, H.M.; Ossola, B.; Schendzielorz, N.; Wilson, B.C.; Chu, C.H.; Chen, S.L.; Wang, Q.; Zhang, D.; Qian, L.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, protects dopaminergic neurons from neurotoxin-induced damage. Br. J. Pharmacol. 2012, 165, 494–505. [Google Scholar] [CrossRef]
- Jin, H.; Kanthasamy, A.; Harischandra, D.S.; Kondru, N.; Ghosh, A.; Panicker, N.; Anantharam, V.; Rana, A.; Kanthasamy, A.G. Histone hyperacetylation up-regulates protein kinase Cdelta in dopaminergic neurons to induce cell death: Relevance to epigenetic mechanisms of neurodegeneration in Parkinson disease. J. Biol. Chem. 2014, 289, 34743–34767. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Liu, L.; Wang, X. HDAC inhibitor trichostatin A-inhibited survival of dopaminergic neuronal cells. Neurosci. Lett. 2009, 467, 212–216. [Google Scholar] [CrossRef]
- Cortes, H.; Reyes-Hernandez, O.D.; Gonzalez-Torres, M.; Vizcaino-Dorado, P.A.; Del Prado-Audelo, M.L.; Alcala-Alcala, S.; Sharifi-Rad, J.; Figueroa-Gonzalez, G.; Gonzalez-Del Carmen, M.; Floran, B.N.; et al. Curcumin for parkinson s disease: Potential therapeutic effects, molecular mechanisms, and nanoformulations to enhance its efficacy. Cell. Mol. Biol. 2021, 67, 101–105. [Google Scholar] [CrossRef]
- Xie, C.; Zhu, J.; Yang, X.; Huang, C.; Zhou, L.; Meng, Z.; Li, X.; Zhong, C. TAp63alpha Is Involved in Tobacco Smoke-Induced Lung Cancer EMT and the Anti-cancer Activity of Curcumin via miR-19 Transcriptional Suppression. Front. Cell Dev. Biol. 2021, 9, 645402. [Google Scholar] [CrossRef]
- Nicholas, A.P.; Lubin, F.D.; Hallett, P.J.; Vattem, P.; Ravenscroft, P.; Bezard, E.; Zhou, S.; Fox, S.H.; Brotchie, J.M.; Sweatt, J.D.; et al. Striatal histone modifications in models of levodopa-induced dyskinesia. J. Neurochem. 2008, 106, 486–494. [Google Scholar] [CrossRef]
- Hong, C.T.; Chan, L.; Bai, C.H. The Effect of Caffeine on the Risk and Progression of Parkinson’s Disease: A Meta-Analysis. Nutrients 2020, 12, 1860. [Google Scholar] [CrossRef]
- Machado-Filho, J.A.; Correia, A.O.; Montenegro, A.B.; Nobre, M.E.; Cerqueira, G.S.; Neves, K.R.; Naffah-Mazzacoratti Mda, G.; Cavalheiro, E.A.; de Castro Brito, G.A.; de Barros Viana, G.S. Caffeine neuroprotective effects on 6-OHDA-lesioned rats are mediated by several factors, including pro-inflammatory cytokines and histone deacetylase inhibitions. Behav. Brain Res. 2014, 264, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Epigenetic Modifications | Reference |
|---|---|
| DNA methylation | |
| - DNMT1 is downregulated in the brain of PD patients | [20] |
| - The SNCA gene is hypomethylated in the brain and blood of PD patients | [17] |
| - Altered DNA methylation patterns on gene variants of PARK16/1q32, GPNMB/7p15, and STX1B/16p11 loci in post-mortem brain samples have also been identified between PD patients and controls | [21] |
| - The DAT gene is hypermethylated in PD patients | [22] |
| - The TNF-α gene is hypomethylated in the SN of PD patients | [23] |
| - The CYP2E1 gene is hypomethylated in the brain of PD patients | [24] |
| - The COMT, PRNP and DCTN1 genes are differentially methylated in PD | [25] |
| - The DNA methylation status is different in blood samples from PD patients compared to controls in mitochondria-related genes, including LARS2, MIR1977, and DDAH2 | [26] |
| Histone modifications | |
| - The acetylation levels of histones H2A, H,3 and H4 are higher in the dopaminergic neurons from the midbrain of PD patients - HDACs are reduced in MPP(+)-treated neuronal cells, and the brain of MPTP-treated mice and | [27] |
| α-synuclein accumulation is associated with H3 hypoacetylation | [28] |
| - SIRT2, a HDAC, is implicated in α-synuclein aggregation, autophagy, oxidative stress, and neuroinflammation, although with conflicting results | [29] |
| - Under oxidative stress, nuclear α-synuclein can bind to the promoter of the PGC-1α gene, which leads to its hypoacetylation and the downregulation of its expression, finally resulting in mitochondrial impairment and neurotoxicity | [30] |
| Non-coding RNAs | |
| - A total of 125 miRs are differentially expressed in the prefrontal cortex of PD patients compared to controls | [31] |
| - MiR-7, miR-203a-3p, and miR-153 bind to and downregulate the expression of the SNCA gene | [32] |
| - MiR-132 is downregulated in rat models of PD, accompanied by lower levels of Nurr1, its molecular target | [33] |
| - MiR-133b levels are reduced in the midbrain of PD patients | [34] |
| - MiR-205 can bind to the 3′ UTR of LRRK2 and downregulate its expression | [35] |
| - MiR-214 levels are reduced after the treatment of cells or mice with MPP+ or MPTP, respectively, accompanied by increased α-synuclein levels in dopaminergic cells | [36] |
| - Mir-124 modulates dopaminergic neuronal loss, mitochondrial function, autophagy, oxidative stress, and neuroinflammation in PD animal models via several signaling pathways | [33] |
| - Mir-26 is upregulated in the SN and CSF of PD patients and downregulated in the blood of PD patients | [37] |
| - MiR-30, miR-34, miR-99, miR-124, miR-125, miR-146, miR-219, and miR-222 are differentially expressed in PD | [32] |
| - MiR-144 and miR-15b-5p are associated with PD | [32] |
| - A total of 87 lncRNAs are differentially expressed in the SN of PD patients | [38] |
| Environmental Factors | Reference |
|---|---|
| Tobacco smoking | |
| - Smoking-induced DMRs may display diverse distribution patterns in both hypomethylated and hypermethylated regions between smokers and non-smokers - The differentially expressed genes are implicated in “immunosuppression” pathways | [52] |
| - The DNA hypermethylation of CYP2D6, which is observed more commonly in poor metabolizers, is associated with a lower risk of heavy smoking | [53] |
| - Tobacco smoking is associated with the reduced methylation of the promoter region of LINE-1 retrotransposons in the blood mononuclear cells only in controls, but not in the PD cases - The inverse relationship between smoking and PD is stronger in low levels of LINE-1 methylation and is less evident as LINE-1 methylation levels are increased | [54] |
| - Methylation levels of the first intron of SLC6A3 gene are potentially related to nicotine dependence, an increased tendency to start smoking, and an impaired ability to quit | [55] |
| - ANKK1/DRD2 genetic region variants are associated with nicotine dependence in males | [56] |
| - MiR-124 and let-7a are differentially expressed between smokers and non-smokers | [50] |
| - Nicotine attenuates inflammation by upregulating miR-124 | [57] |
| - MiR-26, miR-30, miR-34, miR-99, miR-124, miR-125, miR-146, miR-219, and miR-222 are among the most significantly downregulated miRs in the lungs of rats exposed to smoking | [58] |
| Pesticides exposure | |
| - Dieldrin increases H3 and H4 acetylation, leading to proteosomal dysfunction and the accumulation of the cAMP response element-binding protein in dopaminergic neurons - Treatment with the HAT inhibitor anacardic acid prevents against dieldrin-induced histone hyper-acetylation, DNA fragmentation, and dopaminergic degeneration | [59] |
| - Exposure to the herbicide paraquat induces H3 acetylation in dopaminergic cells, and is associated with reduced HDAC levels - Anacardic acid protects against these effects | [60] |
| - In paraquat-treated mice, α-synuclein is accumulated in the nucleus near acetylated H3, and α-synuclein can directly bind to H1 and form a 2:1 complex | [61] |
| - Rotenone promotes H3K9 acetylation by downregulating SIRT1 and upregulating p53, thus promoting neurodegeneration | [62] |
| - SIRT3, a HDAC, can deacetylate SOD2, resulting in protection against MPTP-induced ROS accumulation and dopaminergic neurodegeneration | [63] |
| - SIRT5, another HDAC, is associated with increased SOD2 levels and improved mitochondrial function in MPTP-treated mice, thereby preventing nigrostriatal degeneration | [64] |
| - The miR-380-3p/Sp3-mRNA pathway is involved in MPTP-induced neurodegeneration | [65] |
| - Rotenone is associated with increased miR-26a and miR-34a levels and reduced miR-7 and let7a levels in rat models of PD | [66] |
| - MiR-34a, miR-141, and miR-9 are differentially expressed in MPP+-treated PC12 cells | [67] |
| - MiR-384-5p, which targets and downregulates SIRT1 expression, is increased in rotenone-induced mice and SH-SY5Y cell models of PD | [68] |
| - Differential expression levels of MiR-34a-5p are detected in the plasma of PD patients | [69] |
| Coffee consumption | |
| - Association between the DNA methylation status of CpG sites and coffee consumption in some genes causing familial PD, such as GBA, Parkin, and PINK1 in the blood of non-PD individuals | [70] |
| - Caffeine may increase the expression of DAT, P450 1A2, and the adenosine A2A receptor in the striatum of MPTP-treated mice | [71] |
| - Theacrine protects against dopaminergic degeneration in in vitro and in vivo models of PD by directly activating SIRT3, resulting in SOD2 deacetylation, the prevention of apoptosis, a reduction in ROS accumulation, and the restoration of mitochondrial dysfunction | [72] |
| - Mir-144 and miR-15b-5p are upregulated following treatment with coffee compounds | [73] |
| - Coffee has been demonstrated to upregulate miR-30 | [74] |
| Exposure to heavy metals | |
| - Manganese chloride can inhibit H3 and H4 acetylation, increase HDAC3 and HDAC4 expression, and reduce HAT expression | [75] |
| - Manganese is associated with lower levels of histone acetylation and expression levels of GLT-1 and astrocytic GLAST, thereby promoting neurotoxicity | [76] |
| - Parkin and PINK1 gene activities are affected by increased DNA methylation in vitro upon exposure to manganese | [77] |
| - Reduced methylation levels of the promoter of LINE-1 are related to exposure to lead | [78] |
| - Arsenic alters the status of LINE-1 methylation | [79] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelopoulou, E.; Paudel, Y.N.; Papageorgiou, S.G.; Piperi, C. Environmental Impact on the Epigenetic Mechanisms Underlying Parkinson’s Disease Pathogenesis: A Narrative Review. Brain Sci. 2022, 12, 175. https://doi.org/10.3390/brainsci12020175
Angelopoulou E, Paudel YN, Papageorgiou SG, Piperi C. Environmental Impact on the Epigenetic Mechanisms Underlying Parkinson’s Disease Pathogenesis: A Narrative Review. Brain Sciences. 2022; 12(2):175. https://doi.org/10.3390/brainsci12020175
Chicago/Turabian StyleAngelopoulou, Efthalia, Yam Nath Paudel, Sokratis G. Papageorgiou, and Christina Piperi. 2022. "Environmental Impact on the Epigenetic Mechanisms Underlying Parkinson’s Disease Pathogenesis: A Narrative Review" Brain Sciences 12, no. 2: 175. https://doi.org/10.3390/brainsci12020175
APA StyleAngelopoulou, E., Paudel, Y. N., Papageorgiou, S. G., & Piperi, C. (2022). Environmental Impact on the Epigenetic Mechanisms Underlying Parkinson’s Disease Pathogenesis: A Narrative Review. Brain Sciences, 12(2), 175. https://doi.org/10.3390/brainsci12020175