
Effects of Genetics and Sex on Acute Gene Expression Changes in the Hippocampus Following Neonatal Ethanol Exposure in BXD Recombinant Inbred Mouse Strains


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
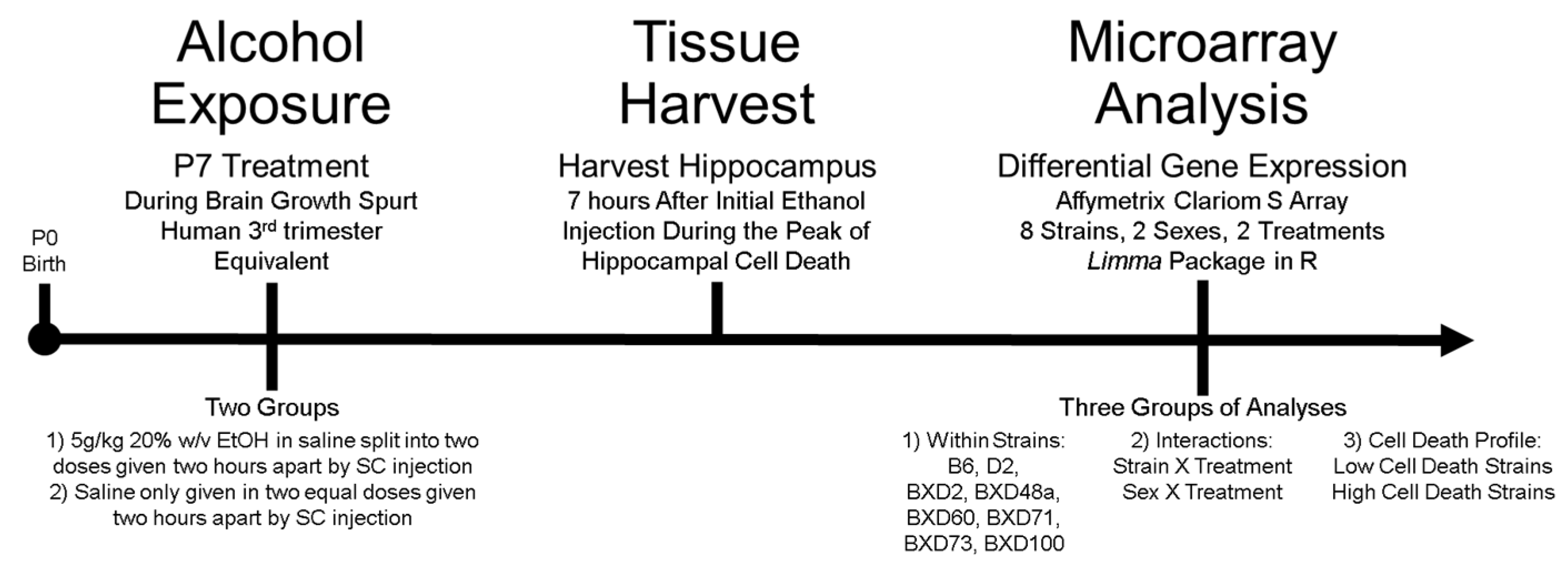
2. Materials and Methods
2.1. Animals and Numbers
2.2. Ethanol Exposure
2.3. Tissue Harvest and RNA Extraction
2.4. Gene Expression Microarray & Data Processing
2.5. Differential Expression and Statistical Analyses
2.5.1. Analysis 1: Ethanol-Induced Differential Expression within Strains
2.5.2. Analysis 2: Treatment Interactions
2.5.3. Analysis 3: Comparison of Strains with Differential Cell Death Phenotype
2.6. Gene Enrichment Analysis
2.7. GeneNetwork Analysis
3. Results
3.1. Ethanol-Induced Differential Expression
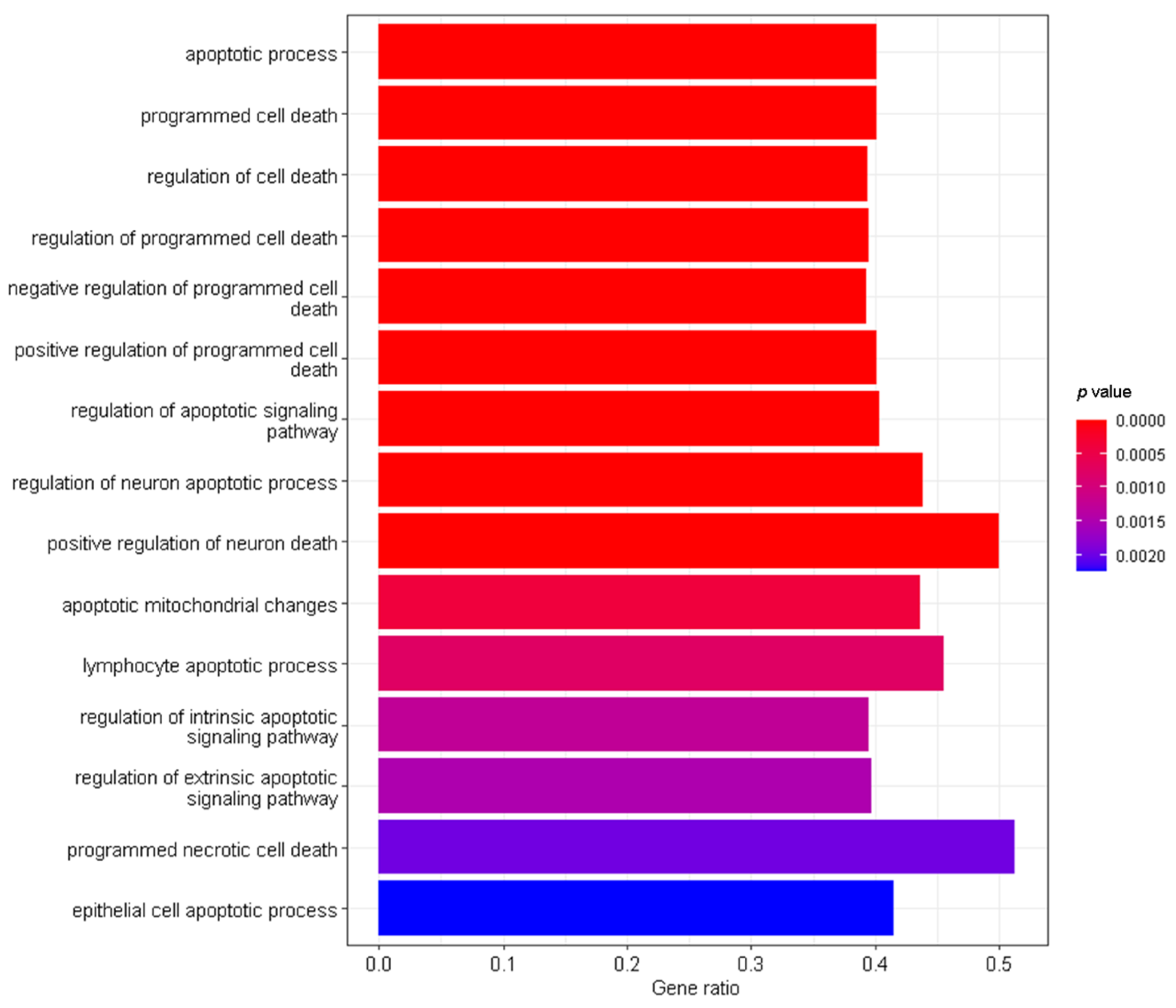
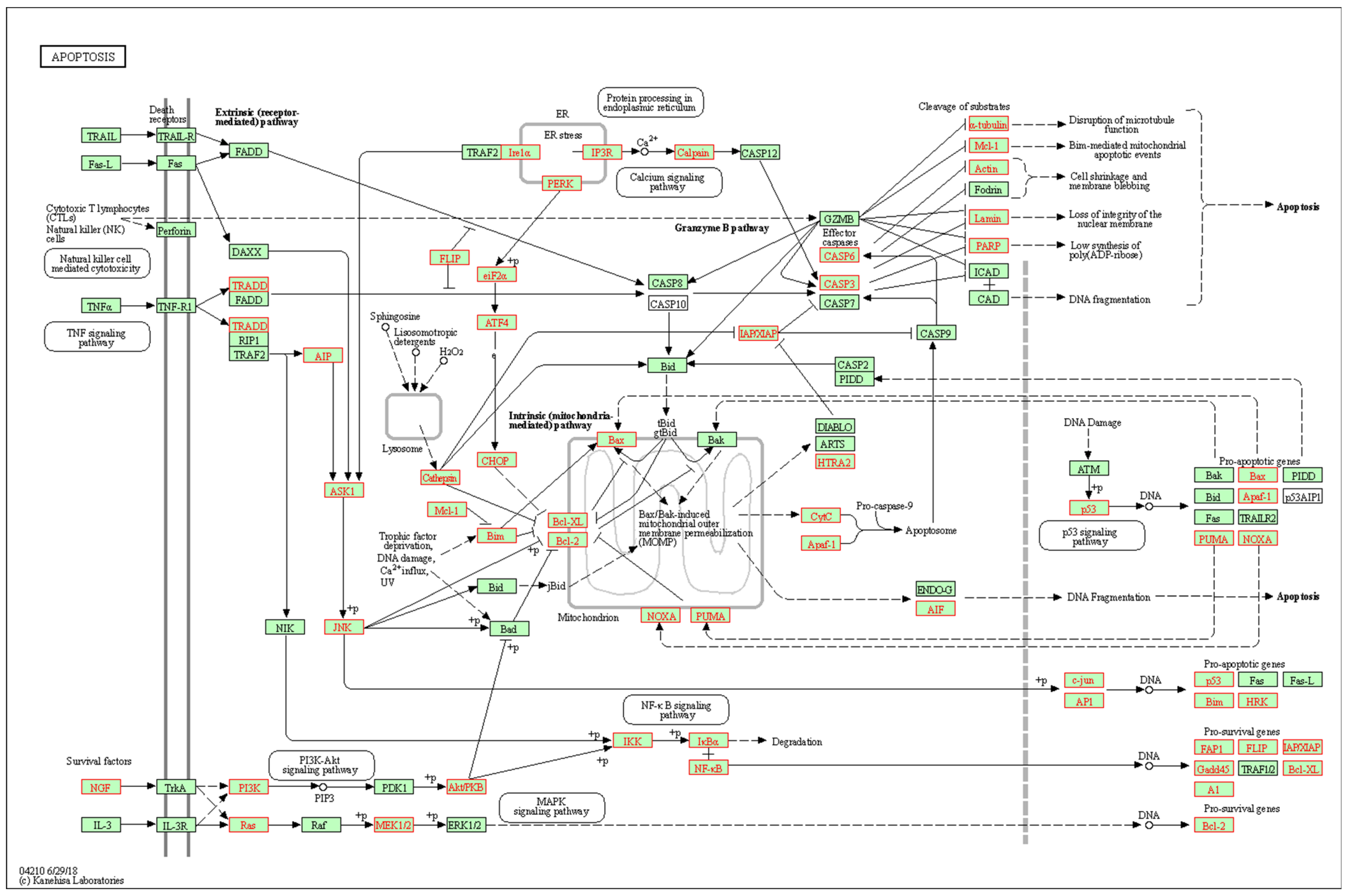
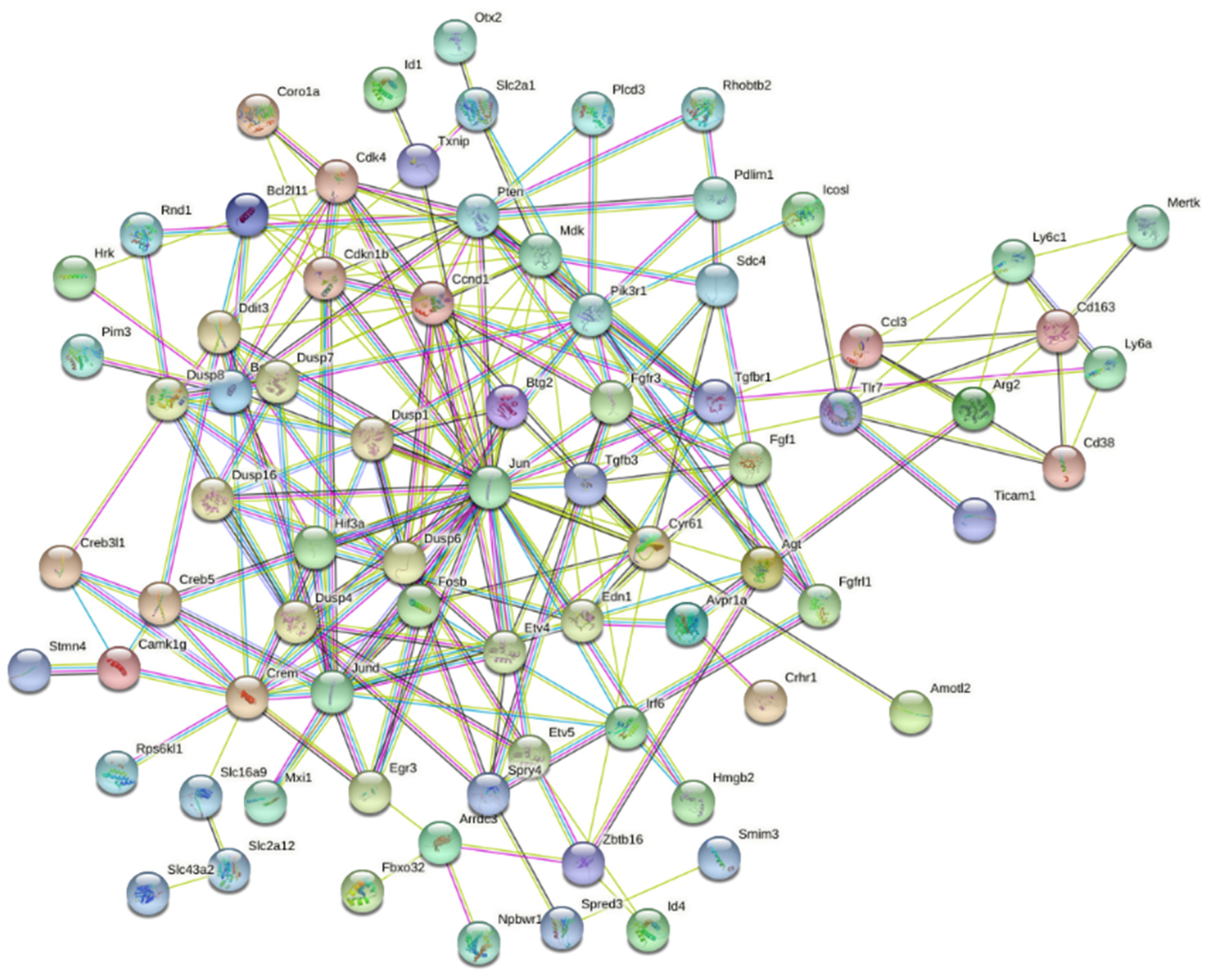
3.2. Interaction Effects and Pathway Analysis of Differentially Exprssed Genes
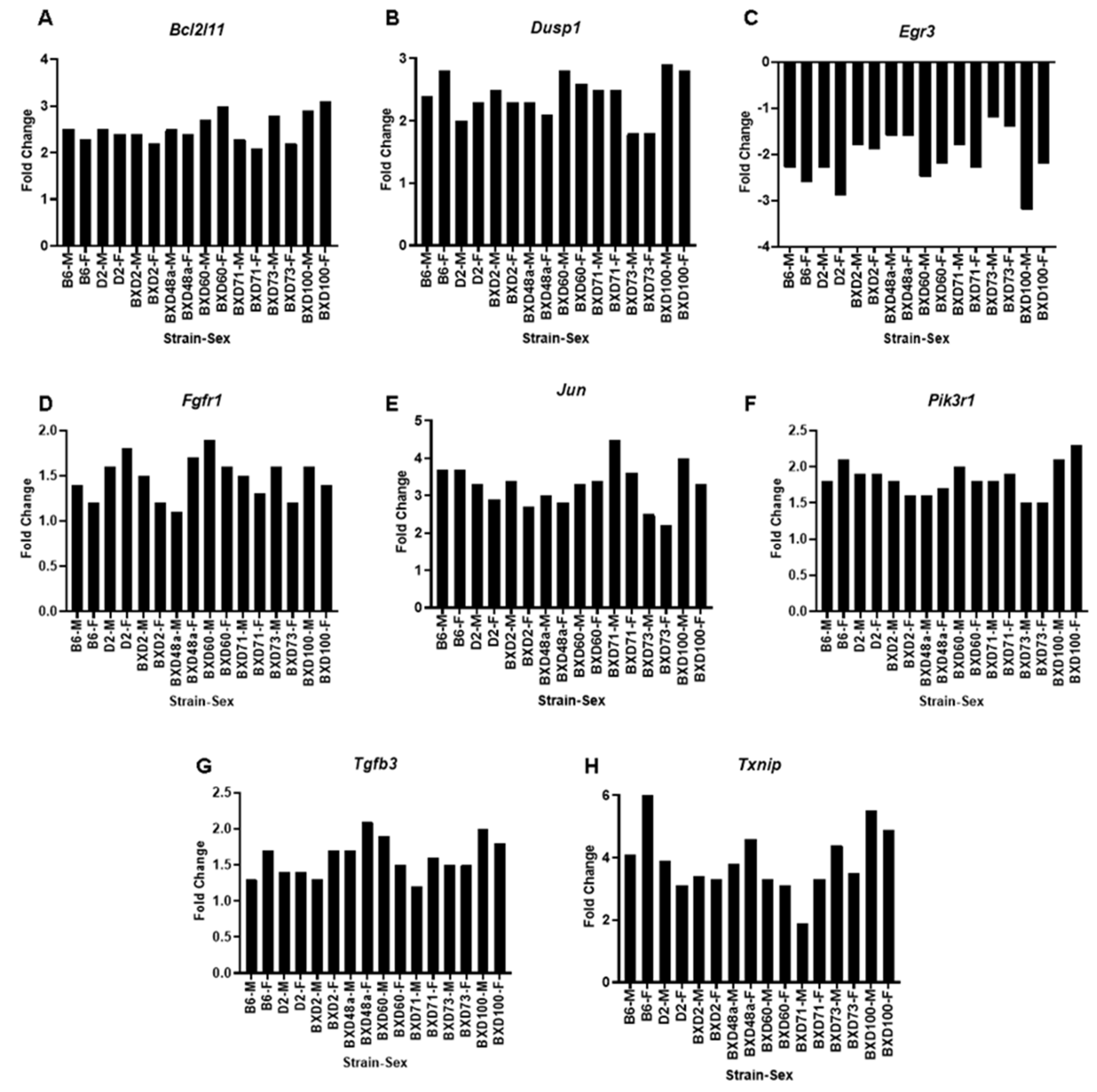
3.3. Genes of Interest within the Previously Identified Quantitative Trait Locus (QTL)
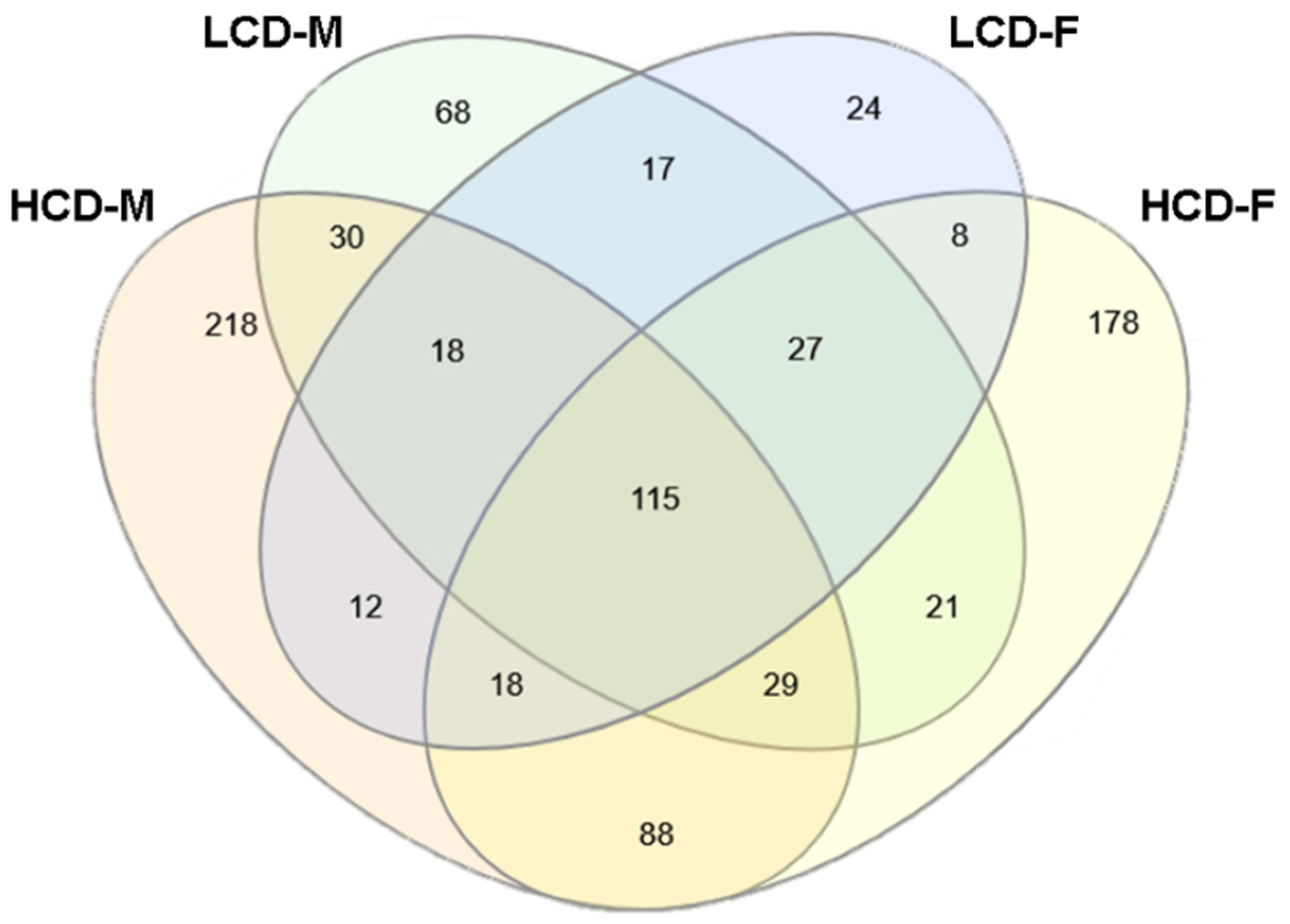
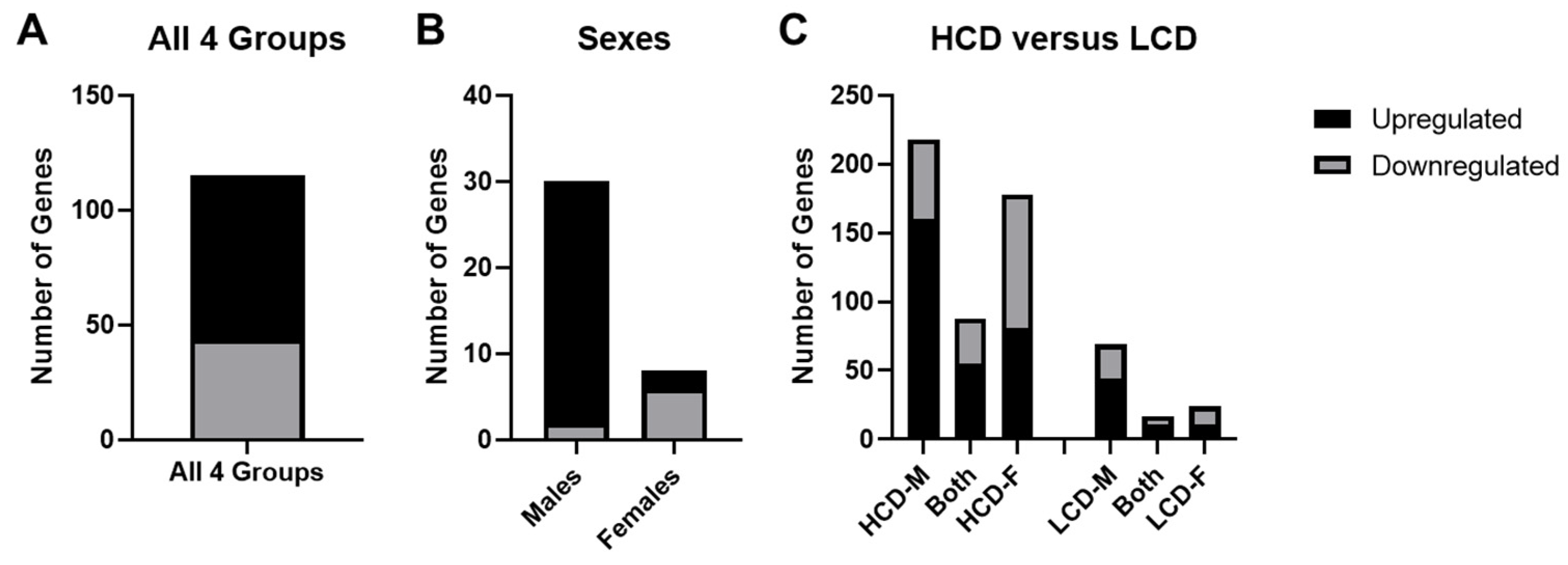
3.4. Comparison between High Cell Death (HCD) Strains and Low Cell Death (LCD) Strains
4. Discussion
4.1. Effects of Sex on Ethanol-Induced Gene Expression Changes
4.2. Effects of Strain on Ethanol-Induced Gene Expression Changes
4.3. Comparison of Ethanol-Inuced Gene Expression Changes between HCD & LCD Strains
4.4. Limitations & Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jones, K.L.; Smith, D.W. Recognition of the fetal alcohol syndrome in early infancy. Lancet 1973, 302, 999–1001. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L.; Smith, D.W.; Ulleland, C.N.; Streissguth, P. Pattern of malformation in offspring of chronic alcoholic mothers. Lancet 1973, 1, 1267–1271. [Google Scholar] [CrossRef] [PubMed]
- May, P.A.; Baete, A.; Russo, J.; Elliott, A.J.; Blankenship, J.; Kalberg, W.O.; Buckley, D.; Brooks, M.; Hasken, J.; Abdul-Rahman, O.; et al. Prevalence and characteristics of fetal alcohol spectrum disorders. Pediatrics 2014, 134, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Roozen, S.; Peters, G.J.; Kok, G.; Townend, D.; Nijhuis, J.; Curfs, L. Worldwide Prevalence of Fetal Alcohol Spectrum Disorders: A Systematic Literature Review Including Meta-Analysis. Alcohol. Clin. Exp. Res. 2016, 40, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, J.; Floyd, L.L.; Weber, M.K.; Fetal Alcohol Syndrome Prevention Team, Division of Birth Defects and Developmental Disabilities, National Center on Birth Defects and Developmental Disabilities, Centers for Disease Control and Prevention (CDC). Guidelines for identifying and referring persons with fetal alcohol syndrome. MMWR Recomm. Rep. 2005, 54, 1–14. [Google Scholar] [PubMed]
- Kable, J.A.; O’Connor, M.J.; Olson, H.C.; Paley, B.; Mattson, S.N.; Anderson, S.M.; Riley, E.P. Neurobehavioral Disorder Associated with Prenatal Alcohol Exposure (ND-PAE): Proposed DSM-5 Diagnosis. Child Psychiatry Hum. Dev. 2016, 47, 335–346. [Google Scholar] [CrossRef]
- Olney, J.W.; Tenkova, T.; Dikranian, K.; Qin, Y.Q.; Labruyere, J.; Ikonomidou, C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res. Dev. Brain Res. 2002, 133, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W. Fetal alcohol syndrome at the cellular level. Addict. Biol. 2004, 9, 137–149, discussion 151. [Google Scholar] [CrossRef] [PubMed]
- Dobbing, J. The later growth of the brain and its vulnerability. Pediatrics 1974, 53, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Chasnoff, I.J. Fetal alcohol syndrome in twin pregnancy. Acta Genet. Med. Gemellol. 1985, 34, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Christoffel, K.K.; Salafsky, I. Fetal alcohol syndrome in dizygotic twins. J. Pediatr. 1975, 87, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Streissguth, A.P.; Dehaene, P. Fetal alcohol syndrome in twins of alcoholic mothers: Concordance of diagnosis and IQ. Am. J. Med. Genet. 1993, 47, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Astley, S.J. Profile of the first 1400 patients receiving diagnostic evaluations for fetal alcohol spectrum disorder at the Washington State Fetal Alcohol Syndrome Diagnostic & Prevention Network. Can. J. Clin. Pharmacol. 2010, 17, e132–e164. [Google Scholar] [PubMed]
- Green, M.L.; Singh, A.V.; Zhang, Y.; Nemeth, K.A.; Sulik, K.K.; Knudsen, T.B. Reprogramming of genetic networks during initiation of the Fetal Alcohol Syndrome. Dev. Dyn. 2007, 236, 613–631. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Debelak, K.A.; Tessmer, L.L.; Cartwright, M.M.; Smith, S.M. Genetic influences on craniofacial outcome in an avian model of prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 2001, 25, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ozturk, N.C.; Ni, L.; Goodlett, C.; Zhou, F.C. Strain differences in developmental vulnerability to alcohol exposure via embryo culture in mice. Alcohol. Clin. Exp. Res. 2011, 35, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Goodlett, C.R.; Gilliam, D.M.; Nichols, J.M.; West, J.R. Genetic influences on brain growth restriction induced by development exposure to alcohol. Neurotoxicology 1989, 10, 321–334. [Google Scholar] [PubMed]
- Ogawa, T.; Kuwagata, M.; Ruiz, J.; Zhou, F.C. Differential teratogenic effect of alcohol on embryonic development between C57BL/6 and DBA/2 mice: A new view. Alcohol. Clin. Exp. Res. 2005, 29, 855–863. [Google Scholar] [CrossRef]
- Debelak, K.A.; Smith, S.M. Avian genetic background modulates the neural crest apoptosis induced by ethanol exposure. Alcohol. Clin. Exp. Res. 2000, 24, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Goldowitz, D.; Lussier, A.A.; Boyle, J.K.; Wong, K.; Lattimer, S.L.; Dubose, C.; Lu, L.; Kobor, M.S.; Hamre, K.M. Molecular pathways underpinning ethanol-induced neurodegeneration. Front. Genet. 2014, 5, 203. [Google Scholar] [CrossRef] [PubMed]
- Theberge, E.T.; Baker, J.A.; Dubose, C.; Boyle, J.K.; Balce, K.; Goldowitz, D.; Hamre, K.M. Genetic Influences on the Amount of Cell Death in the Neural Tube of BXD Mice Exposed to Acute Ethanol at Midgestation. Alcohol. Clin. Exp. Res. 2019, 43, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Amiri, S.; Davie, J.R.; Rastegar, M. Chronic Ethanol Exposure Alters DNA Methylation in Neural Stem Cells: Role of Mouse Strain and Sex. Mol. Neurobiol. 2020, 57, 650–667. [Google Scholar] [CrossRef] [PubMed]
- Downing, C.; Flink, S.; Florez-McClure, M.L.; Johnson, T.E.; Tabakoff, B.; Kechris, K.J. Gene expression changes in C57BL/6J and DBA/2J mice following prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 2012, 36, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Lossie, A.C.; Muir, W.M.; Lo, C.L.; Timm, F.; Liu, Y.; Gray, W.; Zhou, F.C. Implications of genomic signatures in the differential vulnerability to fetal alcohol exposure in C57BL/6 and DBA/2 mice. Front. Genet. 2014, 5, 173. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chater-Diehl, E.J.; Laufer, B.I.; Castellani, C.A.; Alberry, B.L.; Singh, S.M. Alteration of Gene Expression, DNA Methylation, and Histone Methylation in Free Radical Scavenging Networks in Adult Mouse Hippocampus following Fetal Alcohol Exposure. PLoS ONE 2016, 11, e0154836. [Google Scholar] [CrossRef] [PubMed]
- Hard, M.L.; Abdolell, M.; Robinson, B.H.; Koren, G. Gene-expression analysis after alcohol exposure in the developing mouse. J. Lab. Clin. Med. 2005, 145, 47–54. [Google Scholar] [CrossRef]
- Kleiber, M.L.; Mantha, K.; Stringer, R.L.; Singh, S.M. Neurodevelopmental alcohol exposure elicits long-term changes to gene expression that alter distinct molecular pathways dependent on timing of exposure. J. Neurodev. Disord. 2013, 5, 6. [Google Scholar] [CrossRef]
- Lunde-Young, R.; Ramirez, J.; Naik, V.; Orzabal, M.; Lee, J.; Konganti, K.; Hillhouse, A.; Threadgill, D.; Ramadoss, J. Hippocampal transcriptome reveals novel targets of FASD pathogenesis. Brain Behav. 2019, 9, e01334. [Google Scholar] [CrossRef] [PubMed]
- Lussier, A.A.; Stepien, K.A.; Neumann, S.M.; Pavlidis, P.; Kobor, M.S.; Weinberg, J. Prenatal alcohol exposure alters steady-state and activated gene expression in the adult rat brain. Alcohol. Clin. Exp. Res. 2015, 39, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Mandal, C.; Park, K.S.; Jung, K.H.; Chai, Y.G. Ethanol-related alterations in gene expression patterns in the developing murine hippocampus. Acta Biochim. Biophys. Sin. 2015, 47, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Marjonen, H.; Sierra, A.; Nyman, A.; Rogojin, V.; Grohn, O.; Linden, A.M.; Hautaniemi, S.; Kaminen-Ahola, N. Early maternal alcohol consumption alters hippocampal DNA methylation, gene expression and volume in a mouse model. PLoS ONE 2015, 10, e0124931. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.A.; Wnek, C.; Kotlus, B.S.; Roemer, N.; MacTaggart, T.; Phillips, S.J. Genotyping new BXD recombinant inbred mouse strains and comparison of BXD and consensus maps. Mamm. Genome 1999, 10, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pandey, A.K.; Mulligan, M.K.; Williams, E.G.; Mozhui, K.; Li, Z.; Jovaisaite, V.; Quarles, L.D.; Xiao, Z.; Huang, J.; et al. Joint mouse-human phenome-wide association to test gene function and disease risk. Nat. Commun. 2016, 7, 10464. [Google Scholar] [CrossRef] [PubMed]
- Downing, C.; Balderrama-Durbin, C.; Kimball, A.; Biers, J.; Wright, H.; Gilliam, D.; Johnson, T.E. Quantitative trait locus mapping for ethanol teratogenesis in BXD recombinant inbred mice. Alcohol. Clin. Exp. Res. 2012, 36, 1340–1354. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Wei, L.; Peirce, J.L.; Wang, X.; Zhou, J.; Homayouni, R.; Williams, R.W.; Airey, D.C. Using gene expression databases for classical trait QTL candidate gene discovery in the BXD recombinant inbred genetic reference population: Mouse forebrain weight. BMC Genom. 2008, 9, 444. [Google Scholar] [CrossRef]
- Penaloza, C.; Estevez, B.; Orlanski, S.; Sikorska, M.; Walker, R.; Smith, C.; Smith, B.; Lockshin, R.A.; Zakeri, Z. Sex of the cell dictates its response: Differential gene expression and sensitivity to cell death inducing stress in male and female cells. FASEB J. 2009, 23, 1869–1879. [Google Scholar] [CrossRef]
- Nijboer, C.H.; Kavelaars, A.; van Bel, F.; Heijnen, C.J.; Groenendaal, F. Gender-dependent pathways of hypoxia-ischemia-induced cell death and neuroprotection in the immature P3 rat. Dev. Neurosci. 2007, 29, 385–392. [Google Scholar] [CrossRef]
- Du, L.; Bayir, H.; Lai, Y.; Zhang, X.; Kochanek, P.M.; Watkins, S.C.; Graham, S.H.; Clark, R.S. Innate gender-based proclivity in response to cytotoxicity and programmed cell death pathway. J. Biol. Chem. 2004, 279, 38563–38570. [Google Scholar] [CrossRef]
- Kim, T.; Chelluboina, B.; Chokkalla, A.K.; Vemuganti, R. Age and sex differences in the pathophysiology of acute CNS injury. Neurochem. Int. 2019, 127, 22–28. [Google Scholar] [CrossRef]
- Dobbing, J.; Sands, J. Comparative aspects of the brain growth spurt. Early Hum. Dev. 1979, 3, 79–83. [Google Scholar] [CrossRef]
- Schaffner, S.L.; Lussier, A.A.; Baker, J.A.; Goldowitz, D.; Hamre, K.M.; Kobor, M.S. Neonatal Alcohol Exposure in Mice Induces Select Differentiation- and Apoptosis-Related Chromatin Changes Both Independent of and Dependent on Sex. Front. Genet. 2020, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Boehm, S.L., 2nd; Lundahl, K.R.; Caldwell, J.; Gilliam, D.M. Ethanol teratogenesis in the C57BL/6J, DBA/2J, and A/J inbred mouse strains. Alcohol 1997, 14, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.B.; Zhang, L.; Kircher, D.; Wang, S.; Mayfield, R.D.; Crabbe, J.C.; Morrisett, R.A.; Harris, R.A.; Ponomarev, I. Dissecting Brain Networks Underlying Alcohol Binge Drinking Using a Systems Genomics Approach. Mol. Neurobiol. 2019, 56, 2791–2810. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Chipman, H.; Tibshirani, R. Hybrid hierarchical clustering with applications to microarray data. Biostatistics 2006, 7, 286–301. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Huang, Y.; Jiao, Y.; Chen, H.; Cao, Y.; Bennett, B.; Wang, Y.; Gu, W. Expression of alcoholism-relevant genes in the liver are differently correlated to different parts of the brain. Gene Expr. 2013, 16, 31–38. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed]
- Terenina, E.E.; Cavigelli, S.; Mormede, P.; Zhao, W.; Parks, C.; Lu, L.; Jones, B.C.; Mulligan, M.K. Genetic Factors Mediate the Impact of Chronic Stress and Subsequent Response to Novel Acute Stress. Front. Neurosci. 2019, 13, 438. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M. KEGG mapping tools for uncovering hidden features in biological data. Protein. Sci. 2022, 31, 47–53. [Google Scholar] [CrossRef]
- Smith, C.L.; Eppig, J.T. The mammalian phenotype ontology: Enabling robust annotation and comparative analysis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 390–399. [Google Scholar] [CrossRef]
- Kohler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef]
- Hoyme, H.E.; Kalberg, W.O.; Elliott, A.J.; Blankenship, J.; Buckley, D.; Marais, A.S.; Manning, M.A.; Robinson, L.K.; Adam, M.P.; Abdul-Rahman, O.; et al. Updated Clinical Guidelines for Diagnosing Fetal Alcohol Spectrum Disorders. Pediatrics 2016, 138, e20154256. [Google Scholar] [CrossRef]
- Dunty, W.C., Jr.; Chen, S.Y.; Zucker, R.M.; Dehart, D.B.; Sulik, K.K. Selective vulnerability of embryonic cell populations to ethanol-induced apoptosis: Implications for alcohol-related birth defects and neurodevelopmental disorder. Alcohol. Clin. Exp. Res. 2001, 25, 1523–1535. [Google Scholar] [CrossRef]
- Olney, J.W.; Tenkova, T.; Dikranian, K.; Muglia, L.J.; Jermakowicz, W.J.; D’Sa, C.; Roth, K.A. Ethanol-induced caspase-3 activation in the in vivo developing mouse brain. Neurobiol. Dis. 2002, 9, 205–219. [Google Scholar] [CrossRef]
- Bannigan, J.; Cottell, D. Ethanol teratogenicity in mice: An electron microscopic study. Teratology 1984, 30, 281–290. [Google Scholar] [CrossRef]
- Smith, S.M. Alcohol-induced cell death in the embryo. Alcohol Health Res. World 1997, 21, 287–297. [Google Scholar] [PubMed]
- Kleiber, M.L.; Laufer, B.I.; Stringer, R.L.; Singh, S.M. Third trimester-equivalent ethanol exposure is characterized by an acute cellular stress response and an ontogenetic disruption of genes critical for synaptic establishment and function in mice. Dev. Neurosci. 2014, 36, 499–519. [Google Scholar] [CrossRef] [PubMed]
- Mantha, K.; Laufer, B.I.; Singh, S.M. Molecular changes during neurodevelopment following second-trimester binge ethanol exposure in a mouse model of fetal alcohol spectrum disorder: From immediate effects to long-term adaptation. Dev. Neurosci. 2014, 36, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Basavarajappa, B.S. Binge-like Prenatal Ethanol Exposure Causes Impaired Cellular Differentiation in the Embryonic Forebrain and Synaptic and Behavioral Defects in Adult Mice. Brain Sci. 2022, 12, 793. [Google Scholar] [CrossRef]
- Buckley, D.M.; Sidik, A.; Kar, R.D.; Eberhart, J.K. Differentially sensitive neuronal subpopulations in the central nervous system and the formation of hindbrain heterotopias in ethanol-exposed zebrafish. Birth Defects Res. 2019, 111, 700–713. [Google Scholar] [CrossRef]
- Boschen, K.E.; Ptacek, T.S.; Berginski, M.E.; Simon, J.M.; Parnell, S.E. Transcriptomic analyses of gastrulation-stage mouse embryos with differential susceptibility to alcohol. Dis. Model. Mech. 2021, 14, dmm049012. [Google Scholar] [CrossRef]
- Takahashi, Y.; Okano, H.; Takashima, K.; Ojiro, R.; Tang, Q.; Ozawa, S.; Ogawa, B.; Woo, G.H.; Yoshida, T.; Shibutani, M. Oral exposure to high-dose ethanol for 28 days in rats reduces neural stem cells and immediate nascent neural progenitor cells as well as FOS-expressing newborn granule cells in adult hippocampal neurogenesis. Toxicol. Lett. 2022, 360, 20–32. [Google Scholar] [CrossRef]
- Luo, S.; Rubinsztein, D.C. BCL2L11/BIM: A novel molecular link between autophagy and apoptosis. Autophagy 2013, 9, 104–105. [Google Scholar] [CrossRef]
- Trollmann, R.; Rehrauer, H.; Schneider, C.; Krischke, G.; Huemmler, N.; Keller, S.; Rascher, W.; Gassmann, M. Late-gestational systemic hypoxia leads to a similar early gene response in mouse placenta and developing brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1489–R1499. [Google Scholar] [CrossRef][Green Version]
- Shao, L.L.; Gao, M.M.; Gong, J.X.; Yang, L.Y. DUSP1 regulates hippocampal damage in epilepsy rats via ERK1/2 pathway. J. Chem. Neuroanat. 2021, 118, 102032. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, Y.; Li, J.; Wang, Y.; Tian, Y.; Guo, Q.; Cheng, Z. DUSP1 Promotes Microglial Polarization toward M2 Phenotype in the Medial Prefrontal Cortex of Neuropathic Pain Rats via Inhibition of MAPK Pathway. ACS Chem. Neurosci. 2021, 12, 966–978. [Google Scholar] [CrossRef] [PubMed]
- Bossy-Wetzel, E.; Bakiri, L.; Yaniv, M. Induction of apoptosis by the transcription factor c-Jun. EMBO J. 1997, 16, 1695–1709. [Google Scholar] [CrossRef] [PubMed]
- Winnay, J.N.; Solheim, M.H.; Dirice, E.; Sakaguchi, M.; Noh, H.L.; Kang, H.J.; Takahashi, H.; Chudasama, K.K.; Kim, J.K.; Molven, A.; et al. PI3-kinase mutation linked to insulin and growth factor resistance in vivo. J. Clin. Investig. 2016, 126, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Cipriano, R.; Miskimen, K.L.; Bryson, B.L.; Foy, C.R.; Bartel, C.A.; Jackson, M.W. FAM83B-mediated activation of PI3K/AKT and MAPK signaling cooperates to promote epithelial cell transformation and resistance to targeted therapies. Oncotarget 2013, 4, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zuo, D.; Yu, L.; Zhang, L.; Tang, J.; Cui, C.; Bao, L.; Zan, K.; Zhang, Z.; Yang, X.; et al. ROS/TXNIP pathway contributes to thrombin induced NLRP3 inflammasome activation and cell apoptosis in microglia. Biochem. Biophys. Res. Commun. 2017, 485, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Dong, Y.; Zhou, Y.; Li, H. Overexpression of miR-874-3p alleviates LPS-induced apoptosis and inflammation in alveolar epithelial cell by targeting EGR3/NF-kappaB. Acta Biochim. Pol. 2021, 68, 231–238. [Google Scholar] [CrossRef]
- Ghazavi, H.; Hoseini, S.J.; Ebrahimzadeh-Bideskan, A.; Mashkani, B.; Mehri, S.; Ghorbani, A.; Sadri, K.; Mahdipour, E.; Ghasemi, F.; Forouzanfar, F.; et al. Fibroblast Growth Factor Type 1 (FGF1)-Overexpressed Adipose-Derived Mesenchaymal Stem Cells (AD-MSC(FGF1)) Induce Neuroprotection and Functional Recovery in a Rat Stroke Model. Stem. Cell Rev. Rep. 2017, 13, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Shen, C.; Zhao, S.L.; Hu, Y.J.; Song, Y.; Zhong, Q.J. MicroRNA-126 affects cell apoptosis, proliferation, cell cycle and modulates VEGF/TGF-beta levels in pulmonary artery endothelial cells. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3058–3069. [Google Scholar] [CrossRef]
- Ke, C.Y.; Mei, H.H.; Wong, F.H.; Lo, L.J. IRF6 and TAK1 coordinately promote the activation of HIPK2 to stimulate apoptosis during palate fusion. Sci. Signal. 2019, 12, eaav7666. [Google Scholar] [CrossRef] [PubMed]
- Krieglstein, K.; Strelau, J.; Schober, A.; Sullivan, A.; Unsicker, K. TGF-beta and the regulation of neuron survival and death. J. Physiol. Paris 2002, 96, 25–30. [Google Scholar] [CrossRef]
- de Luca, A.; Weller, M.; Fontana, A. TGF-beta-induced apoptosis of cerebellar granule neurons is prevented by depolarization. J. Neurosci. 1996, 16, 4174–4185. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Deehan, G.A., Jr.; Hauser, S.R.; Getachew, B.; Waeiss, R.A.; Engleman, E.A.; Knight, C.P.; McBride, W.J.; Truitt, W.A.; Bell, R.L.; Rodd, Z.A. Selective breeding for high alcohol consumption and response to nicotine: Locomotor activity, dopaminergic in the mesolimbic system, and innate genetic differences in male and female alcohol-preferring, non-preferring, and replicate lines of high-alcohol drinking and low-alcohol drinking rats. Psychopharmacology 2018, 235, 2755–2769. [Google Scholar] [CrossRef] [PubMed]
- McBride, W.J.; Rodd, Z.A.; Bell, R.L.; Lumeng, L.; Li, T.K. The alcohol-preferring (P) and high-alcohol-drinking (HAD) rats--animal models of alcoholism. Alcohol 2014, 48, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Stewart, R.B.; Bell, R.L.; Badia-Elder, N.E.; Carr, L.G.; McBride, W.J.; Lumeng, L.; Li, T.K. Phenotypic and genotypic characterization of the Indiana University rat lines selectively bred for high and low alcohol preference. Behav. Genet. 2002, 32, 363–388. [Google Scholar] [CrossRef] [PubMed]
- Oberlin, B.; Best, C.; Matson, L.; Henderson, A.; Grahame, N. Derivation and characterization of replicate high- and low-alcohol preferring lines of mice and a high-drinking crossed HAP line. Behav. Genet. 2011, 41, 288–302. [Google Scholar] [CrossRef]
- Han, J.H.; Heo, K.S.; Myung, C.S. Cytokine-induced apoptosis inhibitor 1 (CIAPIN1) accelerates vascular remodelling via p53 and JAK2-STAT3 regulation in vascular smooth muscle cells. Br. J. Pharmacol. 2021, 178, 4533–4551. [Google Scholar] [CrossRef]
- Wu, L.; Ma, C.A.; Zhao, Y.; Jain, A. Aurora B interacts with NIR-p53, leading to p53 phosphorylation in its DNA-binding domain and subsequent functional suppression. J. Biol. Chem. 2011, 286, 2236–2244. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, H.; Takai, E.; Matsumura, I.; Kouno, M.; Morii, E.; Kitamura, Y.; Takeda, J.; Kanakura, Y. Identification of a cytokine-induced antiapoptotic molecule anamorsin essential for definitive hematopoiesis. J. Exp. Med. 2004, 199, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Hublitz, P.; Kunowska, N.; Mayer, U.P.; Muller, J.M.; Heyne, K.; Yin, N.; Fritzsche, C.; Poli, C.; Miguet, L.; Schupp, I.W.; et al. NIR is a novel INHAT repressor that modulates the transcriptional activity of p53. Genes Dev. 2005, 19, 2912–2924. [Google Scholar] [CrossRef]
- Salvador, J.M.; Brown-Clay, J.D.; Fornace, A.J., Jr. Gadd45 in stress signaling, cell cycle control, and apoptosis. In Gadd45 Stress Sensor Genes; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2013; Volume 793, pp. 1–19. [Google Scholar] [CrossRef]
- Calenic, B.; Yaegaki, K.; Ishkitiev, N.; Kumazawa, Y.; Imai, T.; Tanaka, T. p53-Pathway activity and apoptosis in hydrogen sulfide-exposed stem cells separated from human gingival epithelium. J. Periodontal. Res. 2013, 48, 322–330. [Google Scholar] [CrossRef]
- Sultan, F.A.; Sweatt, J.D. The role of the Gadd45 family in the nervous system: A focus on neurodevelopment, neuronal injury, and cognitive neuroepigenetics. Adv. Exp. Med. Biol. 2013, 793, 81–119. [Google Scholar] [CrossRef]
- Grassi, D.; Franz, H.; Vezzali, R.; Bovio, P.; Heidrich, S.; Dehghanian, F.; Lagunas, N.; Belzung, C.; Krieglstein, K.; Vogel, T. Neuronal Activity, TGFbeta-Signaling and Unpredictable Chronic Stress Modulate Transcription of Gadd45 Family Members and DNA Methylation in the Hippocampus. Cereb. Cortex 2017, 27, 4166–4181. [Google Scholar] [CrossRef] [PubMed]
- Sepp, M.; Vihma, H.; Nurm, K.; Urb, M.; Page, S.C.; Roots, K.; Hark, A.; Maher, B.J.; Pruunsild, P.; Timmusk, T. The Intellectual Disability and Schizophrenia Associated Transcription Factor TCF4 Is Regulated by Neuronal Activity and Protein Kinase A. J. Neurosci. 2017, 37, 10516–10527. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.C.; Chen, C.H.; Chang, S.H.; Huang, C.T.; Hwang, W.C.; Yang, D.I. Id1 and Sonic Hedgehog Mediate Cell Cycle Reentry and Apoptosis Induced by Amyloid Beta-Peptide in Post-mitotic Cortical Neurons. Mol. Neurobiol. 2019, 56, 465–489. [Google Scholar] [CrossRef] [PubMed]
- Takada, S.; Sakakima, H.; Matsuyama, T.; Otsuka, S.; Nakanishi, K.; Norimatsu, K.; Itashiki, Y.; Tani, A.; Kikuchi, K. Disruption of Midkine gene reduces traumatic brain injury through the modulation of neuroinflammation. J. Neuroinflamm. 2020, 17, 40. [Google Scholar] [CrossRef]
- Qiao, X.R.; Wang, L.; Liu, M.; Tian, Y.; Chen, T. MiR-210-3p attenuates lipid accumulation and inflammation in atherosclerosis by repressing IGF2. Biosci. Biotechnol. Biochem. 2020, 84, 321–329. [Google Scholar] [CrossRef]
- Suzuki, S.; Yamashita, T.; Tanaka, K.; Hattori, H.; Sawamoto, K.; Okano, H.; Suzuki, N. Activation of cytokine signaling through leukemia inhibitory factor receptor (LIFR)/gp130 attenuates ischemic brain injury in rats. J. Cereb. Blood Flow Metab. 2005, 25, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Hu, J.; Chen, F.; Lecomte, N.; Basnet, H.; David, C.J.; Witkin, M.D.; Allen, P.J.; Leach, S.D.; Hollmann, T.J.; et al. ID1 Mediates Escape from TGFbeta Tumor Suppression in Pancreatic Cancer. Cancer Discov. 2020, 10, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Jonson, T.; Albrechtsson, E.; Axelson, J.; Heidenblad, M.; Gorunova, L.; Johansson, B.; Hoglund, M. Altered expression of TGFB receptors and mitogenic effects of TGFB in pancreatic carcinomas. Int. J. Oncol. 2001, 19, 71–81. [Google Scholar] [CrossRef]
- Raineki, C.; Bodnar, T.S.; Holman, P.J.; Baglot, S.L.; Lan, N.; Weinberg, J. Effects of early-life adversity on immune function are mediated by prenatal environment: Role of prenatal alcohol exposure. Brain Behav. Immun. 2017, 66, 210–220. [Google Scholar] [CrossRef]
- Maras, P.M.; Baram, T.Z. Sculpting the hippocampus from within: Stress, spines, and CRH. Trends Neurosci. 2012, 35, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Jegou, S.; El Ghazi, F.; de Lendeu, P.K.; Marret, S.; Laudenbach, V.; Uguen, A.; Marcorelles, P.; Roy, V.; Laquerriere, A.; Gonzalez, B.J. Prenatal alcohol exposure affects vasculature development in the neonatal brain. Ann. Neurol. 2012, 72, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Cobben, J.M.; Krzyzewska, I.M.; Venema, A.; Mul, A.N.; Polstra, A.; Postma, A.V.; Smigiel, R.; Pesz, K.; Niklinski, J.; Chomczyk, M.A.; et al. DNA methylation abundantly associates with fetal alcohol spectrum disorder and its subphenotypes. Epigenomics 2019, 11, 767–785. [Google Scholar] [CrossRef]
- Bodnar, T.S.; Raineki, C.; Wertelecki, W.; Yevtushok, L.; Plotka, L.; Granovska, I.; Zymak-Zakutnya, N.; Pashtepa, A.; Wells, A.; Honerkamp-Smith, G.; et al. Immune network dysregulation associated with child neurodevelopmental delay: Modulatory role of prenatal alcohol exposure. J. Neuroinflamm. 2020, 17, 39. [Google Scholar] [CrossRef]
- Sathyan, P.; Golden, H.B.; Miranda, R.C. Competing interactions between micro-RNAs determine neural progenitor survival and proliferation after ethanol exposure: Evidence from an ex vivo model of the fetal cerebral cortical neuroepithelium. J. Neurosci. 2007, 27, 8546–8557. [Google Scholar] [CrossRef]
- Tunc-Ozcan, E.; Ferreira, A.B.; Redei, E.E. Modeling Fetal Alcohol Spectrum Disorder: Validating an Ex Vivo Primary Hippocampal Cell Culture System. Alcohol. Clin. Exp. Res. 2016, 40, 1273–1282. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, Y.; Balaraman, Y.; Wang, G.; Nephew, K.P.; Zhou, F.C. Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation. Epigenetics 2009, 4, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Shen, Z.; Cai, S.; Chen, L.; Wang, D. FGF21 promotes wound healing of rat brain microvascular endothelial cells through facilitating TNF-alpha-mediated VEGFA and ERK1/2 signaling pathway. Adv. Clin. Exp. Med. 2021, 30, 711–772. [Google Scholar] [CrossRef] [PubMed]
- Tyler, C.R.; Allan, A.M. Prenatal alcohol exposure alters expression of neurogenesis-related genes in an ex vivo cell culture model. Alcohol 2014, 48, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Toledano, L.; Andreu-Fernandez, V.; Aras-Lopez, R.; Garcia-Algar, O.; Martinez, L.; Gomez-Roig, M.D. Epigallocatechin Gallate Ameliorates the Effects of Prenatal Alcohol Exposure in a Fetal Alcohol Spectrum Disorder-Like Mouse Model. Int. J. Mol. Sci. 2021, 22, 715. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Wang, J.; Morishita, J.; Ueda, N. Biosynthetic pathways of the endocannabinoid anandamide. Chem. Biodivers. 2007, 4, 1842–1857. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Marin, L.; Gavito, A.L.; Decara, J.; Pastor, A.; Castilla-Ortega, E.; Suarez, J.; de la Torre, R.; Pavon, F.J.; Rodriguez de Fonseca, F.; Serrano, A. Impact of intermittent voluntary ethanol consumption during adolescence on the expression of endocannabinoid system and neuroinflammatory mediators. Eur. Neuropsychopharmacol. 2020, 33, 126–138. [Google Scholar] [CrossRef]
- Subbanna, S.; Shivakumar, M.; Psychoyos, D.; Xie, S.; Basavarajappa, B.S. Anandamide-CB1 receptor signaling contributes to postnatal ethanol-induced neonatal neurodegeneration, adult synaptic, and memory deficits. J. Neurosci. 2013, 33, 6350–6366. [Google Scholar] [CrossRef]
- Zhou, C.; Chen, J.; Zhang, X.; Costa, L.G.; Guizzetti, M. Prenatal Ethanol Exposure Up-Regulates the Cholesterol Transporters ATP-Binding Cassette A1 and G1 and Reduces Cholesterol Levels in the Developing Rat Brain. Alcohol Alcohol. 2014, 49, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Guizzetti, M.; Chen, J.; Oram, J.F.; Tsuji, R.; Dao, K.; Moller, T.; Costa, L.G. Ethanol induces cholesterol efflux and up-regulates ATP-binding cassette cholesterol transporters in fetal astrocytes. J. Biol. Chem. 2007, 282, 18740–18749. [Google Scholar] [CrossRef] [PubMed]
- Ramadoss, J.; Magness, R.R. Multiplexed digital quantification of binge-like alcohol-mediated alterations in maternal uterine angiogenic mRNA transcriptome. Physiol. Genom. 2012, 44, 622–628. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, G.; Zhou, J.; Huang, W.; Fang, M.; Yu, L.; Wang, H.; Zhang, Y. Prenatal ethanol exposure-induced a low level of foetal blood cholesterol and its mechanism of IGF1-related placental cholesterol transport dysfunction. Toxicology 2019, 424, 152237. [Google Scholar] [CrossRef]
- Quazi, F.; Molday, R.S. Differential phospholipid substrates and directional transport by ATP-binding cassette proteins ABCA1, ABCA7, and ABCA4 and disease-causing mutants. J. Biol. Chem. 2013, 288, 34414–34426. [Google Scholar] [CrossRef]
- Alt, A.; Miguel-Romero, L.; Donderis, J.; Aristorena, M.; Blanco, F.J.; Round, A.; Rubio, V.; Bernabeu, C.; Marina, A. Structural and functional insights into endoglin ligand recognition and binding. PLoS ONE 2012, 7, e29948. [Google Scholar] [CrossRef] [PubMed]
- Castonguay, R.; Werner, E.D.; Matthews, R.G.; Presman, E.; Mulivor, A.W.; Solban, N.; Sako, D.; Pearsall, R.S.; Underwood, K.W.; Seehra, J.; et al. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J. Biol. Chem. 2011, 286, 30034–30046. [Google Scholar] [CrossRef]
- Nagao, S.; Murao, K.; Imachi, H.; Cao, W.M.; Yu, X.; Li, J.; Matsumoto, K.; Nishiuchi, T.; Ahmed, R.A.; Wong, N.C.; et al. Platelet derived growth factor regulates ABCA1 expression in vascular smooth muscle cells. FEBS Lett. 2006, 580, 4371–4376. [Google Scholar] [CrossRef]
- Kapustin, R.V.; Kopteeva, E.V.; Alekseenkova, E.N.; Tral, T.G.; Tolibova, G.K.; Arzhanova, O.N. Placental expression of endoglin, placental growth factor, leptin, and hypoxia-inducible factor-1 in diabetic pregnancy and pre-eclampsia. Gynecol. Endocrinol. 2021, 37, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Bellon, T.; Corbi, A.; Lastres, P.; Cales, C.; Cebrian, M.; Vera, S.; Cheifetz, S.; Massague, J.; Letarte, M.; Bernabeu, C. Identification and expression of two forms of the human transforming growth factor-beta-binding protein endoglin with distinct cytoplasmic regions. Eur. J. Immunol. 1993, 23, 2340–2345. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Labinaz, M.; Goldstein, J.; Miller, H.; Keon, W.J.; Letarte, M.; O’Brien, E. Endoglin is overexpressed after arterial injury and is required for transforming growth factor-beta-induced inhibition of smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2546–2552. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.L.; Huang, R.L.; Shay, J.; Chen, L.Y.; Lin, S.J.; Yan, P.S.; Chao, W.T.; Lai, Y.H.; Lai, Y.L.; Chao, T.K.; et al. Hypermethylation of the TGF-beta target, ABCA1 is associated with poor prognosis in ovarian cancer patients. Clin. Epigenetics 2015, 7, 1. [Google Scholar] [CrossRef]
- Yinon, Y.; Nevo, O.; Xu, J.; Many, A.; Rolfo, A.; Todros, T.; Post, M.; Caniggia, I. Severe intrauterine growth restriction pregnancies have increased placental endoglin levels: Hypoxic regulation via transforming growth factor-beta 3. Am. J. Pathol. 2008, 172, 77–85. [Google Scholar] [CrossRef][Green Version]
- Bus, P.; Gerrits, T.; Heemskerk, S.A.C.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Baelde, H.J.; Scharpfenecker, M. Endoglin Mediates Vascular Endothelial Growth Factor-A-Induced Endothelial Cell Activation by Regulating Akt Signaling. Am. J. Pathol. 2018, 188, 2924–2935. [Google Scholar] [CrossRef]
- Gundogan, F.; Elwood, G.; Longato, L.; Tong, M.; Feijoo, A.; Carlson, R.I.; Wands, J.R.; de la Monte, S.M. Impaired placentation in fetal alcohol syndrome. Placenta 2008, 29, 148–157. [Google Scholar] [CrossRef]
- Mandal, C.; Park, J.H.; Lee, H.T.; Seo, H.; Chung, I.Y.; Choi, I.G.; Jung, K.H.; Chai, Y.G. Reduction of Nfia gene expression and subsequent target genes by binge alcohol in the fetal brain. Neurosci. Lett. 2015, 598, 73–78. [Google Scholar] [CrossRef]
- Dobson, C.C.; Thevasundaram, K.; Mongillo, D.L.; Winterborn, A.; Holloway, A.C.; Brien, J.F.; Reynolds, J.N. Chronic prenatal ethanol exposure alters expression of central and peripheral insulin signaling molecules in adult guinea pig offspring. Alcohol 2014, 48, 687–693. [Google Scholar] [CrossRef]
- Alimov, A.; Wang, H.; Liu, M.; Frank, J.A.; Xu, M.; Ou, X.; Luo, J. Expression of autophagy and UPR genes in the developing brain during ethanol-sensitive and resistant periods. Metab. Brain Dis. 2013, 28, 667–676. [Google Scholar] [CrossRef]
- Yang, F.; Luo, J. Endoplasmic Reticulum Stress and Ethanol Neurotoxicity. Biomolecules 2015, 5, 2538–2553. [Google Scholar] [CrossRef] [PubMed]
- Veazey, K.J.; Carnahan, M.N.; Muller, D.; Miranda, R.C.; Golding, M.C. Alcohol-induced epigenetic alterations to developmentally crucial genes regulating neural stemness and differentiation. Alcohol. Clin. Exp. Res. 2013, 37, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Veazey, K.J.; Wang, H.; Bedi, Y.S.; Skiles, W.M.; Chang, R.C.; Golding, M.C. Disconnect between alcohol-induced alterations in chromatin structure and gene transcription in a mouse embryonic stem cell model of exposure. Alcohol 2017, 60, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Patten, A.R.; Fontaine, C.J.; Christie, B.R. A comparison of the different animal models of fetal alcohol spectrum disorders and their use in studying complex behaviors. Front. Pediatr. 2014, 2, 93. [Google Scholar] [CrossRef] [PubMed]
- Ethen, M.K.; Ramadhani, T.A.; Scheuerle, A.E.; Canfield, M.A.; Wyszynski, D.F.; Druschel, C.M.; Romitti, P.A.; National Birth Defects Prevention, S. Alcohol consumption by women before and during pregnancy. Matern. Child. Health J. 2009, 13, 274–285. [Google Scholar] [CrossRef]
- Fontaine, C.J.; Patten, A.R.; Sickmann, H.M.; Helfer, J.L.; Christie, B.R. Effects of pre-natal alcohol exposure on hippocampal synaptic plasticity: Sex, age and methodological considerations. Neurosci. Biobehav. Rev. 2016, 64, 12–34. [Google Scholar] [CrossRef]
- Mantione, K.J.; Kream, R.M.; Kuzelova, H.; Ptacek, R.; Raboch, J.; Samuel, J.M.; Stefano, G.B. Comparing bioinformatic gene expression profiling methods: Microarray and RNA-Seq. Med. Sci. Monit. Basic Res. 2014, 20, 138–142. [Google Scholar] [CrossRef]
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baker, J.A.; Brettin, J.T.; Mulligan, M.K.; Hamre, K.M. Effects of Genetics and Sex on Acute Gene Expression Changes in the Hippocampus Following Neonatal Ethanol Exposure in BXD Recombinant Inbred Mouse Strains. Brain Sci. 2022, 12, 1634. https://doi.org/10.3390/brainsci12121634
Baker JA, Brettin JT, Mulligan MK, Hamre KM. Effects of Genetics and Sex on Acute Gene Expression Changes in the Hippocampus Following Neonatal Ethanol Exposure in BXD Recombinant Inbred Mouse Strains. Brain Sciences. 2022; 12(12):1634. https://doi.org/10.3390/brainsci12121634
Chicago/Turabian StyleBaker, Jessica A., Jacob T. Brettin, Megan K. Mulligan, and Kristin M. Hamre. 2022. "Effects of Genetics and Sex on Acute Gene Expression Changes in the Hippocampus Following Neonatal Ethanol Exposure in BXD Recombinant Inbred Mouse Strains" Brain Sciences 12, no. 12: 1634. https://doi.org/10.3390/brainsci12121634
APA StyleBaker, J. A., Brettin, J. T., Mulligan, M. K., & Hamre, K. M. (2022). Effects of Genetics and Sex on Acute Gene Expression Changes in the Hippocampus Following Neonatal Ethanol Exposure in BXD Recombinant Inbred Mouse Strains. Brain Sciences, 12(12), 1634. https://doi.org/10.3390/brainsci12121634