
A Systematic Review of Unexplained Early Regression in Adolescents and Adults with Down Syndrome

Abstract
:1. Introduction
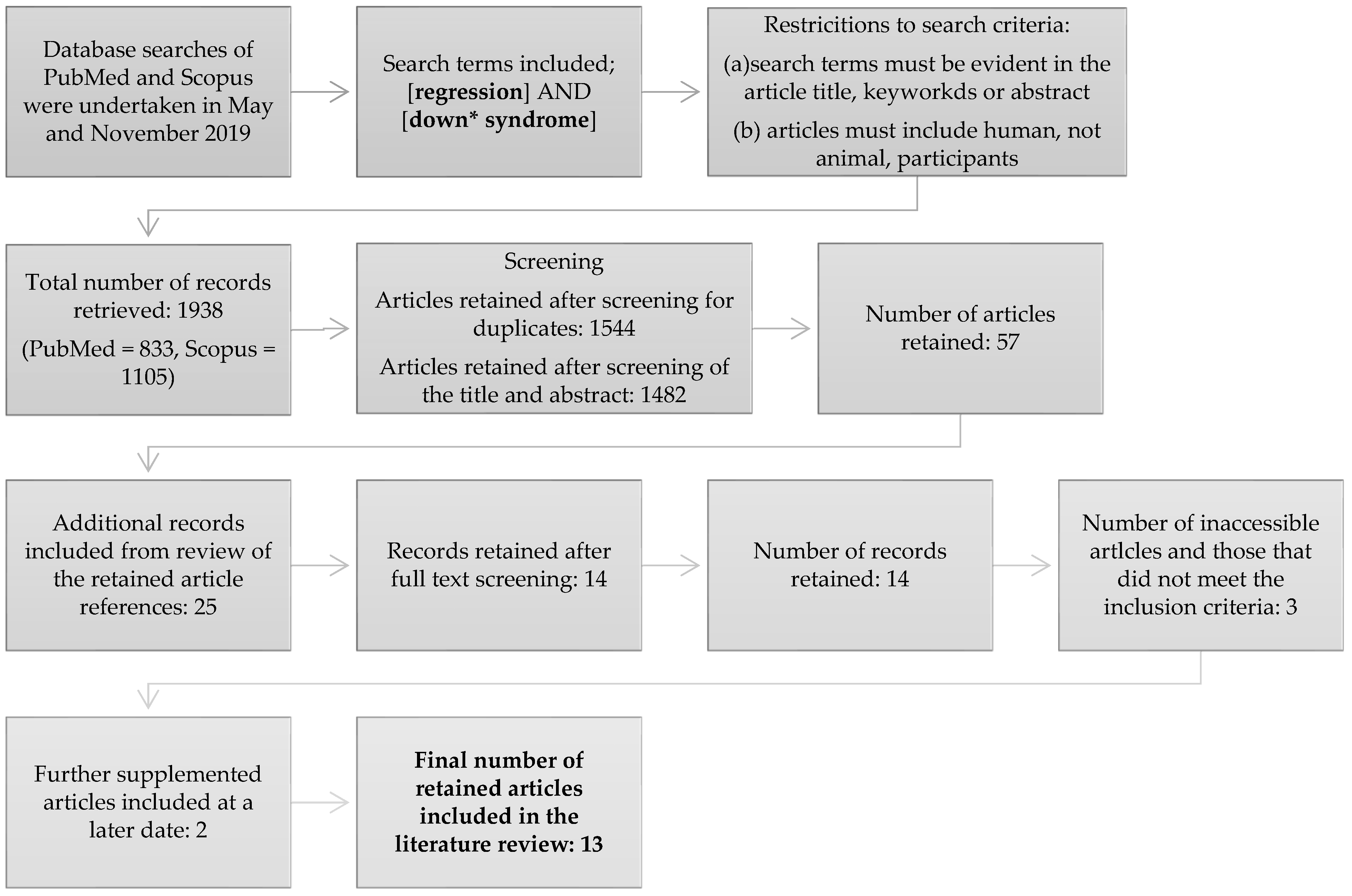
2. Methodology
2.1. Identification of Articles
2.2. Inclusion Criteria
- Research article involving at least one individual with DS.
- Age of patient under 35 years.
- Evidence of at least one regressive period that included changes to cognition, functioning and/or behaviour and personality.
- Regression identified did not progress to a clinical diagnosis of AD.
2.3. Data Collection Process
3. Methodology
3.1. Article Search Results
3.2. Additional Comments and Exclusions
4. Results
4.1. Patient Demographics
4.2. Descriptive Terminology
4.3. Symptom Severity and Diversity
4.4. Events Preceding Regression
4.5. Brain Abnormalities
4.6. Medications, Interventions and Outcomes
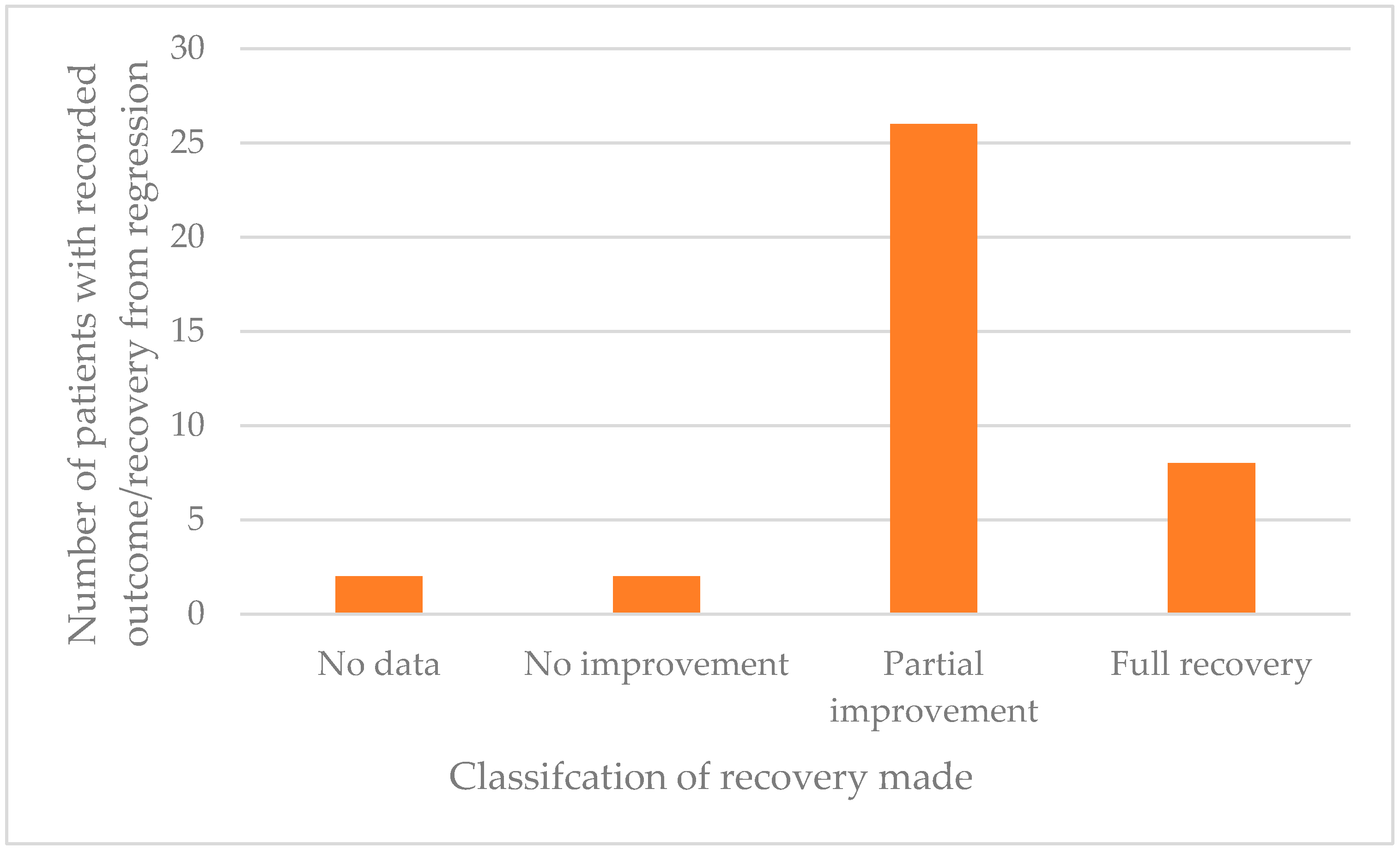
4.7. Prognosis
5. Discussion
Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment/Intervention | Administered n | Positive Response | Negative Response | No Response |
|---|---|---|---|---|
| SSRI—fluvoxamine | 5 | 3 “Symptoms improved” “Moderate improvement” “Significantly better” | 0 | 2 “No improvement” “No change” |
| Amantadine | 2 | 2 “Partial improvement” “Partial improvement” | ||
| Levomepromazine | 2 | 2 “Partial improvement” “Partial improvement” | ||
| Haloperidol | 4 | 3 “Improvement” “Partially improved” “Improvement for 6 months” | 1 “Unsuccessful” | |
| Mexazolam | 1 | 1 “Improvement” | ||
| Bromazepam | 1 | 1 “Partial improvement” | ||
| Carbamazepine | 2 | 1 “Partial improvement” | 1 “Unsuccessful” | |
| Clomipramine | 4 | 3 “Partial improvement” “Partial improvement” “Complete recovery” “Slight improvement, side effects” | ||
| Romethazine | 1 | 1 “Partial improvement” | ||
| Lorazepam | 10 | 4 “Partial improvement” “80% return to baseline” “Responded to” “Showed increase” “Significant improvement” | 1 “Negative effects” | 5 “No consistent improvement” “No consistent improvement” “No consistent improvement” “No change” “Ineffective” |
| Methylprednisolon | 3 | 3 “Dramatic improvement” “Immediate improvement” “Many behaviours resolved” | ||
| IVIG | 4 | 4 “Full recovery” “Steady improvement” “Lots of symptoms resolved” “Resolution of everything except insomnia” | ||
| Mycephenolate | 1 | 1 Discontinued no data | ||
| Oral steroid | 2 | 2 “Immediate improvement” “Lots of resolved symptoms” | ||
| Rituximab | 1 | 1 “Improvement” | ||
| Electro-convulsive therapy | 10 | 10 “Some behaviours completely resolved” “Complete recovery” “Complete recovery” “Complete recovery” “Complete recovery” “Robust response” “Significant improvement” “Return to almost baseline” “Excellent response” Strong response” | ||
| Benzodiazepines | 1 | 1 “Partial improvement” | ||
| Anti depressants | 1 | 1 “Steady return to baseline” | ||
| Positive airway pressure for obstructive sleep apnoea | 1 | 1 “Steady return to baseline” | ||
| Psychological support | 2 | 2 “Steady return to baseline” “Moderate improvement for 6 months” | ||
| Donepezil | 1 | 1 “Return to baseline” | ||
| Acetylcholinesterase inhibitor | 1 | 1 “Return to baseline” | ||
| Bupropion | 1 | 1 “Worsening of catatonia and further decline” | ||
| Trazodone | 4 | 1 “Some improvement” | 2 “Worsening of catatonia and further decline” “Worsening and decline” | 1 “Unsuccessful” |
| Olanzapine | 1 | 1 “Worsening of catatonia and further decline” | ||
| Aripiprazole | 1 | 1 “Worsening of catatonia and further decline” | ||
| Ziprasidone | 1 | 1 “Worsening of catatonia and further decline” | ||
| Lithium | 3 | 1 “Improvement” | 1 “Worsening of catatonia and further decline” | 1 “No improvement” |
| Clozapine | 1 | 1 “85% return to baseline” | ||
| Desipramine | 6 | 1 “Moderate improvement for 6 months” | 1 “Worsening of symptoms” | 4 “No change” “No effect” “Unsuccessful” “Unsuccessful” |
| Thiothixine | 2 | 1 “Complete recovery” | 1 “No change” | |
| Amitriptyline | 1 | 1 “Complete recovery” | ||
| Nortriptyline | 1 | 1 “Unsuccessful” | ||
| Clonazepam | 2 | 1 “Partial improvement” | 1 “Unsuccessful” | |
| Ethosuximide | 1 | 1 “No effect” | ||
| Lidexamfetamine | 1 | 1 “Worsening” | ||
| SSRI citalopram | 1 | 1 “Worsening” | ||
| Amiloride | 1 | 1 “Improvement” | ||
| Lamotrigine | 1 | 1 “No effect” | ||
| Antipsychotic treatment | 1 | 1 “Good response” |
Appendix B
| Sleep (n = 22) | % | Language (n = 14) | % | Withdrawal and disinterest (n = 14) | % | Slowness and immobility (n = 14) | % | Weight loss and anorexia (n = 12) | % | Depression (n = 11) | % | Hallucinations (n = 11) | % | Abulia (n = 10) | % | Skill loss (n = 10) | % | Catatonia (n = 8) | % | Aggression (n = 7) | % | Irritability (n = 6) | % | Obsessive compulsions (n = 6) | % | Fatigue (n = 5) | % | Abnormal blinking and gaze (n = 4) | % | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sleep | 0 | 10 | 71 | 9 | 64 | 8 | 57 | 10 | 83 | 9 | 82 | 7 | 64 | 7 | 70 | 6 | 60 | 6 | 75 | 6 | 86 | 2 | 33 | 5 | 83 | 4 | 80 | 2 | 50 | |
| Language | 10 | 45 | 0 | 3 | 21 | 7 | 50 | 4 | 33 | 7 | 64 | 6 | 55 | 2 | 20 | 6 | 60 | 6 | 75 | 4 | 57 | 2 | 33 | 1 | 17 | 0 | 0 | 2 | 50 | |
| Withdrawal and disinterest | 9 | 41 | 3 | 21 | 0 | 6 | 43 | 5 | 42 | 5 | 45 | 4 | 36 | 8 | 80 | 2 | 20 | 0 | 0 | 2 | 29 | 3 | 50 | 4 | 67 | 3 | 60 | 0 | 0 | |
| Slowness and immobility | 8 | 36 | 7 | 50 | 6 | 43 | 0 | 7 | 58 | 5 | 45 | 4 | 36 | 2 | 20 | 4 | 40 | 5 | 63 | 2 | 29 | 1 | 17 | 3 | 50 | 2 | 40 | 4 | 100 | |
| Weight loss and anorexia | 10 | 45 | 4 | 29 | 5 | 36 | 7 | 50 | 0 | 3 | 3 | 4 | 36 | 2 | 20 | 2 | 20 | 2 | 25 | 2 | 29 | 1 | 17 | 1 | 17 | 4 | 80 | 2 | 50 | |
| Depression | 9 | 41 | 7 | 50 | 5 | 36 | 5 | 36 | 3 | 25 | 0 | 4 | 36 | 0 | 0 | 2 | 20 | 6 | 75 | 2 | 29 | 1 | 17 | 0 | 0 | 0 | 0 | 2 | 50 | |
| Hallucinations | 7 | 32 | 6 | 43 | 4 | 29 | 4 | 29 | 4 | 33 | 4 | 36 | 0 | 0 | 0 | 4 | 40 | 0 | 0 | 2 | 29 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 75 | |
| Abulia | 7 | 32 | 2 | 14 | 8 | 57 | 2 | 14 | 2 | 17 | 4 | 36 | 0 | 0 | 0 | 0 | 0 | 2 | 25 | 2 | 29 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 75 | |
| Skill loss | 6 | 27 | 6 | 43 | 2 | 14 | 4 | 29 | 2 | 17 | 2 | 18 | 6 | 55 | 2 | 20 | 0 | 2 | 25 | 0 | 0 | 1 | 17 | 0 | 0 | 2 | 40 | 0 | 0 | |
| Catatonia | 6 | 27 | 6 | 43 | 0 | 0 | 5 | 36 | 2 | 17 | 2 | 18 | 0 | 0 | 2 | 20 | 6 | 60 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 60 | 0 | 0 | |
| Aggression | 6 | 27 | 4 | 29 | 2 | 14 | 2 | 14 | 2 | 17 | 2 | 18 | 2 | 18 | 2 | 20 | 2 | 20 | 0 | 0 | 0 | 2 | 33 | 0 | 0 | 1 | 20 | 0 | 0 | |
| Irritability | 2 | 9 | 2 | 14 | 3 | 21 | 1 | 7 | 1 | 8 | 1 | 9 | 0 | 0 | 3 | 30 | 1 | 10 | 2 | 25 | 2 | 29 | 0 | 2 | 33 | 0 | 0 | 2 | 50 | |
| Obsessive compulsions | 5 | 23 | 1 | 7 | 4 | 29 | 3 | 21 | 1 | 8 | 0 | 0 | 0 | 0 | 4 | 40 | 0 | 0 | 0 | 0 | 4 | 57 | 2 | 33 | 0 | 0 | 0 | 0 | 0 | |
| Fatigue | 4 | 18 | 0 | 0 | 3 | 21 | 2 | 14 | 4 | 33 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 29 | 2 | 33 | 0 | 0 | 0 | 1 | 25 | |
| Abnormal blinking and gaze | 2 | 9 | 2 | 14 | 0 | 0 | 4 | 29 | 2 | 17 | 2 | 18 | 3 | 27 | 1 | 10 | 2 | 20 | 1 | 13 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 20 | 0 |
References
- Antonakaris, S.E.; Stotko, B.G.; Rafii, M.S.; Strydom, A.; Pape, S.E.; Bianchi, D.W.; Sherman, S.L.; Reeves, R.H. Down Syndrome. Nat. Rev. Dis. Primers 2020, 6, 1–20. [Google Scholar] [CrossRef]
- Rasmussen, S.A.; Whitehead, N.; Collier, S.A.; Frías, J.L. Setting a public health research agenda for Down syndrome: Summary of a meeting sponsored by the centers for disease control and prevention and the national down syndrome society. Am. J. Med. Genet. Part. A 2008, 146, 2998–3010. [Google Scholar] [CrossRef] [PubMed]
- Dykens, E.M.; Shah, B.; Davis, B.; Baker, C.; Fife, T.; Fitzpatrick, J. Psychiatric disorders in adolescents and young adults with Down syndrome and other intellectual disabilities. J. Neurodev. Disord. 2015, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Santoro, S.L.; Cannon, S.; Capone, G.; Franklin, C.; Hart, S.J.; Hobensack, V.; Kishnani, P.S.; Macklin, E.A.; Manickam, K.; McCormick, A.; et al. Unexplained regression in Down syndrome: 35 cases from an international Down syndrome database. Genet. Med. 2019, 22, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.M.; Esiri, M.M. The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down’s syndrome. J. Neurol. Sci. 1989, 89, 169–179. [Google Scholar] [CrossRef]
- Annus, T.; Wilson, L.R.; Hong, Y.T.; Acosta-Cabronero, J.; Fryer, T.D.; Cardenas-Blanco, A.; Smith, R.; Boros, I.; Coles, J.P.; Aigbirhio, F.I.; et al. The pattern of amyloid accumulation in the brains of adults with Down syndrome. Alzheimer’s Dement. 2016, 12, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2012, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Rosso, M.; Fremion, E.; Santoro, S.L.; Oreskovic, N.M.; Chitnis, T.; Skotko, B.G.; Santoro, J.D. Down Syndrome Disintegrative Disorder: A Clinical Regression Syndrome of Increasing Importance. Pediatrics 2020, 145, e20192939. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Altman, D.; Antes, G.; Atkins, D.; Barbour, V.; Barrowman, N.; Berlin, J.A.; et al. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. J. Chin. Integr. Med. 2009, 7, 889–896. [Google Scholar] [CrossRef]
- Myers, B.A.; Pueschel, S.M. Major depression in a small group of adults with down syndrome. Res. Dev. Disabil. 1995, 16, 285–299. [Google Scholar] [CrossRef]
- Capone, G.T.; Aidikoff, J.M.; Goyal, P. Adolescents and Young Adults With Down Syndrome Presenting to a Medical Clinic With Depression: Phenomenology and Characterization Using the Reiss Scales and Aberrant Behavior Checklist. J. Ment. Health Res. Intellect. Disabil. 2011, 4, 244–264. [Google Scholar] [CrossRef]
- Akahoshi, K.; Matsuda, H.; Funahashi, M.; Hanaoka, T.; Suzuki, Y. Acute neuropsychiatric disorders in adolescents and young adults with down syndrome: Japanese case reports. Neuropsychiatr. Dis. Treat. 2012, 8, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Stein, D.S.; Munir, K.M.; Karweck, A.J.; Davidson, E.J.; Stein, M.T. Developmental regression, depression, and psychosocial stress in an adolescent with Down syndrome. J. Dev. Behav. Pediatr. 2013, 34, 216–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capone, G.T.; Aidikoff, J.M.; Taylor, K.; Rykiel, N. Adolescents and young adults with down syndrome presenting to a medical clinic with depression: Co-morbid obstructive sleep apnea. Am. J. Med. Genet. Part A 2013, 161, 2188–2196. [Google Scholar] [CrossRef] [PubMed]
- Ghaziuddin, N.; Nassiri, A.; Miles, J.H. Catatonia in down syndrome; a treatable cause of regression. Neuropsychiatr. Dis. Treat. 2015, 11, 941–949. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, J.; Schwartz, A.; McDougle, C.J.; Skotko, B.G. Rapid clinical deterioration in an individual with Down syndrome. Am. J. Med. Genet. Part. A 2016, 170, 1899–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamasaki, A.; Saito, Y.; Ueda, R.; Ohno, K.; Yokoyama, K.; Satake, T.; Sakuma, H.; Takahashi, Y.; Kondoh, T.; Maegaki, Y. Effects of donepezil and serotonin reuptake inhibitor on acute regression during adolescence in Down syndrome. Brain Dev. 2016, 38, 113–117. [Google Scholar] [CrossRef]
- Mircher, C.; Cieuta-Walti, C.; Marey, I.; Rebillat, A.-S.; Cretu, L.; Milenko, E.; Conte, M.; Sturtz, F.; Rethore, M.-O.; Ravel, A. Acute Regression in Young People with Down Syndrome. Brain Sci. 2017, 7, 57. [Google Scholar] [CrossRef]
- Cardinale, K.M.; Bocharnikov, A.; Hart, S.J.; Baker, J.A.; Eckstein, C.; Jasien, J.M.; Gallentine, W.; Worley, G.; Kishnani, P.S.; Van Mater, H. Immunotherapy in selected patients with Down syndrome disintegrative disorder. Dev. Med. Child. Neurol. 2018, 61, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.H.; Takahashi, N.; Muckerman, J.; Nowell, K.P.; Ithman, M. Catatonia in down syndrome: Systematic approach to diagnosis, treatment and outcome assessment based on a case series of seven patients. Neuropsychiatr. Dis. Treat. 2019, 15, 2723–2741. [Google Scholar] [CrossRef] [Green Version]
- Castillo, H.; Patterson, B.; Hickey, F.; Kinsman, A.; Howard, J.M.; Mitchell, T.; Molloy, C.A. Difference in age at regression in children with autism with and without Down syndrome. J. Dev. Behav. Pediatr. 2008, 29, 89–93. [Google Scholar] [CrossRef]
- Worley, G.; Crissman, B.G.; Cadogan, E.; Milleson, C.; Adkins, D.W.; Kishnani, P.S. Down syndrome disintegrative disorder: New-onset autistic regression, dementia, and insomnia in older children and adolescents with down syndrome. J. Child. Neurol. 2015, 30, 1147–1152. [Google Scholar] [CrossRef]
- Prasher, V. Disintegrative syndrome in young adults. Int. J. Psychiatr. Med. 2002, 19, 101–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, S.-A.; Collacott, R.A. Clinical Features and Diagnostic Criteria of Depression in Down Syndrome. Br. J. Psychiatry 1994, 165, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Brodtmann, A. Hashimoto encephalopathy and down syndrome. Arch. Neurol. 2009, 66, 663–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattman, A.; Jarvis-Selinger, S.; Mezei, M.M.; Salvarinova-Zivkovic, R.; Alfadhel, M.; Lillquist, Y. Mitochondrial disease clinical manifestations: An overview. Br. Columbia Med. J. 2011, 53, 183–187. [Google Scholar]
- Kusters, M.A.A.; Verstegen, R.H.J.; Gemen, E.F.A.; De Vries, E. Intrinsic defect of the immune system in children with Down syndrome: A review. Clin. Exp. Immunol. 2009, 156, 189–193. [Google Scholar] [CrossRef]
- Kohlenberg, T.M.; Trelles, M.P.; McLarney, B.; Betancur, C.; Thurm, A.; Kolevzon, A. Psychiatric illness and regression in individuals with Phelan-McDermid syndrome. J. Neurodev. Disord. 2020, 12, 7. [Google Scholar] [CrossRef] [Green Version]
- Bey, A.L.; Gorman, M.P.; Gallentine, W.; Kohlenberg, T.M.; Frankovich, J.; Jiang, Y.-H.; Van Haren, K. Subacute Neuropsychiatric Syndrome in Girls With SHANK3 Mutations Responds to Immunomodulation. Pediatrics 2020, 145, e20191490. [Google Scholar] [CrossRef] [Green Version]
- Soni, S.; Whittington, J.; Holland, A.J.; Webb, T.; Maina, E.N.; Boer, H.; Clarke, D. The phenomenology and diagnosis of psychiatric illness in people with Prader–Willi syndrome. Psychol. Med. 2008, 38, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.P.T.; Altman, D.G.; Gøtzsche, P.C.; Jüni, P.; Moher, D.; Oxman, A.D.; Savović, J.; Schulz, K.F.; Weeks, L.; Sterne, J.A.C. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ 2011, 343, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]





| Author | Number of Case Study Patients (Group A) | Number of Cohort Study Patients (Group B) | Gender (Female: Male) | Age |
|---|---|---|---|---|
| Myers and Pueschel (1995) [10] | 8 | 8 | 4:4 | Range 21–44 years |
| Capone, Aidikoff and Goyal (2011) [11] | 0 | 33 | 14:19 | Range 13–35 years Mean 22 years |
| Akahoshi et al. (2012) [12] | 12 | 12 | 6:6 | Range 13–29 years |
| Stein et al. (2013) [13] | 1 | 1 | Female | 13 years |
| Capone et al. (2013) [14] | 0 | 28 | 14:14 | Male mean 21.8 years Female mean 20.3 years |
| Dykens et al. (2015) [3] | 1 | 49 | 49% male | Range 13–29 years |
| Ghaziuddin, Nassiri and Miles (2015) [15] | 4 | 4 | 2:2 | Range 14–18 years |
| Jacobs et al. (2016) [16] | 1 | 1 | Male | 19 years |
| Tamasaki et al. (2016) [17] | 1 | 1 | Male | 15 years |
| Mircher et al. (2017) [18] | 0 | 30 | 20:10 | Range 12–30 years |
| Cardinale et al. (2018) [19] | 4 | 4 | 3:1 | Range 17–25 years |
| Santoro et al. (2019) [4] | 0 | 35 | 53% female | 9–34 years |
| Miles et al. (2020) [20] | 7 | 0 | 6:1 | 18–33 years |
| Regression Related Terminology | Times Used | Disorder Related Terminology | Times Used | Function Related Terminology | Times Used |
|---|---|---|---|---|---|
| Regression | 2 | Psychiatric disorders | 1 | Deterioration | 1 |
| Developmental regression | 1 | Down syndrome disintegrative Disorder | 2 | Clinical deterioration | 1 |
| Cognitive regression | 1 | New-onset mood disorder | 1 | Functional decline | 1 |
| Unexplained regression | 2 | Acute neuropsychiatric disorders | 1 | ||
| Rapid regression | 2 | Depression/major depression | 3 | ||
| Acute regression | 2 | ||||
| Total | 10 | Total | 8 | Total | 3 |
| Symptom | Moderate Symptoms | Severe Symptoms |
|---|---|---|
| Sleep | Restless sleep Poor sleep Disturbed sleep | Insomnia |
| Language | Vocal stereotypies Language decline Incoherent speech | Mutism |
| Weight loss | Weight loss Appetite loss | Anorexia nervosa |
| Slowing of movement | Slowness Slow movement | Immobility Becoming bedridden |
| Core Symptoms and Signs | Potential Triggers for Regression | Exclusions |
|---|---|---|
| New onset poor sleep | Transitions | Autism spectrum disorder presents in 5 years and above |
| Change in language output | (e.g., changes in an individual’s home/school/college routine) | Medical causes (incl. thyroid dysfunction and other conditions with autoimmune aetiology) |
| Abulia, withdrawal, disinterest, personality changes | Life events | New onset sensory impairment |
| Mood changes, loss of appetite and weight loss | Stressors | Age-related decrease in activity |
| Motor features–catatonia, stereotypies, extra-pyramidal signs | Other mental illness (e.g., depression) | |
| Loss of skills (adaptive functioning) | Unlikely over the age of 40 years (dementia is possible) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walpert, M.; Zaman, S.; Holland, A. A Systematic Review of Unexplained Early Regression in Adolescents and Adults with Down Syndrome. Brain Sci. 2021, 11, 1197. https://doi.org/10.3390/brainsci11091197
Walpert M, Zaman S, Holland A. A Systematic Review of Unexplained Early Regression in Adolescents and Adults with Down Syndrome. Brain Sciences. 2021; 11(9):1197. https://doi.org/10.3390/brainsci11091197
Chicago/Turabian StyleWalpert, Madeleine, Shahid Zaman, and Anthony Holland. 2021. "A Systematic Review of Unexplained Early Regression in Adolescents and Adults with Down Syndrome" Brain Sciences 11, no. 9: 1197. https://doi.org/10.3390/brainsci11091197
APA StyleWalpert, M., Zaman, S., & Holland, A. (2021). A Systematic Review of Unexplained Early Regression in Adolescents and Adults with Down Syndrome. Brain Sciences, 11(9), 1197. https://doi.org/10.3390/brainsci11091197