
Brain AVMs-Related microRNAs: Machine Learning Algorithm for Expression Profiles of Target Genes
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Sources and Inclusion Criteria
2.2. Bioinformatic Analyses
3. Results
3.1. Literature Review and Data Extraction
3.2. MicroRNA Sequencing Analyses
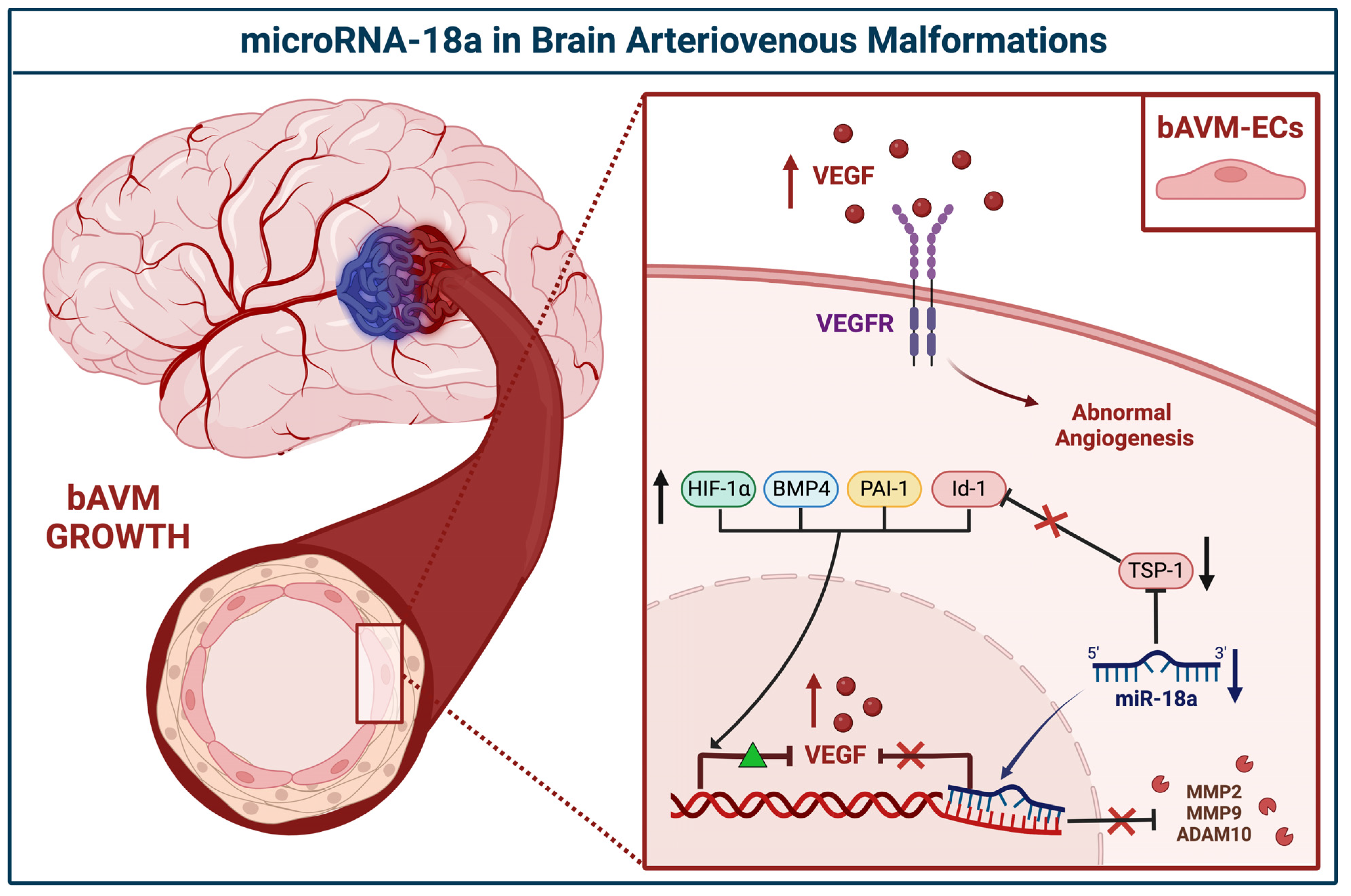
3.2.1. miR-18a
3.2.2. miR-137 and miR-195*
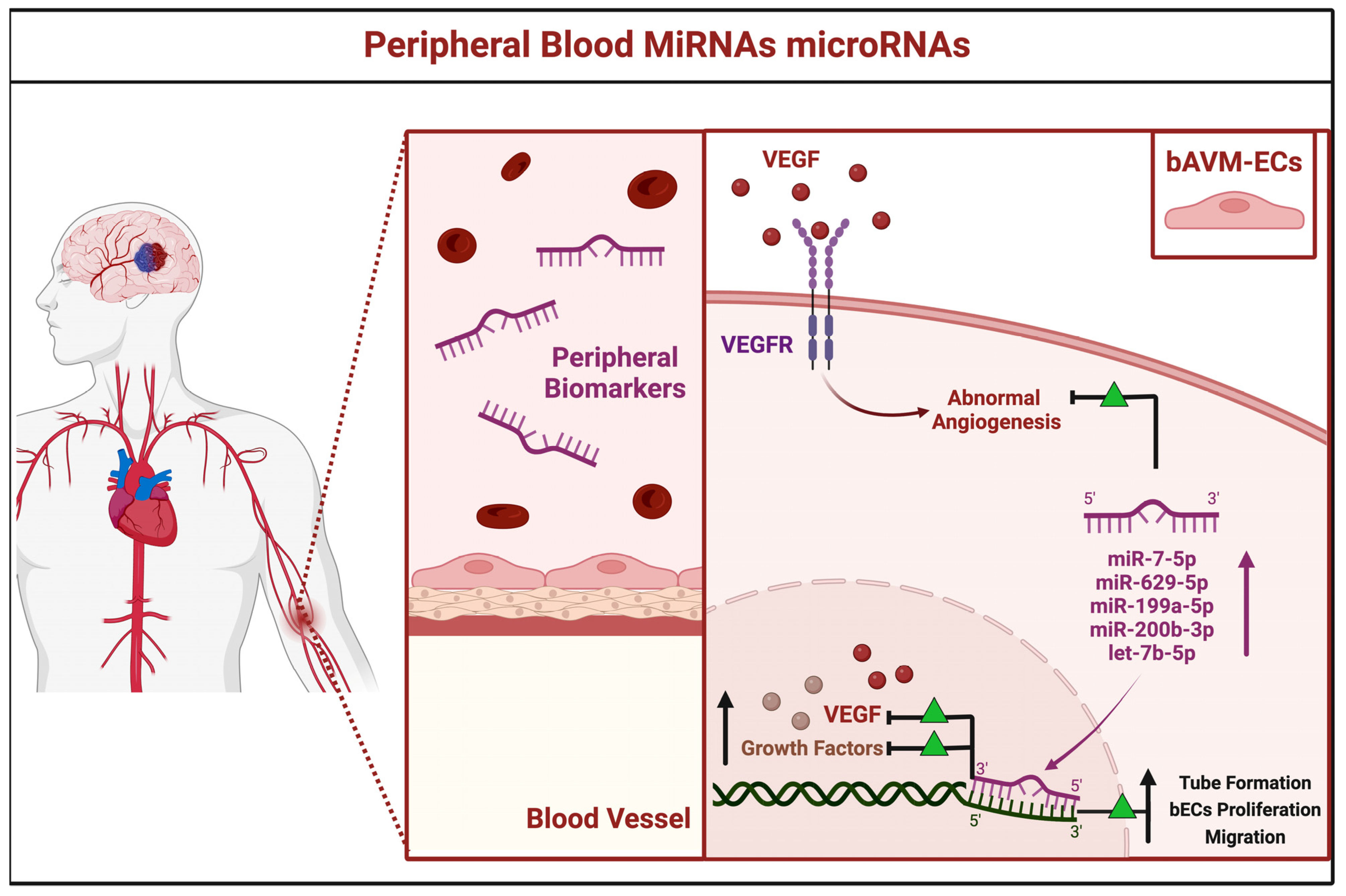
3.2.3. Peripheral Blood miRNAs
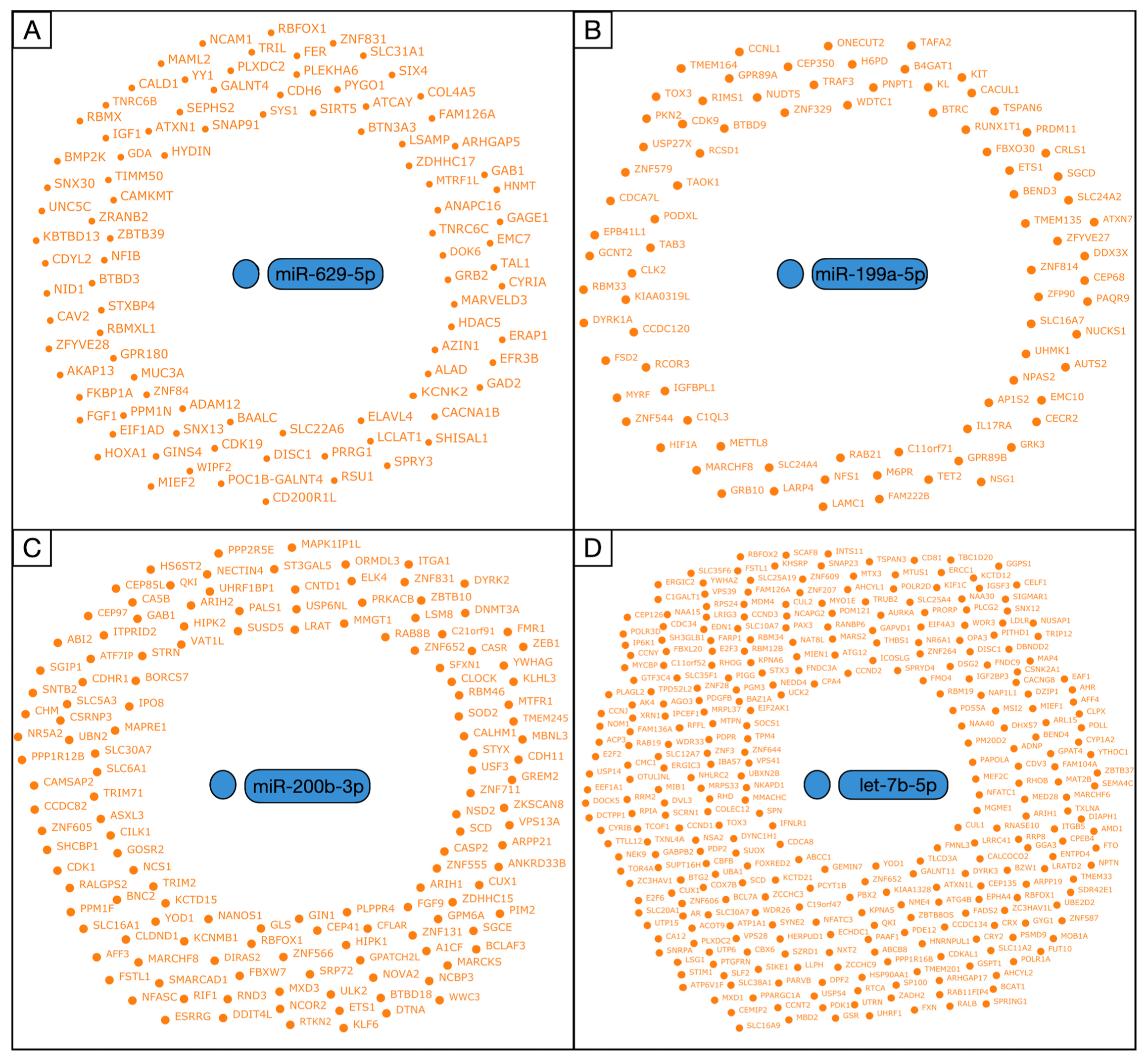
3.3. Prediction of miRNA-Related Targets
4. Discussion
4.1. Experimental Strategies and Future Perspectives for bAVMs
4.2. Emerging miRNA-Based Therapies
4.3. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Crowley, R.W.; Ducruet, A.F.; McDougall, C.G.; Albuquerque, F.C. Endovascular advances for brain arteriovenous malformations. Neurosurgery 2014, 74 (Suppl. 1), S74–S82. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, R.L.; Lazzaro, M.A.; Castonguay, A.C.; Zaidat, O.O. The diagnosis and management of brain arteriovenous malformations. Neurol. Clin. 2013, 31, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Stapf, C.; Mast, H.; Sciacca, R.R.; Berenstein, A.; Nelson, P.K.; Gobin, Y.P.; Pile-Spellman, J.; Mohr, J.P. The New York Islands AVM Study: Design, study progress, and initial results. Stroke 2003, 34, e29–e33. [Google Scholar] [CrossRef]
- Morris, Z.; Whiteley, W.N.; Longstreth, W.T., Jr.; Weber, F.; Lee, Y.C.; Tsushima, Y.; Alphs, H.; Ladd, S.C.; Warlow, C.; Wardlaw, J.M.; et al. Incidental findings on brain magnetic resonance imaging: Systematic review and meta-analysis. BMJ 2009, 339, b3016. [Google Scholar] [CrossRef] [PubMed]
- Lawton, M.T.; Rutledge, W.C.; Kim, H.; Stapf, C.; Whitehead, K.J.; Li, D.Y.; Krings, T.; terBrugge, K.; Kondziolka, D.; Morgan, M.K.; et al. Brain arteriovenous malformations. Nat. Rev. Dis. Prim. 2015, 1, 15008. [Google Scholar] [CrossRef]
- Rutledge, C.; Cooke, D.L.; Hetts, S.W.; Abla, A.A. Brain arteriovenous malformations. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2021; Volume 176, pp. 171–178. [Google Scholar] [CrossRef]
- Crawford, P.M.; West, C.R.; Chadwick, D.W.; Shaw, M.D. Arteriovenous malformations of the brain: Natural history in unoperated patients. J. Neurol. Neurosurg. Psychiatry 1986, 49, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Graf, C.J.; Perret, G.E.; Torner, J.C. Bleeding from cerebral arteriovenous malformations as part of their natural history. J. Neurosurg. 1983, 58, 331–337. [Google Scholar] [CrossRef]
- Mast, H.; Young, W.L.; Koennecke, H.C.; Sciacca, R.R.; Osipov, A.; Pile-Spellman, J.; Hacein-Bey, L.; Duong, H.; Stein, B.M.; Mohr, J.P. Risk of spontaneous haemorrhage after diagnosis of cerebral arteriovenous malformation. Lancet 1997, 350, 1065–1068. [Google Scholar] [CrossRef]
- Gross, B.A.; Du, R. Diagnosis and treatment of vascular malformations of the brain. Curr. Treat. Options Neurol. 2014, 16, 279. [Google Scholar] [CrossRef]
- Kim, H.; Al-Shahi Salman, R.; McCulloch, C.E.; Stapf, C.; Young, W.L. Untreated brain arteriovenous malformation: Patient-level meta-analysis of hemorrhage predictors. Neurology 2014, 83, 590–597. [Google Scholar] [CrossRef]
- Stapf, C.; Mast, H.; Sciacca, R.R.; Choi, J.H.; Khaw, A.V.; Connolly, E.S.; Pile-Spellman, J.; Mohr, J.P. Predictors of hemorrhage in patients with untreated brain arteriovenous malformation. Neurology 2006, 66, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Castilla, L.; Russin, J.J.; Martinez-Del-Campo, E.; Soriano-Baron, H.; Spetzler, R.F.; Nakaji, P. Molecular and cellular biology of cerebral arteriovenous malformations: A review of current concepts and future trends in treatment. Neurosurg. Focus 2014, 37, E1. [Google Scholar] [CrossRef]
- Florian, I.A.; Timiș, T.L.; Ungureanu, G.; Florian, I.S.; Bălașa, A.; Berindan-Neagoe, I. Deciphering the vascular labyrinth: Role of microRNAs and candidate gene SNPs in brain AVM development—Literature review. Neurol. Res. 2020, 42, 1043–1054. [Google Scholar] [CrossRef]
- Thomas, J.M.; Surendran, S.; Abraham, M.; Rajavelu, A.; Kartha, C.C. Genetic and epigenetic mechanisms in the development of arteriovenous malformations in the brain. Clin. Epigenet. 2016, 8, 78. [Google Scholar] [CrossRef]
- Bameri, O.; Salarzaei, M.; Parooie, F. KRAS/BRAF mutations in brain arteriovenous malformations: A systematic review and meta-analysis. Interv. Neuroradiol. 2021, 27, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Schotland, H.; Denstaedt, S. Hereditary Hemorrhagic Telangiectasia. N. Engl. J. Med. 2019, 381, 2552. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.; Carter, M.T.; Latino, G.A.; Dirks, P.; Ratjen, F. Brain arteriovenous malformations in patients with hereditary hemorrhagic telangiectasia: Clinical presentation and anatomical distribution. Pediatr. Neurol. 2013, 49, 445–450. [Google Scholar] [CrossRef]
- Chen, W.; Choi, E.J.; McDougall, C.M.; Su, H. Brain arteriovenous malformation modeling, pathogenesis, and novel therapeutic targets. Transl. Stroke Res. 2014, 5, 316–329. [Google Scholar] [CrossRef]
- Ota, T.; Komiyama, M. Pathogenesis of non-hereditary brain arteriovenous malformation and therapeutic implications. Interv. Neuroradiol. 2020, 26, 244–253. [Google Scholar] [CrossRef]
- Araldi, E.; Suárez, Y. MicroRNAs as regulators of endothelial cell functions in cardiometabolic diseases. Biochim. Biophys. Acta 2016, 1861, 2094–2103. [Google Scholar] [CrossRef]
- Zammar, S.G.; El Tecle, N.E.; El Ahmadieh, T.Y.; Mcclendon, J., Jr.; Comair, Y.G.; Bendok, B.R. A Biological Approach to Treating Brain Arteriovenous Malformations. Neurosurgery 2014, 74, N15–N17. [Google Scholar] [CrossRef] [PubMed]
- Florian, I.A.; Buruiana, A.; Timis, T.L.; Susman, S.; Florian, I.S.; Balasa, A.; Berindan-Neagoe, I. An Insight into the microRNAs Associated with Arteriovenous and Cavernous Malformations of the Brain. Cells 2021, 10, 1373. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.J.; Searles, C.D. Development of MicroRNA-Based Therapeutics for Vascular Disease. Circ. Res. 2020, 127, 1179–1181. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Group, P. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. Int. J. Surg. 2010, 8, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Gretz, N.; Sticht, C. miRWalk database for miRNA-target interactions. Methods Mol. Biol. 2014, 1182, 289–305. [Google Scholar] [CrossRef]
- Huang, H.Y.; Lin, Y.C.; Li, J.; Huang, K.Y.; Shrestha, S.; Hong, H.C.; Tang, Y.; Chen, Y.G.; Jin, C.N.; Yu, Y.; et al. miRTarBase 2020: Updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Santos, T.; Amar, A.; Tahara, S.M.; Chen, T.C.; Giannotta, S.L.; Hofman, F.M. MicroRNA-18a improves human cerebral arteriovenous malformation endothelial cell function. Stroke 2014, 45, 293–297. [Google Scholar] [CrossRef]
- Marín-Ramos, N.I.; Thein, T.Z.; Ghaghada, K.B.; Chen, T.C.; Giannotta, S.L.; Hofman, F.M. miR-18a Inhibits BMP4 and HIF-1α Normalizing Brain Arteriovenous Malformations. Circ. Res. 2020, 127, e210–e231. [Google Scholar] [CrossRef]
- Huang, J.; Song, J.; Qu, M.; Wang, Y.; An, Q.; Song, Y.; Yan, W.; Wang, B.; Wang, X.; Zhang, S.; et al. MicroRNA-137 and microRNA-195* inhibit vasculogenesis in brain arteriovenous malformations. Ann. Neurol. 2017, 82, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, Z.; Shi, Y.; Huang, G.; Chen, L.; Tan, H.; Wang, Z.; Yin, C.; Hu, J. Deep Sequencing of Small RNAs in Blood of Patients with Brain Arteriovenous Malformations. World Neurosurg. 2018, 115, e570–e579. [Google Scholar] [CrossRef]
- Xu, M.; Xu, H.; Qin, Z.; Zhang, J.; Yang, X.; Xu, F. Increased expression of angiogenic factors in cultured human brain arteriovenous malformation endothelial cells. Cell Biochem. Biophys. 2014, 70, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, D.; Wang, Z.; Li, X.; Xia, W.; Han, Y.; Su, L.; Fan, X. MiR-18a-5p acts as a novel serum biomarker for venous malformation and promotes angiogenesis by regulating the thrombospondin-1/P53 signaling axis. Am. J. Transl. Res. 2021, 13, 11271–11286. [Google Scholar] [PubMed]
- Dogar, A.M.; Semplicio, G.; Guennewig, B.; Hall, J. Multiple microRNAs derived from chemically synthesized precursors regulate thrombospondin 1 expression. Nucleic Acid Ther. 2014, 24, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Gao, Y.; Li, F.; Pan, Q.; Liu, Z.; Lu, X.; Song, C.; Diao, X. MicroRNA-18a regulates invasive meningiomas via hypoxia-inducible factor-1α. Exp. Ther. Med. 2015, 10, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Uranishi, R.; Baev, N.I.; Kim, J.H.; Awad, I.A. Vascular smooth muscle cell differentiation in human cerebral vascular malformations. Neurosurgery 2001, 49, 671–679; discussion 679–680. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Ding, D.; Derdeyn, C.P.; Lanzino, G.; Friedlander, R.M.; Southerland, A.M.; Lawton, M.T.; Sheehan, J.P. Brain arteriovenous malformations: A review of natural history, pathobiology, and interventions. Neurology 2020, 95, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Hafez, A.; Koroknay-Pál, P.; Oulasvirta, E.; Elseoud, A.A.; Lawton, M.T.; Niemelä, M.; Laakso, A. The Application of the Novel Grading Scale (Lawton-Young Grading System) to Predict the Outcome of Brain Arteriovenous Malformation. Neurosurgery 2019, 84, 529–536. [Google Scholar] [CrossRef]
- Rutledge, W.C.; Ko, N.U.; Lawton, M.T.; Kim, H. Hemorrhage rates and risk factors in the natural history course of brain arteriovenous malformations. Transl. Stroke Res. 2014, 5, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.D., Jr.; Wiebers, D.O.; Forbes, G.; O’Fallon, W.M.; Piepgras, D.G.; Marsh, W.R.; Maciunas, R.J. The natural history of unruptured intracranial arteriovenous malformations. J. Neurosurg. 1988, 68, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Bayrak-Toydemir, P.; Mao, R.; Lewin, S.; McDonald, J. Hereditary hemorrhagic telangiectasia: An overview of diagnosis and management in the molecular era for clinicians. Genet. Med. 2004, 6, 175–191. [Google Scholar] [CrossRef]
- Berg, J.N.; Gallione, C.J.; Stenzel, T.T.; Johnson, D.W.; Allen, W.P.; Schwartz, C.E.; Jackson, C.E.; Porteous, M.E.; Marchuk, D.A. The activin receptor-like kinase 1 gene: Genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am. J. Hum. Genet. 1997, 61, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Nishino, K.; Ito, Y.; Sorimachi, T.; Shimbo, J.; Fujii, Y. Sturge-Weber syndrome associated with arteriovenous malformation in a patient presenting with progressive brain edema and cyst formation. J. Neurosurg. Pediatr. 2010, 5, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Luzzi, S.; Del Maestro, M.; Elbabaa, S.K.; Galzio, R. Letter to the Editor Regarding “One and Done: Multimodal Treatment of Pediatric Cerebral Arteriovenous Malformations in a Single Anesthesia Event”. World Neurosurg. 2020, 134, 660. [Google Scholar] [CrossRef] [PubMed]
- Brinjikji, W.; Iyer, V.N.; Wood, C.P.; Lanzino, G. Prevalence and characteristics of brain arteriovenous malformations in hereditary hemorrhagic telangiectasia: A systematic review and meta-analysis. J. Neurosurg. 2017, 127, 302–310. [Google Scholar] [CrossRef] [PubMed]
- McAllister, K.A.; Grogg, K.M.; Johnson, D.W.; Gallione, C.J.; Baldwin, M.A.; Jackson, C.E.; Helmbold, E.A.; Markel, D.S.; McKinnon, W.C.; Murrel, J.; et al. Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 1994, 8, 345–351. [Google Scholar] [CrossRef]
- Johnson, D.W.; Berg, J.N.; Baldwin, M.A.; Gallione, C.J.; Marondel, I.; Yoon, S.J.; Stenzel, T.T.; Speer, M.; Pericak-Vance, M.A.; Diamond, A.; et al. Mutations in the activin receptor–like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat. Genet. 1996, 13, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Jiang, W.; Lin, W.; Wang, Y.; Chen, H.; Zou, H.; Huang, S.; Zhu, N.; Han, S. A novel BMPR2 mutation in a patient with heritable pulmonary arterial hypertension and suspected hereditary hemorrhagic telangiectasia: A case report. Medicine 2020, 99, e21342. [Google Scholar] [CrossRef] [PubMed]
- Gallione, C.J.; Repetto, G.M.; Legius, E.; Rustgi, A.K.; Schelley, S.L.; Tejpar, S.; Mitchell, G.; Drouin, E.; Westermann, C.J.; Marchuk, D.A. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004, 363, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Chen, G.; Li, Y.-P. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef] [PubMed]
- Burks, T.N.; Cohn, R.D. Role of TGF-β signaling in inherited and acquired myopathies. Skelet. Muscle 2011, 1, 19. [Google Scholar] [CrossRef] [PubMed]
- Alsina-Sanchís, E.; García-Ibáñez, Y.; Figueiredo, A.M.; Riera-Domingo, C.; Figueras, A.; Matias-Guiu, X.; Casanovas, O.; Botella, L.M.; Pujana, M.A.; Riera-Mestre, A.; et al. ALK1 Loss Results in Vascular Hyperplasia in Mice and Humans Through PI3K Activation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1216–1229. [Google Scholar] [CrossRef]
- Ola, R.; Dubrac, A.; Han, J.; Zhang, F.; Fang, J.S.; Larrivée, B.; Lee, M.; Urarte, A.A.; Kraehling, J.R.; Genet, G.; et al. PI3 kinase inhibition improves vascular malformations in mouse models of hereditary haemorrhagic telangiectasia. Nat. Commun. 2016, 7, 13650. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Grossman, S.R.; Takahashi, Y.; Rokas, M.V.; Nakamura, N.; Sellers, W.R. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J. Biol. Chem. 2001, 276, 48627–48630. [Google Scholar] [CrossRef] [PubMed]
- Bourdeau, A.; Dumont, D.J.; Letarte, M. A murine model of hereditary hemorrhagic telangiectasia. J. Clin. Investig. 1999, 104, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Torsney, E.; Charlton, R.; Diamond, A.G.; Burn, J.; Soames, J.V.; Arthur, H.M. Mouse model for hereditary hemorrhagic telangiectasia has a generalized vascular abnormality. Circulation 2003, 107, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Hanes, M.A.; Dickens, T.; Porteous, M.E.M.; Oh, S.P.; Hale, L.P.; Marchuk, D.A. A mouse model for hereditary hemorrhagic telangiectasia (HHT) type 2. Hum. Mol. Genet. 2003, 12, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Sturiale, C.L.; Puca, A.; Sebastiani, P.; Gatto, I.; Albanese, A.; Di Rocco, C.; Maira, G.; Pola, R. Single nucleotide polymorphisms associated with sporadic brain arteriovenous malformations: Where do we stand? Brain 2013, 136, 665–681. [Google Scholar] [CrossRef] [PubMed]
- Pawlikowska, L.; Tran, M.N.; Achrol, A.S.; McCulloch, C.E.; Ha, C.; Lind, D.L.; Hashimoto, T.; Zaroff, J.; Lawton, M.T.; Marchuk, D.A.; et al. Polymorphisms in genes involved in inflammatory and angiogenic pathways and the risk of hemorrhagic presentation of brain arteriovenous malformations. Stroke 2004, 35, 2294–2300. [Google Scholar] [CrossRef]
- Nikolaev, S.I.; Vetiska, S.; Bonilla, X.; Boudreau, E.; Jauhiainen, S.; Rezai Jahromi, B.; Khyzha, N.; DiStefano, P.V.; Suutarinen, S.; Kiehl, T.R.; et al. Somatic Activating KRAS Mutations in Arteriovenous Malformations of the Brain. N. Engl. J. Med. 2018, 378, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Kushamae, M.; Aoki, T.; Yamaguchi, T.; Kitazato, K.; Abekura, Y.; Kawamata, T.; Mizutani, T.; Miyamoto, S.; Takagi, Y. KRAS G12D or G12V Mutation in Human Brain Arteriovenous Malformations. World Neurosurg. 2019, 126, e1365–e1373. [Google Scholar] [CrossRef] [PubMed]
- Al-Olabi, L.; Polubothu, S.; Dowsett, K.; Andrews, K.A.; Stadnik, P.; Joseph, A.P.; Knox, R.; Pittman, A.; Clark, G.; Baird, W.; et al. Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J. Clin. Investig. 2018, 128, 1496–1508. [Google Scholar] [CrossRef]
- Eerola, I.; Boon, L.M.; Mulliken, J.B.; Burrows, P.E.; Dompmartin, A.; Watanabe, S.; Vanwijck, R.; Vikkula, M. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am. J. Hum. Genet. 2003, 73, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Limaye, N.; Kangas, J.; Mendola, A.; Godfraind, C.; Schlögel, M.J.; Helaers, R.; Eklund, L.; Boon, L.M.; Vikkula, M. Somatic Activating PIK3CA Mutations Cause Venous Malformation. Am. J. Hum. Genet. 2015, 97, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Priemer, D.S.; Vortmeyer, A.O.; Zhang, S.; Chang, H.Y.; Curless, K.L.; Cheng, L. Activating KRAS mutations in arteriovenous malformations of the brain: Frequency and clinicopathologic correlation. Hum. Pathol. 2019, 89, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Jin, Y.; Muhl, L.; Burmakin, M.; Wang, Y.; Duchez, A.C.; Betsholtz, C.; Arthur, H.M.; Jakobsson, L. Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through VEGFR2 signalling. Nat. Cell Biol. 2017, 19, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Ola, R.; Künzel, S.H.; Zhang, F.; Genet, G.; Chakraborty, R.; Pibouin-Fragner, L.; Martin, K.; Sessa, W.; Dubrac, A.; Eichmann, A. SMAD4 Prevents Flow Induced Arteriovenous Malformations by Inhibiting Casein Kinase 2. Circulation 2018, 138, 2379–2394. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, M.; Zhao, S.; Xie, Z.; Zhang, Y.; Liu, J.; Zhang, Y.; Yang, X.; Wu, N. Mutational spectrum of syndromic genes in sporadic brain arteriovenous malformation. Chin. Neurosurg. J. 2022, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.; Fish, J.E.; D’Abreo, C.; Lin, S.; Robb, G.B.; Teichert, A.M.; Karantzoulis-Fegaras, F.; Keightley, A.; Steer, B.M.; Marsden, P.A. The cell-specific expression of endothelial nitric-oxide synthase: A role for DNA methylation. J. Biol. Chem. 2004, 279, 35087–35100. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.J.; Wei, S.Y.; Chiu, J.J. Mechanical regulation of epigenetics in vascular biology and pathobiology. J. Cell. Mol. Med. 2013, 17, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.J.; Chien, S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef]
- Illi, B.; Nanni, S.; Scopece, A.; Farsetti, A.; Biglioli, P.; Capogrossi, M.C.; Gaetano, C. Shear stress-mediated chromatin remodeling provides molecular basis for flow-dependent regulation of gene expression. Circ. Res. 2003, 93, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Balta, S. Endothelial Dysfunction and Inflammatory Markers of Vascular Disease. Curr. Vasc. Pharmacol. 2021, 19, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef]
- Giotta Lucifero, A.; Baldoncini, M.; Bruno, N.; Galzio, R.; Hernesniemi, J.; Luzzi, S. Shedding the Light on the Natural History of Intracranial Aneurysms: An Updated Overview. Medicina 2021, 57, 742. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hao, Q.; Zhao, Y.; Guo, Y.; Ge, W. Dysregulation of cell-cell interactions in brain arteriovenous malformations: A quantitative proteomic study. Proteom. Clin. Appl. 2017, 11, 1600093. [Google Scholar] [CrossRef] [PubMed]
- Spetzler, R.F.; Martin, N.A. A proposed grading system for arteriovenous malformations. J. Neurosurg. 1986, 65, 476–483. [Google Scholar] [CrossRef]
- Spetzler, R.F.; Ponce, F.A. A 3-tier classification of cerebral arteriovenous malformations. Clinical article. J. Neurosurg. 2011, 114, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Giotta Lucifero, A.; Luzzi, S. Against the Resilience of High-Grade Gliomas: The Immunotherapeutic Approach (Part I). Brain Sci. 2021, 11, 386. [Google Scholar] [CrossRef]
- Giotta Lucifero, A.; Luzzi, S.; Brambilla, I.; Guarracino, C.; Mosconi, M.; Foiadelli, T.; Savasta, S. Gene therapies for high-grade gliomas: From the bench to the bedside. Acta Biomed. 2020, 91, 32–50. [Google Scholar] [CrossRef] [PubMed]
- Giotta Lucifero, A.; Luzzi, S.; Brambilla, I.; Schena, L.; Mosconi, M.; Foiadelli, T.; Savasta, S. Potential roads for reaching the summit: An overview on target therapies for high-grade gliomas. Acta Biomed. 2020, 91, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Giotta Lucifero, A.; Luzzi, S.; Brambilla, I.; Trabatti, C.; Mosconi, M.; Savasta, S.; Foiadelli, T. Innovative therapies for malignant brain tumors: The road to a tailored cure. Acta Biomed. 2020, 91, 5–17. [Google Scholar] [CrossRef]
- Luzzi, S.; Crovace, A.M.; Del Maestro, M.; Giotta Lucifero, A.; Elbabaa, S.K.; Cinque, B.; Palumbo, P.; Lombardi, F.; Cimini, A.; Cifone, M.G.; et al. The cell-based approach in neurosurgery: Ongoing trends and future perspectives. Heliyon 2019, 5, e02818. [Google Scholar] [CrossRef] [PubMed]
- Luzzi, S.; Giotta Lucifero, A.; Brambilla, I.; Magistrali, M.; Mosconi, M.; Savasta, S.; Foiadelli, T. Adoptive immunotherapies in neuro-oncology: Classification, recent advances, and translational challenges. Acta Biomed. 2020, 91, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Luzzi, S.; Giotta Lucifero, A.; Brambilla, I.; Trabatti, C.; Mosconi, M.; Savasta, S.; Foiadelli, T. The impact of stem cells in neuro-oncology: Applications, evidence, limitations and challenges. Acta Biomed. 2020, 91, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Giotta Lucifero, A.; Luzzi, S. Against the Resilience of High-Grade Gliomas: Gene Therapies (Part II). Brain Sci. 2021, 11, 976. [Google Scholar] [CrossRef] [PubMed]
- Luzzi, S.; Del Maestro, M.; Galzio, R. Letter to the Editor. Preoperative embolization of brain arteriovenous malformations. J. Neurosurg. 2019, 132, 2014–2016. [Google Scholar] [CrossRef] [PubMed]
- Luzzi, S.; Crovace, A.M.; Lacitignola, L.; Valentini, V.; Francioso, E.; Rossi, G.; Invernici, G.; Galzio, R.J.; Crovace, A. Engraftment, neuroglial transdifferentiation and behavioral recovery after complete spinal cord transection in rats. Surg. Neurol. Int. 2018, 9, 19. [Google Scholar] [CrossRef]
- Luzzi, S.; Giotta Lucifero, A.; Del Maestro, M.; Marfia, G.; Navone, S.E.; Baldoncini, M.; Nuñez, M.; Campero, A.; Elbabaa, S.K.; Galzio, R. Anterolateral Approach for Retrostyloid Superior Parapharyngeal Space Schwannomas Involving the Jugular Foramen Area: A 20-Year Experience. World Neurosurg. 2019, 132, e40–e52. [Google Scholar] [CrossRef] [PubMed]
- Bellantoni, G.; Guerrini, F.; Del Maestro, M.; Galzio, R.; Luzzi, S. Simple schwannomatosis or an incomplete Coffin-Siris? Report of a particular case. eNeurologicalSci 2019, 14, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Campanella, R.; Guarnaccia, L.; Cordiglieri, C.; Trombetta, E.; Caroli, M.; Carrabba, G.; La Verde, N.; Rampini, P.; Gaudino, C.; Costa, A.; et al. Tumor-Educated Platelets and Angiogenesis in Glioblastoma: Another Brick in the Wall for Novel Prognostic and Targetable Biomarkers, Changing the Vision from a Localized Tumor to a Systemic Pathology. Cells 2020, 9, 294. [Google Scholar] [CrossRef]
- Walker, E.J.; Su, H.; Shen, F.; Choi, E.J.; Oh, S.P.; Chen, G.; Lawton, M.T.; Kim, H.; Chen, Y.; Chen, W.; et al. Arteriovenous malformation in the adult mouse brain resembling the human disease. Ann. Neurol. 2011, 69, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Choe, S.W.; Kim, Y.H.; Acharya, A.P.; Keselowsky, B.G.; Sorg, B.S.; Lee, Y.J.; Oh, S.P. VEGF neutralization can prevent and normalize arteriovenous malformations in an animal model for hereditary hemorrhagic telangiectasia 2. Angiogenesis 2014, 17, 823–830. [Google Scholar] [CrossRef]
- Kim, H.; Pawlikowska, L.; Chen, Y.; Su, H.; Yang, G.Y.; Young, W.L. Brain arteriovenous malformation biology relevant to hemorrhage and implication for therapeutic development. Stroke 2009, 40, S95–S97. [Google Scholar] [CrossRef]
- Merrill, M.J.; Oldfield, E.H. A reassessment of vascular endothelial growth factor in central nervous system pathology. J. Neurosurg. 2005, 103, 853–868. [Google Scholar] [CrossRef]
- Raper, D.M.S.; Winkler, E.A.; Rutledge, W.C.; Cooke, D.L.; Abla, A.A. An Update on Medications for Brain Arteriovenous Malformations. Neurosurgery 2020, 87, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Z.; Young, W.L. Management of brain arteriovenous malformations. Curr. Opin. Anaesthesiol. 2005, 18, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Liu, M.; Wang, Y.; Chen, X.; Xu, J.; Sun, Y.; Zhao, L.; Qu, H.; Fan, Y.; Wu, C. Antagonism of miR-21 reverses epithelial-mesenchymal transition and cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting PTEN. PLoS ONE 2012, 7, e39520. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Z.; Li, C.; Chen, Q.; Jing, Y.; Carpenter, R.; Jiang, Y.; Kung, H.F.; Lai, L.; Jiang, B.H. MiR-21 induced angiogenesis through AKT and ERK activation and HIF-1α expression. PLoS ONE 2011, 6, e19139. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Patrawala, L.; Brown, D.; Bader, A.G. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010, 70, 5923–5930. [Google Scholar] [CrossRef] [PubMed]
- Tivnan, A.; Orr, W.S.; Gubala, V.; Nooney, R.; Williams, D.E.; McDonagh, C.; Prenter, S.; Harvey, H.; Domingo-Fernández, R.; Bray, I.M.; et al. Inhibition of neuroblastoma tumor growth by targeted delivery of microRNA-34a using anti-disialoganglioside GD2 coated nanoparticles. PLoS ONE 2012, 7, e38129. [Google Scholar] [CrossRef]
- Wong, M.Y.; Yu, Y.; Walsh, W.R.; Yang, J.L. microRNA-34 family and treatment of cancers with mutant or wild-type p53 (Review). Int. J. Oncol. 2011, 38, 1189–1195. [Google Scholar] [CrossRef]
- Jansson, M.D.; Lund, A.H. MicroRNA and cancer. Mol. Oncol. 2012, 6, 590–610. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L. Targeting microRNA-122 to Treat Hepatitis C Virus Infection. Viruses 2010, 2, 1382–1393. [Google Scholar] [CrossRef] [PubMed]
- Jangra, R.K.; Yi, M.; Lemon, S.M. Regulation of hepatitis C virus translation and infectious virus production by the MicroRNA miR-122. J. Virol. 2010, 84, 6615–6625. [Google Scholar] [CrossRef]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Ørum, H. Therapeutic Silencing of MicroRNA-122 in Primates with Chronic Hepatitis C Virus Infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef]
- Hildebrandt-Eriksen, E.S.; Aarup, V.; Persson, R.; Hansen, H.F.; Munk, M.E.; Ørum, H. A Locked Nucleic Acid Oligonucleotide Targeting MicroRNA 122 Is Well-Tolerated in Cynomolgus Monkeys. Nucleic Acid Ther. 2012, 22, 152–161. [Google Scholar] [CrossRef]
- Janssen, H.L.A.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV Infection by Targeting MicroRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Elmén, J.; Lindow, M.; Schütz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjärn, M.; Hansen, H.F.; Berger, U.; et al. LNA-mediated microRNA silencing in non-human primates. Nature 2008, 452, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.J.; Sheedy, F.J.; Esau, C.C.; Hussain, F.N.; Temel, R.E.; Parathath, S.; Van Gils, J.M.; Rayner, A.J.; Chang, A.N.; Suarez, Y.; et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J. Clin. Investig. 2011, 121, 2921–2931. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.J.; Esau, C.C.; Hussain, F.N.; McDaniel, A.L.; Marshall, S.M.; van Gils, J.M.; Ray, T.D.; Sheedy, F.J.; Goedeke, L.; Liu, X.; et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 2011, 478, 404–407. [Google Scholar] [CrossRef]
- Chau, B.N.; Xin, C.; Hartner, J.; Ren, S.; Castano, A.P.; Linn, G.; Li, J.; Tran, P.T.; Kaimal, V.; Huang, X.; et al. MicroRNA-21 Promotes Fibrosis of the Kidney by Silencing Metabolic Pathways. Sci. Transl. Med. 2012, 4, 121ra118. [Google Scholar] [CrossRef] [PubMed]
- Dong, X. Current Strategies for Brain Drug Delivery. Theranostics 2018, 8, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Garg, T.; Bhandari, S.; Rath, G.; Goyal, A.K. Current strategies for targeted delivery of bio-active drug molecules in the treatment of brain tumor. J. Drug Target. 2015, 23, 865–887. [Google Scholar] [CrossRef]
- Simion, V.; Nadim, W.D.; Benedetti, H.; Pichon, C.; Morisset-Lopez, S.; Baril, P. Pharmacomodulation of microRNA Expression in Neurocognitive Diseases: Obstacles and Future Opportunities. Curr. Neuropharmacol. 2017, 15, 276–290. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Site | Status | Level of Gene Expression | Author, Year | Country | |
|---|---|---|---|---|---|---|
| High | Low | |||||
| miR-18a | BECs | Downregulated | TSP-1, VEGF-A | Id-1, VEGF-D | Ferreira et al., 2014 [29] | USA |
| TSP-1 | VEGF, PAI-1, BMP4, HIF-1α MMP2, MMP9, ADAM10 | Marín-Ramos et al., 2020 [30] | USA | |||
| miR-137 | VSMCs | Downregulated | NA | VEGF, PI3K/Akt, MAPK/ERK, P38, NFkB | Huang et al., 2017 [31] | China |
| miR-195* | ||||||
| miR-7-5p | PB | Over-expressed | VEGF | NA | Chen et al., 2018 [32] | China |
| miR-629-5p | ||||||
| miR-199a-5p | ||||||
| miR-200b-3p | ||||||
| let-7b-5p | NA | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giotta Lucifero, A.; Luzzi, S. Brain AVMs-Related microRNAs: Machine Learning Algorithm for Expression Profiles of Target Genes. Brain Sci. 2022, 12, 1628. https://doi.org/10.3390/brainsci12121628
Giotta Lucifero A, Luzzi S. Brain AVMs-Related microRNAs: Machine Learning Algorithm for Expression Profiles of Target Genes. Brain Sciences. 2022; 12(12):1628. https://doi.org/10.3390/brainsci12121628
Chicago/Turabian StyleGiotta Lucifero, Alice, and Sabino Luzzi. 2022. "Brain AVMs-Related microRNAs: Machine Learning Algorithm for Expression Profiles of Target Genes" Brain Sciences 12, no. 12: 1628. https://doi.org/10.3390/brainsci12121628
APA StyleGiotta Lucifero, A., & Luzzi, S. (2022). Brain AVMs-Related microRNAs: Machine Learning Algorithm for Expression Profiles of Target Genes. Brain Sciences, 12(12), 1628. https://doi.org/10.3390/brainsci12121628