
Therapeutic Strategies for Mutant SPAST-Based Hereditary Spastic Paraplegia


Abstract
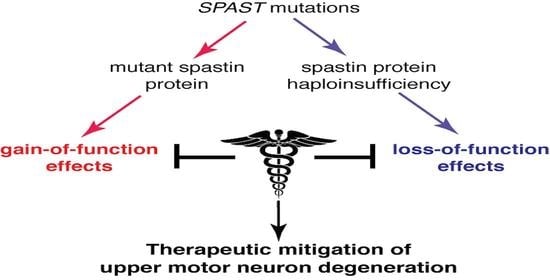
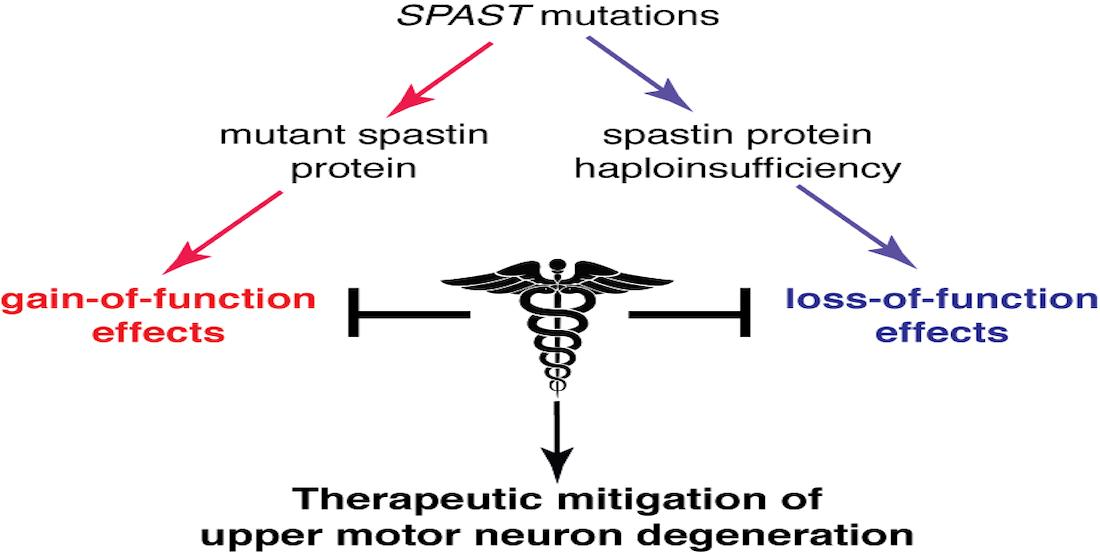
:
{kind=link}
{kind=link}
1. Introduction
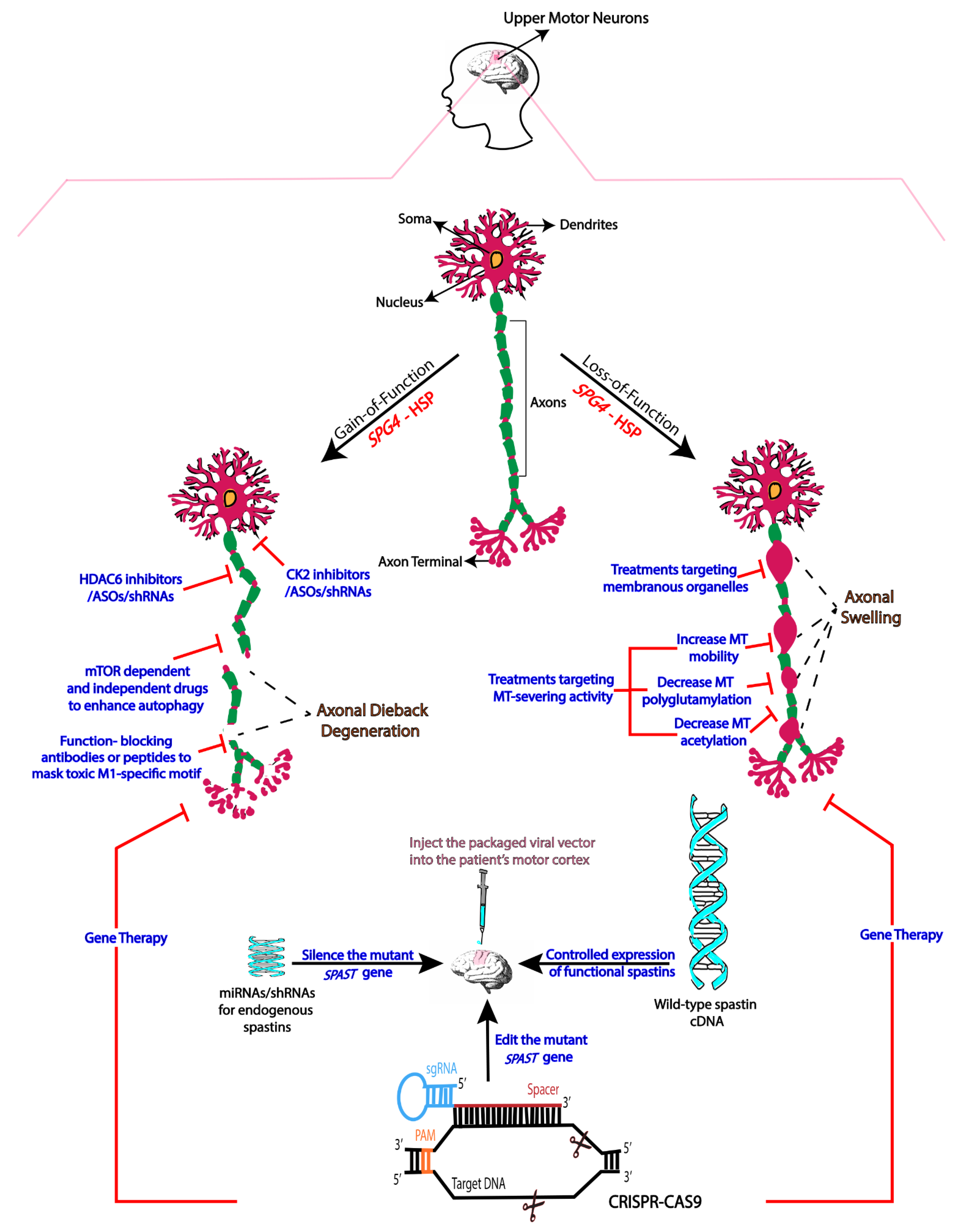
2. Mechanistic Basis of SPG4-HSP
3. Therapies Based on Gain-of-Function Mechanisms
4. Therapies Based on Loss-of-Function
5. Gene Therapy
6. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- McDermott, C.; White, K.; Bushby, K.; Shaw, P. Hereditary spastic paraparesis: A review of new developments. J. Neurol. Neurosurg. Psychiatry 2000, 69, 150–160. [Google Scholar] [CrossRef] [Green Version]
- Denton, K.R.; Xu, C.; Shah, H.; Li, X.J. Modeling Axonal Defects in Hereditary Spastic Paraplegia with Human Pluripotent Stem Cells. Front. Biol. 2016, 11, 339–354. [Google Scholar] [CrossRef] [Green Version]
- Qiang, L.; Piermarini, E.; Baas, P.W. New hypothesis for the etiology of SPAST-based hereditary spastic paraplegia. Cytoskeleton 2019, 76, 289–297. [Google Scholar] [CrossRef]
- Denton, K.R.; Lei, L.; Grenier, J.; Rodionov, V.; Blackstone, C.; Li, X.J. Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem Cells 2014, 32, 414–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazan, J.; Fonknechten, N.; Mavel, D.; Paternotte, C.; Samson, D.; Artiguenave, F.; Davoine, C.S.; Cruaud, C.; Dürr, A.; Wincker, P.; et al. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat. Genet. 1999, 23, 296–303. [Google Scholar] [CrossRef]
- Fonknechten, N.; Mavel, D.; Byrne, P.; Davoine, C.S.; Cruaud, C.; Boentsch, D.; Samson, D.; Coutinho, P.; Hutchinson, M.; Monagle, P.M.; et al. Spectrum of SPG4 mutations in autosomal dominant spastic paraplegia. Hum. Mol. Genet. 2000, 9, 637–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bürger, J.; Fonknechten, N.; Hoeltzenbein, M.; Neumann, L.; Bratanoff, E.; Hazan, J.; Reis, A. Hereditary spastic paraplegia caused by mutations in the SPG4 gene. Eur. J. Hum. Genet. 2000, 8, 771–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depienne, C.; Stevanin, G.; Brice, A.; Durr, A. Hereditary spastic paraplegias: An update. Curr. Opin. Neurol. 2007, 20, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Boone, P.M.; Yuan, B.; Campbell, I.M.; Scull, J.C.; Withers, M.A.; Baggett, B.C.; Beck, C.R.; Shaw, C.J.; Stankiewicz, P.; Moretti, P.; et al. The Alu-rich genomic architecture of SPAST predisposes to diverse and functionally distinct disease-associated CNV alleles. Am. J. Hum. Genet. 2014, 95, 143–161. [Google Scholar] [CrossRef] [Green Version]
- Julien, C.; Lissouba, A.; Madabattula, S.; Fardghassemi, Y.; Rosenfelt, C.; Androschuk, A.; Strautman, J.; Wong, C.; Bysice, A.; O’sullivan, J.; et al. Conserved pharmacological rescue of hereditary spastic paraplegia-related phenotypes across model organisms. Hum. Mol. Genet. 2016, 25, 1088–1099. [Google Scholar] [CrossRef] [Green Version]
- Newton, T.; Allison, R.; Edgar, J.R.; Lumb, J.H.; Rodger, C.E.; Manna, P.T.; Rizo, T.; Kohl, Z.; Nygren, A.O.; Arning, L.; et al. Mechanistic basis of an epistatic interaction reducing age at onset in hereditary spastic paraplegia. Brain 2018, 141, 1286–1299. [Google Scholar] [CrossRef] [Green Version]
- Kawarai, T.; Montecchiani, C.; Miyamoto, R.; Gaudiello, F.; Caltagirone, C.; Izumi, Y.; Kaji, R.; Orlacchio, A. Spastic paraplegia type 4: A novel SPAST splice site donor mutation and expansion of the phenotype variability. J. Neurol. Sci. 2017, 380, 92–97. [Google Scholar] [CrossRef]
- Solowska, J.M.; D’Rozario, M.; Jean, D.C.; Davidson, M.W.; Marenda, D.R.; Baas, P.W. Pathogenic mutation of spastin has gain-of-function effects on microtubule dynamics. J. Neurosci. 2014, 34, 1856–1867. [Google Scholar] [CrossRef] [Green Version]
- Leo, L.; Weissmann, C.; Burns, M.; Kang, M.; Song, Y.; Qiang, L.; Brady, S.T.; Baas, P.W.; Morfini, G. Mutant spastin proteins promote deficits in axonal transport through an isoform-specific mechanism involving casein kinase 2 activation. Hum. Mol. Genet. 2017, 26, 2321–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiang, L.; Piermarini, E.; Muralidharan, H.; Yu, W.; Leo, L.; Hennessy, L.E.; Fernandes, S.; Connors, T.; Yates, P.L.; Swift, M.; et al. Hereditary spastic paraplegia: Gain-of-function mechanisms revealed by new transgenic mouse. Hum. Mol. Genet. 2019, 28, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Svenson, I.K.; Ashley-Koch, A.E.; Gaskell, P.C.; Riney, T.J.; Cumming, W.K.; Kingston, H.M.; Hogan, E.L.; Boustany, R.M.; Vance, J.M.; Nance, M.A.; et al. Identification and expression analysis of spastin gene mutations in hereditary spastic paraplegia. Am. J. Hum. Genet. 2001, 68, 1077–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrahamsen, G.; Fan, Y.; Matigian, N.; Wali, G.; Bellette, B.; Sutharsan, R.; Raju, J.; Wood, S.A.; Veivers, D.; Sue, C.M.; et al. A patient-derived stem cell model of hereditary spastic paraplegia with SPAST mutations. Dis. Models Mech. 2013, 6, 489–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solowska, J.M.; Baas, P.W. Hereditary spastic paraplegia SPG4: What is known and not known about the disease. Brain 2015, 138, 2471–2484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasher, P.R.; De Vos, K.J.; Wharton, S.B.; Manser, C.; Bennett, E.J.; Bingley, M.; Wood, J.D.; Milner, R.; McDermott, C.J.; Miller, C.C.; et al. Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients. J. Neurochem. 2009, 110, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Tarrade, A.; Fassier, C.; Courageot, S.; Charvin, D.; Vitte, J.; Peris, L.; Thorel, A.; Mouisel, E.; Fonknechten, N.; Roblot, N.; et al. A mutation of spastin is responsible for swellings and impairment of transport in a region of axon characterized by changes in microtubule composition. Hum. Mol. Genet. 2006, 15, 3544–3558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claudiani, P.; Riano, E.; Errico, A.; Andolfi, G.; Rugarli, E.I. Spastin subcellular localization is regulated through usage of different translation start sites and active export from the nucleus. Exp. Cell Res. 2005, 309, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Pushing the limits of the scanning mechanism for initiation of translation. Gene 2002, 299, 1–34. [Google Scholar] [CrossRef]
- Solowska, J.M.; Morfini, G.; Falnikar, A.; Himes, B.T.; Brady, S.T.; Huang, D.; Baas, P.W. Quantitative and functional analyses of spastin in the nervous system: Implications for hereditary spastic paraplegia. J. Neurosci. 2008, 28, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Solowska, J.M.; Rao, A.N.; Baas, P.W. Truncating mutations of SPAST associated with hereditary spastic paraplegia indicate greater accumulation and toxicity of the M1 isoform of spastin. Mol. Biol. Cell. 2017, 28, 1728–1737. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.L.; Yardin, C.; Terro, F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Bowles, K.R.; Jones, L. Kinase signalling in Huntington’s disease. J. Huntingt. Dis. 2014, 3, 89–123. [Google Scholar] [CrossRef] [Green Version]
- Kanaan, N.M.; Morfini, G.A.; LaPointe, N.E.; Pigino, G.F.; Patterson, K.R.; Song, Y.; Andreadis, A.; Fu, Y.; Brady, S.T.; Binder, L.I. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J. Neurosci. 2011, 31, 9858–9868. [Google Scholar] [CrossRef] [PubMed]
- Kneynsberg, A.; Combs, B.; Christensen, K.; Morfini, G.; Kanaan, N.M. Axonal Degeneration in Tauopathies: Disease Relevance and Underlying Mechanisms. Front. Neurosci. 2017, 11, 572. [Google Scholar] [CrossRef] [PubMed]
- Janke, C.; Montagnac, G. Causes and Consequences of Microtubule Acetylation. Curr. Biol. 2017, 27, R1287–R1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Taes, I.; Timmers, M.; Hersmus, N.; Bento-Abreu, A.; Van Den Bosch, L.; Van Damme, P.; Auwerx, J.; Robberecht, W. Hdac6 deletion delays disease progression in the SOD1G93A mouse model of ALS. Hum. Mol. Genet. 2013, 22, 1783–1790. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, N.; Rao, P.; Burkhardt, S.; Sananbenesi, F.; Schlüter, O.M.; Bradke, F.; Lu, J.; Fischer, A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 52–63. [Google Scholar] [CrossRef]
- Brunden, K.R.; Lee, V.M.; Smith, A.B., 3rd; Trojanowski, J.Q.; Ballatore, C. Altered microtubule dynamics in neurodegenerative disease: Therapeutic potential of microtubule-stabilizing drugs. Neurobiol. Dis. 2017, 105, 328–335. [Google Scholar] [CrossRef]
- Wali, G.; Liyanage, E.; Blair, N.F.; Sutharsan, R.; Park, J.S.; Mackay-Sim, A.; Sue, C.M. Oxidative Stress-Induced Axon Fragmentation Is a Consequence of Reduced Axonal Transport in Hereditary Spastic Paraplegia SPAST Patient Neurons. Front. Neurosci. 2020, 14, 401. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Wali, G.; Sutharsan, R.; Bellette, B.; Crane, D.I.; Sue, C.M.; Mackay-Sim, A. Low dose tubulin-binding drugs rescue peroxisome trafficking deficit in patient-derived stem cells in Hereditary Spastic Paraplegia. Biol. Open 2014, 3, 494–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genc, B.; Gozutok, O.; Ozdinler, P.H. Complexity of Generating Mouse Models to Study the Upper Motor Neurons: Let Us Shift Focus from Mice to Neurons. Int. J. Mol. Sci. 2019, 20, 3848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Liu, X.; Cai, H.; Le, W. Autophagy in neurodegenerative diseases: Pathogenesis and therapy. Brain Pathol. 2018, 28, 3–13. [Google Scholar] [CrossRef]
- Park, H.; Kang, J.H.; Lee, S. Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates. Int. J. Mol. Sci. 2020, 21, 3369. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Sanchez, M.J.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heckmann, B.L.; Teubner, B.J.; Tummers, B.; Boada-Romero, E.; Harris, L.; Yang, M.; Guy, C.S.; Zakharenko, S.S.; Green, D.R. LC3-Associated Endocytosis Facilitates β-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell 2019, 178, 536–551.e14. [Google Scholar] [CrossRef]
- Yan, C.; Liu, J.; Gao, J.; Sun, Y.; Zhang, L.; Song, H.; Xue, L.; Zhan, L.; Gao, G.; Ke, Z.; et al. IRE1 promotes neurodegeneration through autophagy-dependent neuron death in the Drosophila model of Parkinson’s disease. Cell Death Dis. 2019, 10, 800. [Google Scholar] [CrossRef] [Green Version]
- Stone, M.C.; Rao, K.; Gheres, K.W.; Kim, S.; Tao, J.; La Rochelle, C.; Folker, C.T.; Sherwood, N.T.; Rolls, M.M. Normal spastin gene dosage is specifically required for axon regeneration. Cell Rep. 2012, 2, 1340–1350. [Google Scholar] [CrossRef] [Green Version]
- Fink, J.K. Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013, 126, 307–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, K.; Stone, M.C.; Weiner, A.T.; Gheres, K.W.; Zhou, C.; Deitcher, D.L.; Levitan, E.S.; Rolls, M.M. Spastin, atlastin, and ER relocalization are involved in axon but not dendrite regeneration. Mol. Biol. Cell. 2016, 27, 3245–3256. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.M. Spastin joins LDs and peroxisomes in the interorganelle contact ballet. J. Cell. Biol. 2019, 218, 2439–2441. [Google Scholar] [CrossRef] [Green Version]
- Allison, R.; Lumb, J.H.; Fassier, C.; Connell, J.W.; Ten Martin, D.; Seaman, M.N.; Hazan, J.; Reid, E. An ESCRT-spastin interaction promotes fission of recycling tubules from the endosome. J. Cell. Biol. 2013, 202, 527–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadepalle, N.; Robers, L.; Veronese, M.; Zentis, P.; Babatz, F.; Brodesser, S.; Gruszczyk, A.V.; Schauss, A.; Höning, S.; Rugarli, E.I. Microtubule-dependent and independent roles of spastin in lipid droplet dispersion and biogenesis. Life Sci. Alliance 2020, 3, e202000715. [Google Scholar] [CrossRef]
- Baas, P.W.; Rao, A.N.; Matamoros, A.J.; Leo, L. Stability properties of neuronal microtubules. Cytoskeleton 2016, 73, 442–460. [Google Scholar] [CrossRef] [Green Version]
- Valenstein, M.L.; Roll-Mecak, A. Graded Control of Microtubule Severing by Tubulin Glutamylation. Cell 2016, 164, 911–921. [Google Scholar] [CrossRef] [Green Version]
- Sirajuddin, M.; Rice, L.M.; Vale, R.D. Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat. Cell Biol. 2014, 16, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janke, C.; Kneussel, M. Tubulin post-translational modifications: Encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 2010, 33, 362–372. [Google Scholar] [CrossRef]
- Lacroix, B.; Van Dijk, J.; Gold, N.D.; Guizetti, J.; Aldrian-Herrada, G.; Rogowski, K.; Gerlich, D.W.; Janke, C. Tubulin polyglutamylation stimulates spastin-mediated microtubule severing. J. Cell. Biol. 2010, 189, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.N.; Patil, A.; Black, M.M.; Craig, E.M.; Myers, K.A.; Yeung, H.T.; Baas, P.W. Cytoplasmic Dynein Transports Axonal Microtubules in a Polarity-Sorting Manner. Cell Rep. 2017, 19, 2210–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, A.T.; Hausrat, T.J.; Heisler, F.F.; Gromova, K.V.; Lombino, F.L.; Fischer, T.; Ruschkies, L.; Breiden, P.; Thies, E.; Hermans-Borgmeyer, I.; et al. Spastin depletion increases tubulin polyglutamylation and impairs kinesin-mediated neuronal transport, leading to working and associative memory deficits. PLoS Biol. 2020, 18, e3000820. [Google Scholar] [CrossRef]
- Rao, A.N.; Baas, P.W. Polarity Sorting of Microtubules in the Axon. Trends Neurosci. 2018, 41, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Waldrop, M.A.; Karingada, C.; Storey, M.A.; Powers, B.; Iammarino, M.A.; Miller, N.F.; Alfano, L.N.; Noritz, G.; Rossman, I.; Ginsberg, M.; et al. Gene Therapy for Spinal Muscular Atrophy: Safety and Early Outcomes. Pediatrics 2020, 146, e20200729. [Google Scholar] [CrossRef]
- Kruminis-Kaszkiel, E.; Juranek, J.; Maksymowicz, W.; Wojtkiewicz, J. CRISPR/Cas9 Technology as an Emerging Tool for Targeting Amyotrophic Lateral Sclerosis (ALS). Int. J. Mol. Sci. 2018, 19, 906. [Google Scholar] [CrossRef] [Green Version]
- Pena, S.A.; Iyengar, R.; Eshraghi, R.S.; Bencie, N.; Mittal, J.; Aljohani, A.; Mittal, R.; Eshraghi, A.A. Gene therapy for neurological disorders: Challenges and recent advancements. J. Drug Target 2020, 28, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Wang, Q.; Chen, Z.; Cai, L.; Bao, L.; Qi, Q.; Liu, L.; Wang, X.; Jin, H.; Wang, J.; et al. 3-Anhydro-6-hydroxy-ophiobolin A, a fungal sesterterpene from Bipolaris oryzae induced autophagy and promoted the degradation of α-synuclein in PC12 cells. Bioorganic Med. Chem. Lett. 2015, 25, 1464–1470. [Google Scholar] [CrossRef]
- Dangi, A.; Yu, S.; Luo, X. Emerging approaches and technologies in transplantation: The potential game changers. Cell. Mol. Immunol. 2019, 16, 334–342. [Google Scholar] [CrossRef]
- Zhang, B. CRISPR/Cas gene therapy. J. Cell. Physiol. 2021, 236, 2459–2481. [Google Scholar] [CrossRef] [PubMed]
- Valenti, M.T.; Serena, M.; Carbonare, L.D.; Zipeto, D. CRISPR/Cas system: An emerging technology in stem cell research. World J. Stem Cells 2019, 11, 937–956. [Google Scholar] [CrossRef] [PubMed]
- Uddin, F.; Rudin, C.M.; Sen, T. CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Front. Oncol. 2020, 10, 1387. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohan, N.; Qiang, L.; Morfini, G.; Baas, P.W. Therapeutic Strategies for Mutant SPAST-Based Hereditary Spastic Paraplegia. Brain Sci. 2021, 11, 1081. https://doi.org/10.3390/brainsci11081081
Mohan N, Qiang L, Morfini G, Baas PW. Therapeutic Strategies for Mutant SPAST-Based Hereditary Spastic Paraplegia. Brain Sciences. 2021; 11(8):1081. https://doi.org/10.3390/brainsci11081081
Chicago/Turabian StyleMohan, Neha, Liang Qiang, Gerardo Morfini, and Peter W. Baas. 2021. "Therapeutic Strategies for Mutant SPAST-Based Hereditary Spastic Paraplegia" Brain Sciences 11, no. 8: 1081. https://doi.org/10.3390/brainsci11081081
APA StyleMohan, N., Qiang, L., Morfini, G., & Baas, P. W. (2021). Therapeutic Strategies for Mutant SPAST-Based Hereditary Spastic Paraplegia. Brain Sciences, 11(8), 1081. https://doi.org/10.3390/brainsci11081081