
Cytoplasmic Human TDP-43 Mislocalization Induces Widespread Dendritic Spine Loss in Mouse Upper Motor Neurons


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Cohort
2.2. Tissue Processing
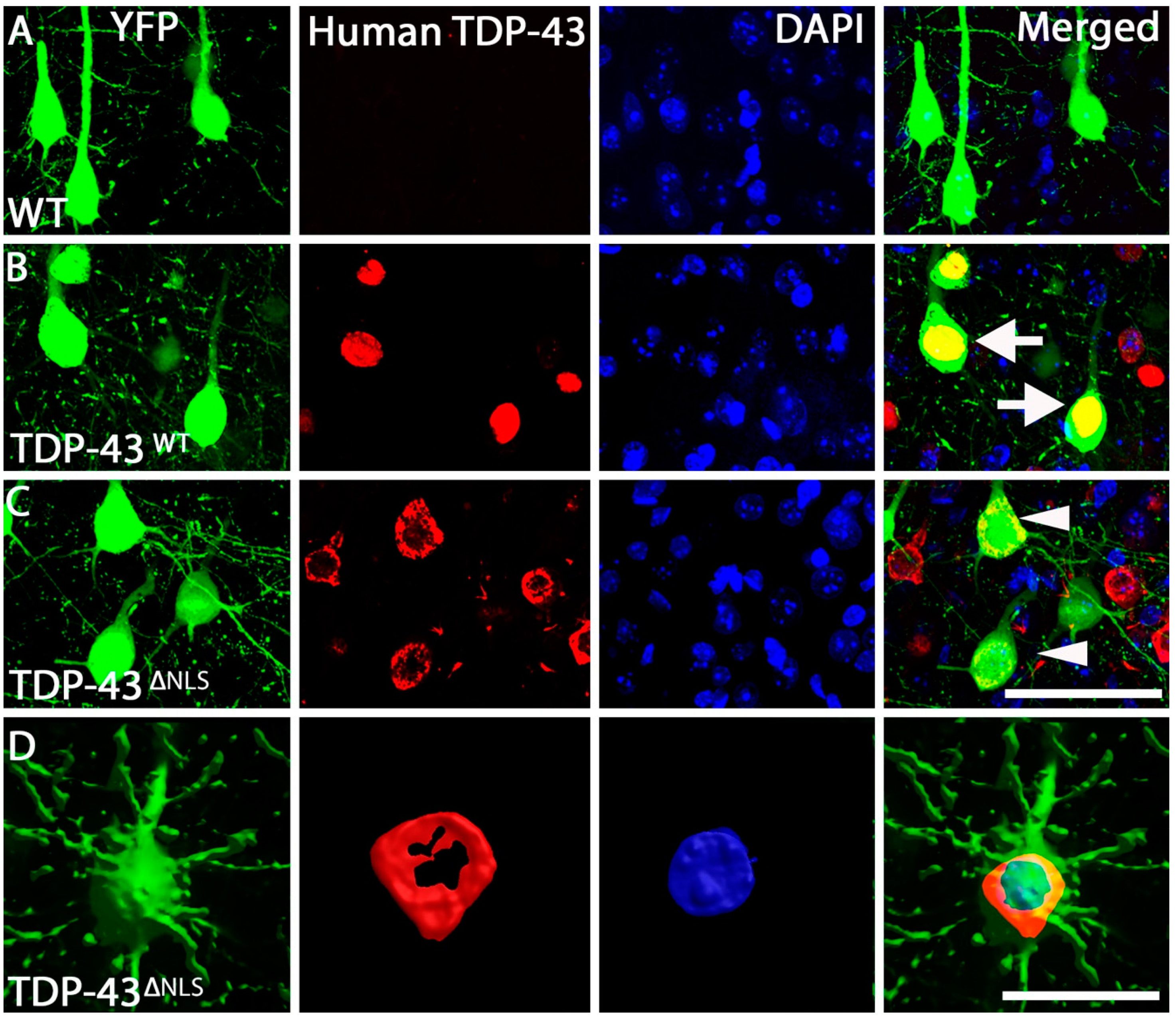
2.3. Immunohistochemistry
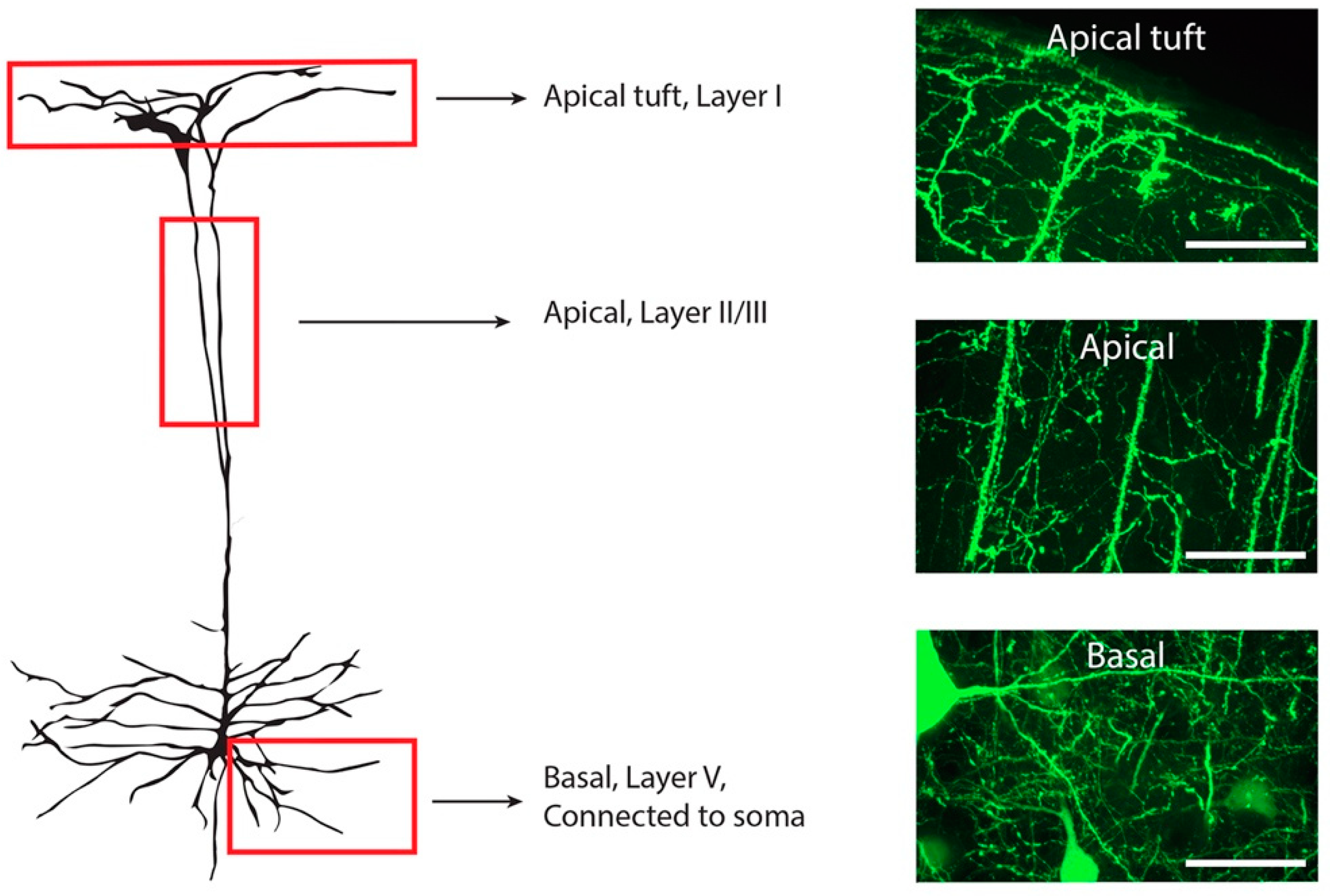
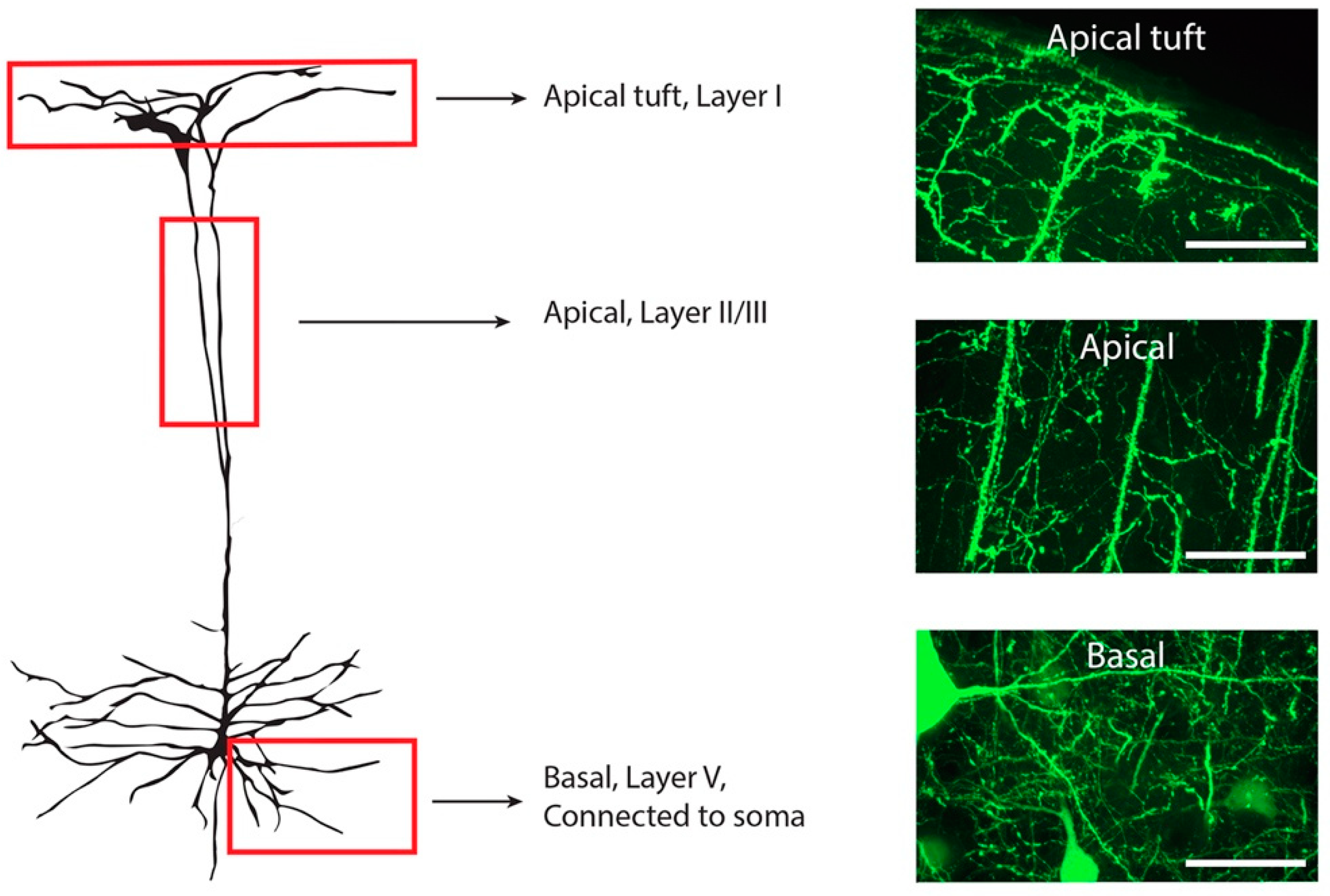
2.4. Microscopy
2.5. Quantification
2.6. Statistical Analysis
3. Results
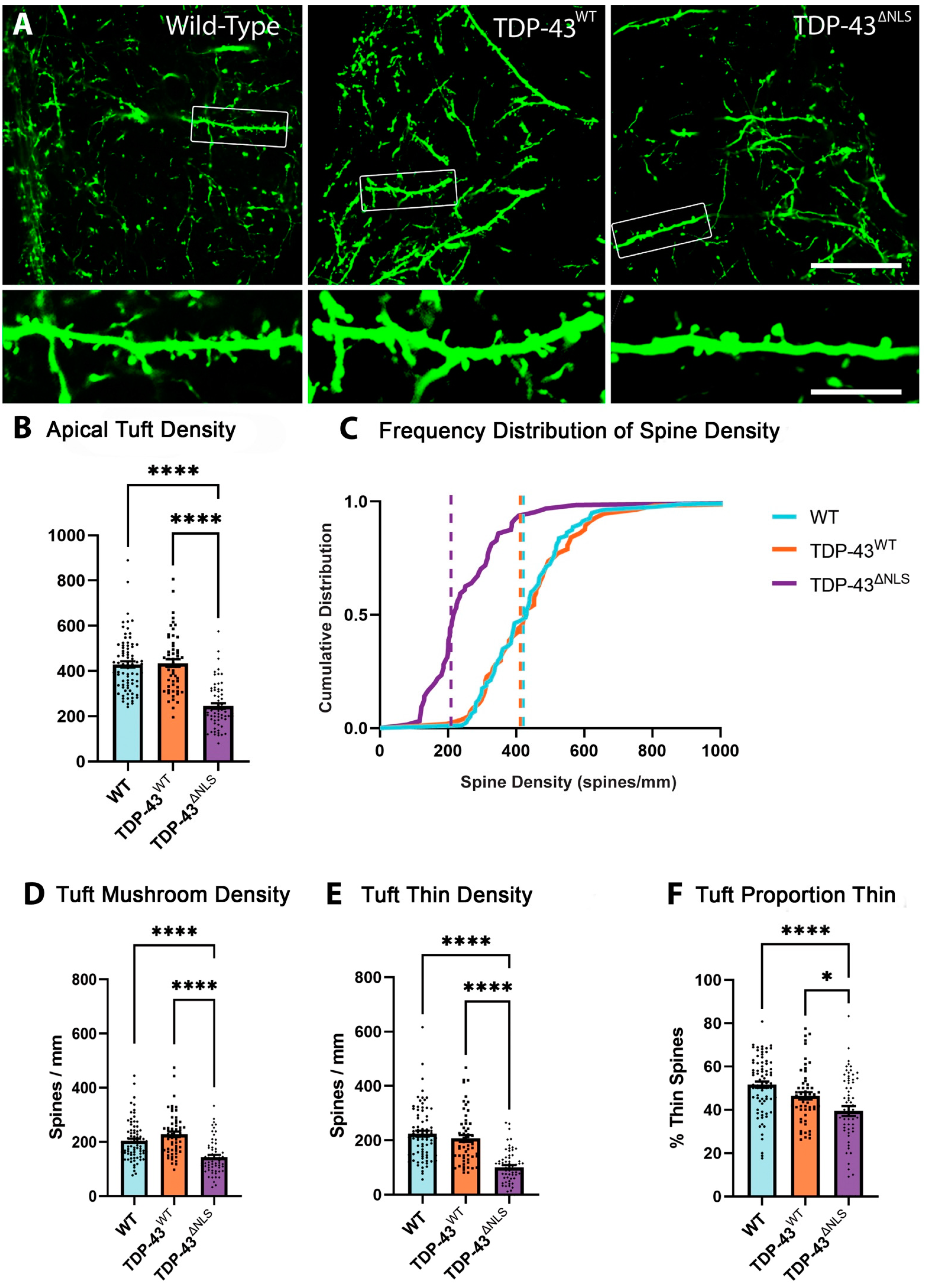
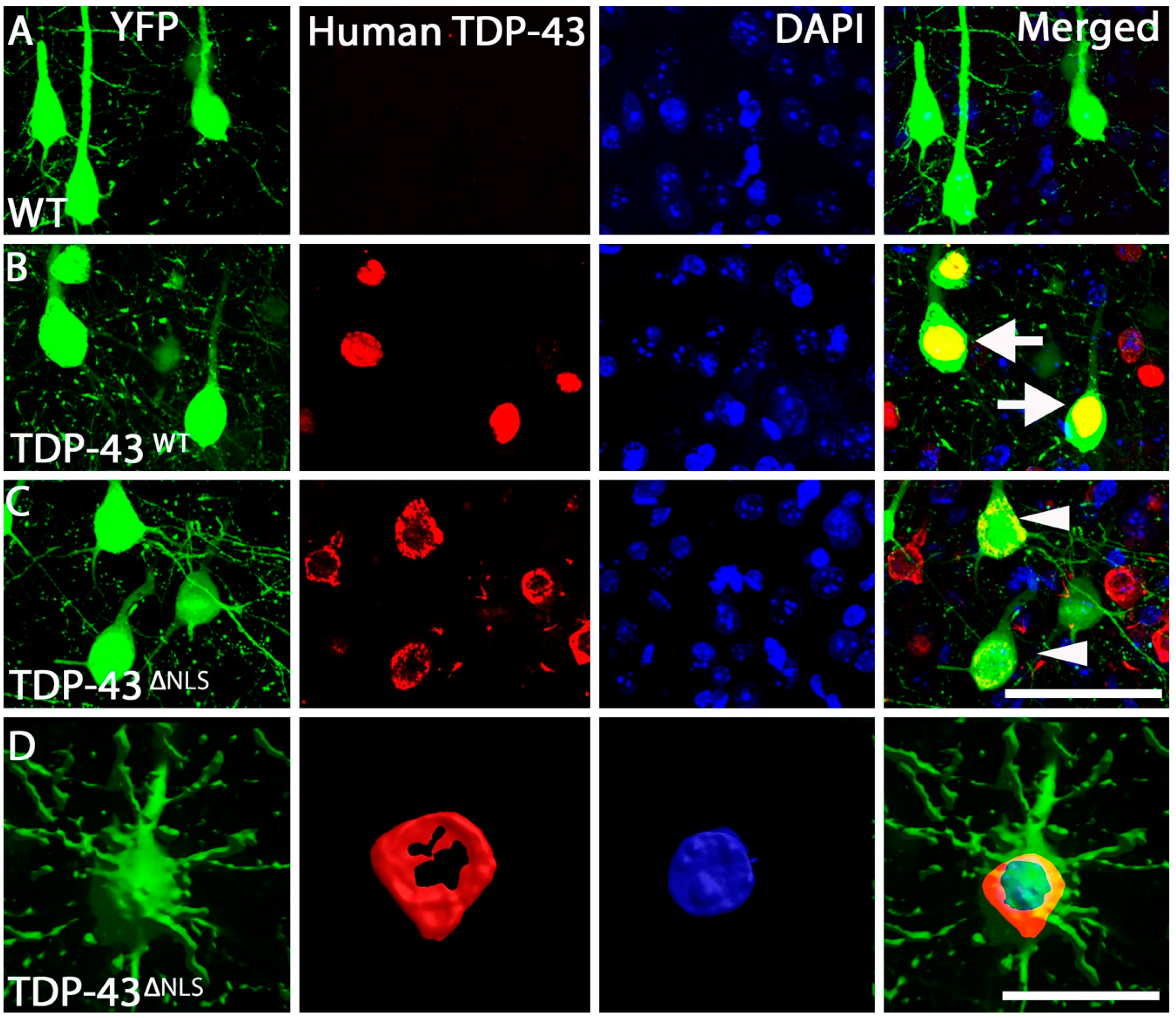
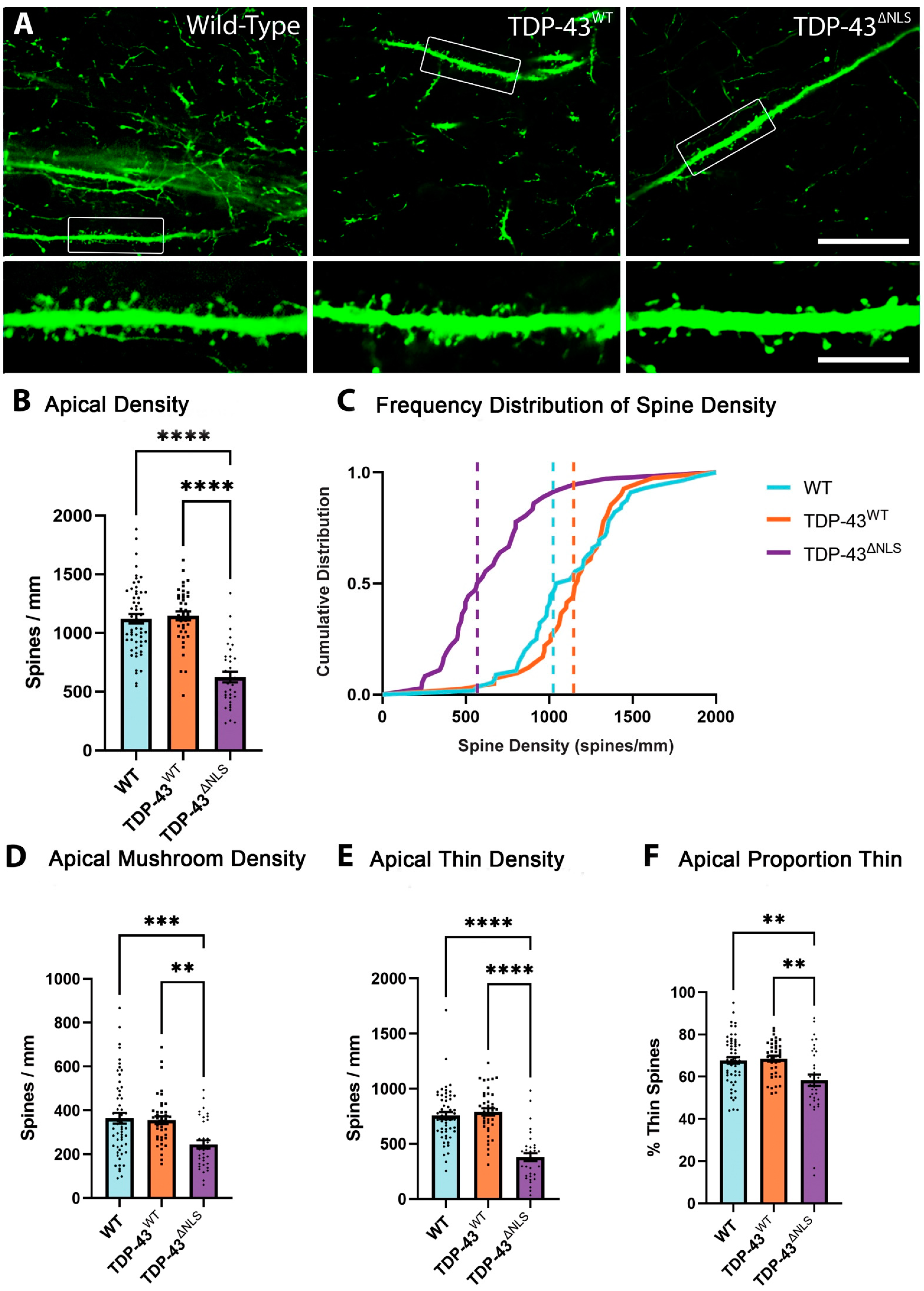
3.1. Expression of Mislocalised TDP-43 Caused a Loss of Spines in Apical Tuft Dendrites
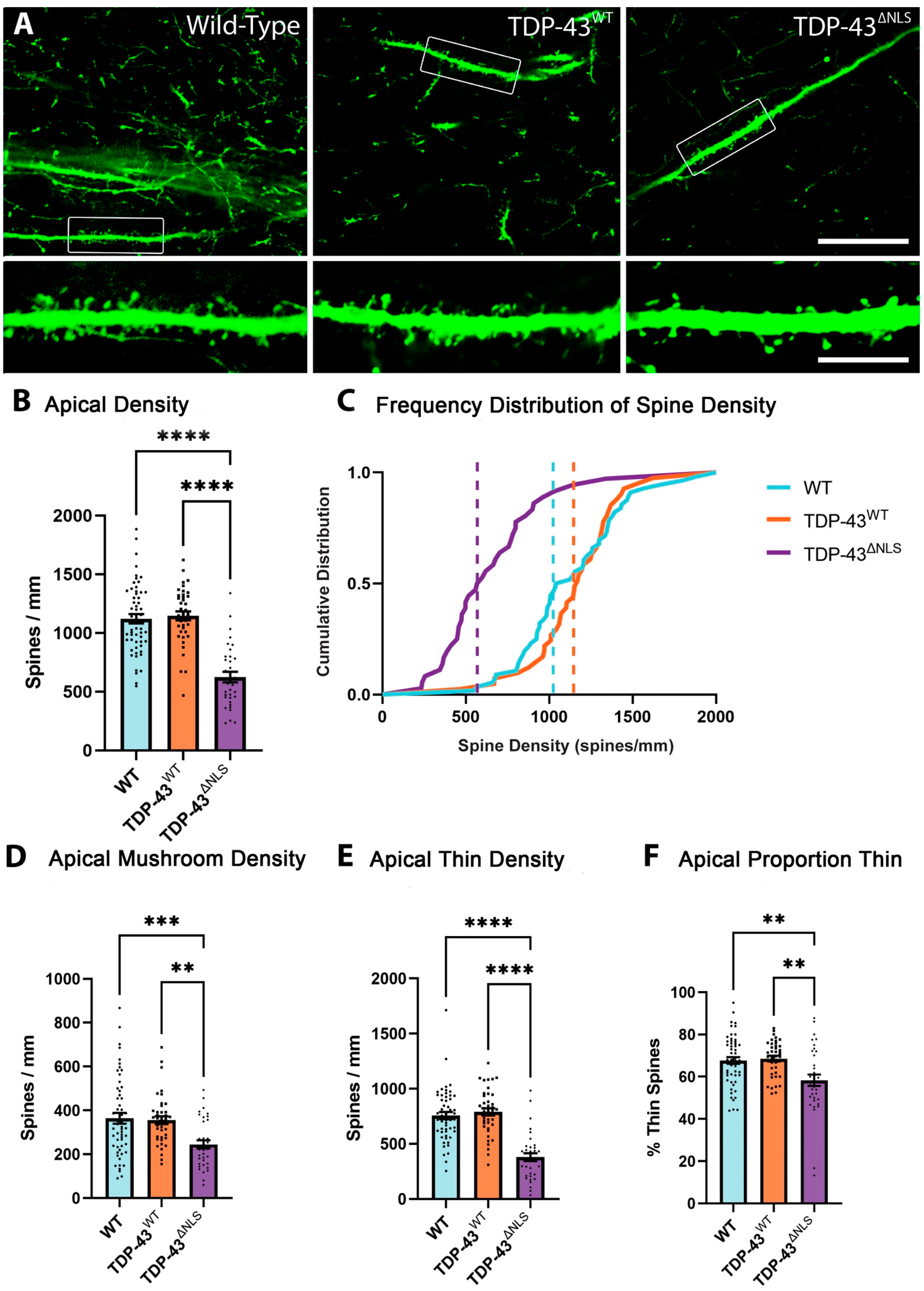
3.2. The Density of Spines Is Reduced in Apical Trunk Dendrites in TDP-43ΔNLS Neurons
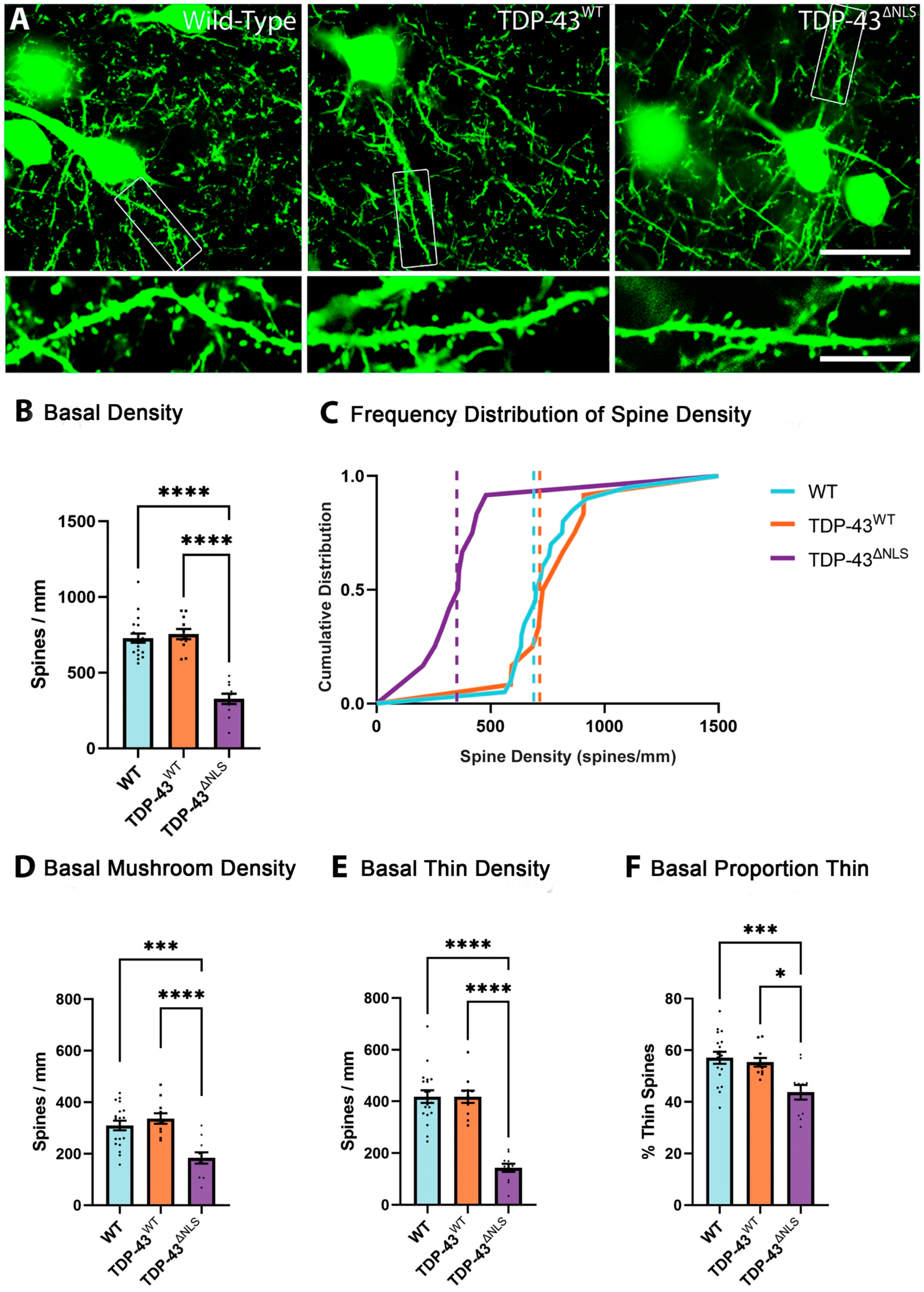
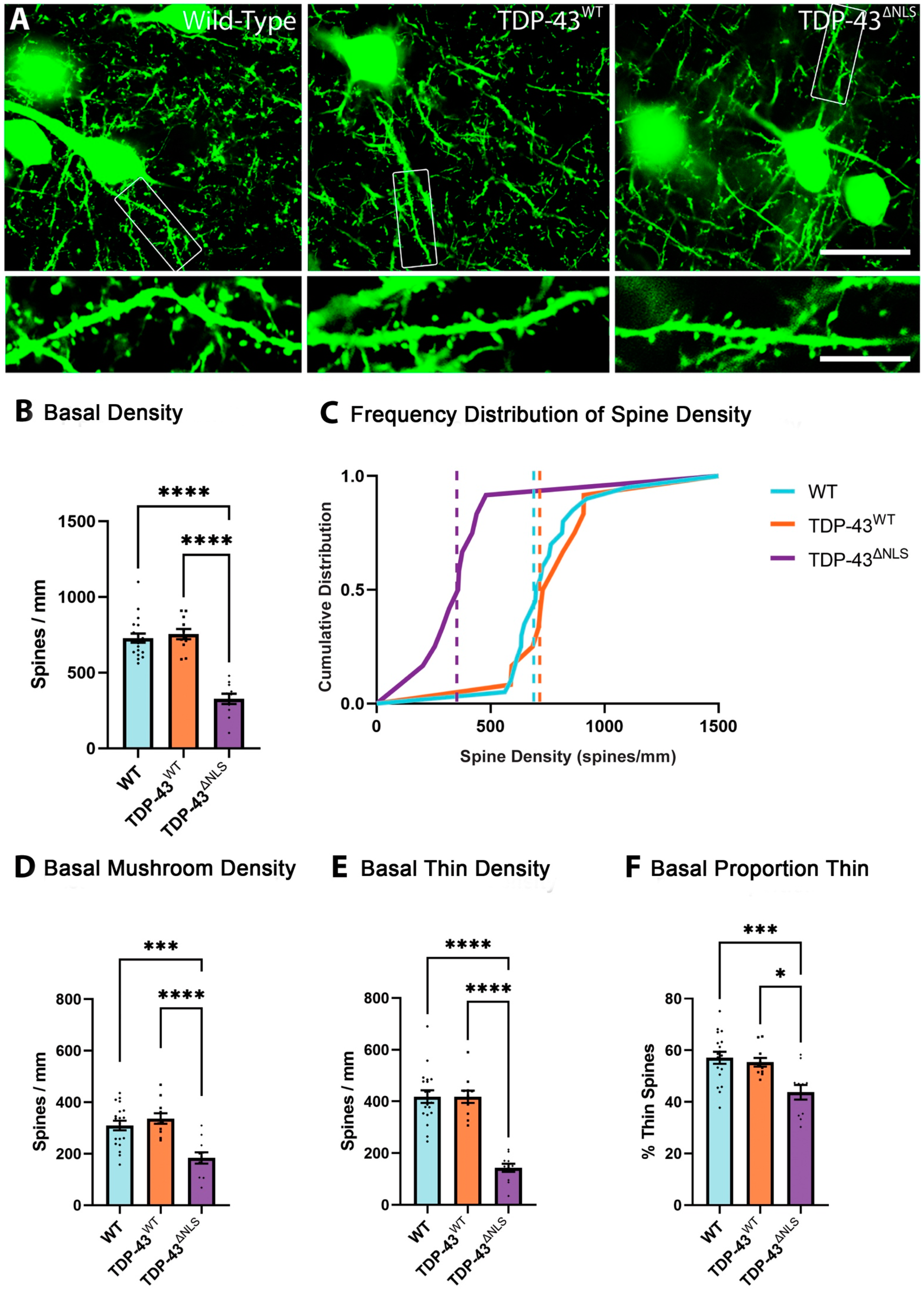
3.3. The Density of Spines Is Reduced in Basal Dendrites in TDP-43ΔNLS Neurons
3.4. Changes to Dendritic Spines Were Dependent on the Localisation of TDP-43 but Not the Location of the Dendritic Spine
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Hardiman, O. Major advances in amyotrophic lateral sclerosis in 2020. Lancet Neurol. 2021, 20, 14–15. [Google Scholar] [CrossRef]
- Shibuya, K.; Park, S.B.; Geevasinga, N.; Menon, P.; Howells, J.; Simon, N.G.; Huynh, W.; Noto, Y.I.; Götz, J.; Kril, J.J.; et al. Motor cortical function determinesprognosis in sporadic ALS. Neurology 2016, 87, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Dharmadasa, T. Cortical Excitability across the ALS Clinical Motor Phenotypes. Brain Sci. 2021, 11, 715. [Google Scholar] [CrossRef]
- Ravits, J.P.; Jorg, C.P. Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 2007, 68, 1571–1575. [Google Scholar] [CrossRef]
- Ragagnin, A.M.; Shadfar, S.; Vidal, M.; Jamali, M.S.; Atkin, J.D. Motor Neuron Susceptibility in ALS/FTD. Front. Neurosci. 2019, 13, 532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiernan, M.C.; Vucic, S.; Talbot, K.; McDermott, C.J.; Hardiman, O.; Shefner, J.M.; Al-Chalabi, A.; Huynh, W.; Cudkowicz, M.; Talman, P.; et al. Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2021, 17, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Menon, P.; Higashihara, M.; van den Bos, M.; Geevasinga, N.; Kiernan, M.C.; Vucic, S. Cortical hyperexcitability evolves with disease progression in ALS. Ann. Clin. Transl. Neurol. 2020, 7, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Menon, P.; Kiernan, M.C.; Vucic, S. Cortical hyperexcitability precedes lower motor neuron dysfunction in ALS. Clin. Neurophysiol. 2015, 126, 803–809. [Google Scholar] [CrossRef]
- Eisen, A. The Dying Forward Hypothesis of ALS: Tracing Its History. Brain Sci. 2021, 11, 300. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Cheah, B.C.; Yiannikas, C.; Kiernan, M.C. Cortical excitability distinguishes ALS from mimic disorders. Clin. Neurophysiol. 2011, 122, 1860–1866. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, M.J.; Klenowski, P.M.; Lee, J.D.; Drieberg-Thompson, J.R.; Bartlett, S.E.; Ngo, S.T.; Hilliard, M.A.; Bellingham, M.C.; Noakes, P.G. Cortical synaptic and dendritic spine abnormalities in a presymptomatic TDP-43 model of amyotrophic lateral sclerosis. Sci. Rep. 2016, 6, 37968. [Google Scholar] [CrossRef] [Green Version]
- Fogarty, M.J.; Noakes, P.G.; Bellingham, M.C. Motor cortex layer V pyramidal neurons exhibit dendritic regression, spine loss, and increased synaptic excitation in the presymptomatic hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2015, 35, 643–647. [Google Scholar] [CrossRef] [Green Version]
- Saba, L.; Viscomi, M.T.; Caioli, S.; Pignataro, A.; Bisicchia, E.; Pieri, M.; Molinari, M.; Ammassari-Teule, M.; Zona, C. Altered Functionality, Morphology, and Vesicular Glutamate Transporter Expression of Cortical Motor Neurons from a Presymptomatic Mouse Model of Amyotrophic Lateral Sclerosis. Cereb. Cortex 2016, 26, 1512–1528. [Google Scholar] [CrossRef] [Green Version]
- Handley, E.E.; Pitman, K.A.; Dawkins, E.; Young, K.M.; Clark, R.M.; Jiang, T.C.; Turner, B.J.; Dickson, T.C.; Blizzard, C.A. Synapse Dysfunction of Layer V Pyramidal Neurons Precedes Neurodegeneration in a Mouse Model of TDP-43 Proteinopathies. Cereb. Cortex 2017, 27, 3630–3647. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhang, L.; Liang, B.; Schroeder, D.; Zhang, Z.W.; Cox, G.A.; Li, Y.; Lin, D.T. Hyperactive somatostatin interneurons contribute to excitotoxicity in neurodegenerative disorders. Nat. Neurosci. 2016, 19, 557–559. [Google Scholar] [CrossRef] [Green Version]
- Hammer, R.; Tomiyasu, U.; Scheibel, A. Degeneration of the Human Betz Cell Due to Amyotrophic Lateral Sclerosis. Exp. Neurol. 1979, 63, 336–346. [Google Scholar] [CrossRef]
- Ghasemi, M.; Brown, R.H., Jr. Genetics of Amyotrophic Lateral Sclerosis. Cold Spring Harb. Perspect Med. 2018, 8, 1465–1470. [Google Scholar] [CrossRef]
- Neumann, M.S.; Kwong, D.M.; Truax, L.K.; Micsenyi, A.C.; Chou, M.C.; Bruce, T.T.; Schuck, J.; Grossman, T.; Clark, M.; McCluskey, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyer, M.S.; Reale, L.A.; Lewis, K.E.; Walker, A.K.; Dickson, T.C.; Woodhouse, A.; Blizzard, C.A. Mislocalisation of TDP-43 to the cytoplasm causes cortical hyperexcitability and reduced excitatory neurotransmission in the motor cortex. J. Neurochem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Weskamp, K.; Tank, E.M.; Miguez, R.; McBride, J.P.; Gómez, N.B.; White, M.; Lin, Z.; Gonzalez, C.M.; Serio, A.; Sreedharan, J.; et al. Shortened TDP43 isoforms upregulated by neuronal hyperactivity drive TDP43 pathology in ALS. J. Clin. Investig. 2020, 130, 1139–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Zuo, Y. Spine plasticity in the motor cortex. Curr. Opin. Neurobiol. 2011, 21, 169–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtmaat, A.; Svoboda, K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci. 2009, 10, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Spruston, N. Pyramidal neurons: Dendritic structure and synaptic integration. Nat. Rev. Neurosci. 2008, 9, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.R.; Stuart, G.J. Role of dendritic synapse location in the control of action potential output. Trends Neurosci. 2003, 26, 147–154. [Google Scholar] [CrossRef]
- Berry, K.P.; Nedivi, E. Spine Dynamics: Are They All the Same? Neuron 2017, 96, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Igaz, L.M.; Kwong, L.K.; Lee, E.B.; Chen-Plotkin, A.; Swanson, E.; Unger, T.; Malunda, J.; Xu, Y.; Winton, M.J.; Trojanowski, J.Q.; et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Investig. 2011, 121, 726–738. [Google Scholar] [CrossRef] [Green Version]
- Porrero, C.; Rubio-Garrido, P.; Avendaño, C.; Clascá, F. Mapping of fluorescent protein-expressing neurons and axon pathways in adult and developing Thy1-eYFP-H transgenic mice. Brain Res. 2010, 1345, 59–72. [Google Scholar] [CrossRef]
- Feng, G.; Mellor, R.H.; Bernstein, M.; Keller-Peck, C.; Nguyen, Q.T.; Wallace, M.; Nerbonne, J.M.; Lichtman, J.W.; Sanes, J.R. Imaging Neuronal Subsetsin Transgenic Mice ExpressingMultiple Spectral Variants of GFP. Neuron 2000, 28, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Paxinos, G.; Franklin, K. The Mouse Brain in Stereotaxic Coordinates; Academic Press: Cambridge, MA, USA, 2007. [Google Scholar]
- Pchitskaya, E.; Bezprozvanny, I. Dendritic Spines Shape Analysis-Classification or Clusterization? Perspective. Front. Synaptic Neurosci. 2020, 12, 31. [Google Scholar] [CrossRef]
- Faul, F.; Erdfelder, E.; Buchner, A.; Lang, A.G. Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behav. Res. Methods 2009, 41, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Phillips, W.A. Cognitive functions of intracellular mechanisms for contextual amplification. Brain Cogn. 2017, 112, 39–53. [Google Scholar] [CrossRef]
- Rubio-Garrido, P.; Pérez-de-Manzo, F.; Porrero, C.; Galazo, M.J.; Clascá, F. Thalamic input to distal apical dendrites in neocortical layer 1 is massive and highly convergent. Cereb. Cortex 2009, 19, 2380–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuste, R.; Majewska, A.; Holthoff, K. From form to function: Calcium compartmentalization in dendritic spines. Nat. Neurosci. 2000, 3, 653–659. [Google Scholar] [CrossRef]
- Sala, C.; Segal, M. Dendritic spines: The locus of structural and functional plasticity. Physiol. Rev. 2014, 94, 141–188. [Google Scholar] [CrossRef]
- Fogarty, M.J. The bigger they are the harder they fall: Size-dependent vulnerability of motor neurons in amyotrophic lateral sclerosis. J. Physiol. 2018, 596, 2471–2472. [Google Scholar] [CrossRef]
- Ravits, J.M.; La Spada, A.R. ALS motor phenotype heterogeneity, focality, and spread. Neurology 2009, 73, 805–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vucic, S.; Nicholson, G.A.; Kiernan, M.C. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 2008, 131 Pt 6, 1540–1550. [Google Scholar] [CrossRef] [Green Version]
- Tonnesen, J.; Nagerl, V.U. Dendritic Spines as Tunable Regulators of Synaptic Signals. Front. Psychiatry 2016, 7, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genç, B.; Jara, J.H.; Lagrimas, A.K.; Pytel, P.; Roos, R.P.; Mesulam, M.M.; Geula, C.; Bigio, E.H.; Özdinler, P.H. Apical dendrite degeneration, a novel cellular pathology for Betz cells in ALS. Sci. Rep. 2017, 7, 41765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, J.; Bellingham, C.M. Neurophysiological Mechanisms Underlying Cortical Hyper-Excitability in Amyotrophic Lateral Sclerosis: A Review. Brain Sci. 2021, 11, 549. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, M.J.; Mu, E.W.; Lavidis, N.A.; Noakes, P.G.; Bellingham, M.C. Motor Areas Show Altered Dendritic Structure in an Amyotrophic Lateral Sclerosis Mouse Model. Front. Neurosci. 2017, 11, 609. [Google Scholar] [CrossRef] [PubMed]
- Sephton, C.F.; Tang, A.A.; Kulkarni, A.; West, J.; Brooks, M.; Stubblefield, J.J.; Liu, Y.; Zhang, M.Q.; Green, C.B.; Huber, K.M.; et al. Activity-dependent FUS dysregulation disrupts synaptic homeostasis. Proc. Natl. Acad. Sci. USA 2014, 111, E4769–E4778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogarty, M.J.; Mu, E.W.; Noakes, P.G.; Lavidis, N.A.; Bellingham, M.C. Marked changes in dendritic structure and spine density precede significant neuronal death in vulnerable cortical pyramidal neuron populations in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2016, 4, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Handley, E.; Brizuela, M.; Dawkins, E.; Lewis, K.E.; Clark, R.M.; Dickson, T.C.; Blizzard, C.A. Amyotrophic lateral sclerosis mutant TDP-43 may cause synaptic dysfunction through altered dendritic spine function. Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Murayama, S.; Bouldin, T.; Suzuki, K. Immunocytochemical and ultrastructural studies of upper motor neurons in amyotrophic lateral sclerosis. Acta Neuropathol. 1992, 83, 518–524. [Google Scholar] [CrossRef]
- Braak, H.; Ludolph, A.C.; Neumann, M.; Ravits, J.; Del Tredici, K. Pathological TDP-43 changes in Betz cells differ from those in bulbar and spinal alpha-motoneurons in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, I.; Arai, T.; Hasegawa, M. The basis of clinicopathological heterogeneity in TDP-43 proteinopathy. Acta Neuropathol. 2019, 138, 751–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hergesheimer, R.C.; Chami, A.A.; de Assis, D.R.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Lanznaster, D.; Blasco, H. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: A resolution in sight? Brain 2019, 142, 1176–1194. [Google Scholar] [CrossRef]
- Wang, I.F.; Wu, L.S.; Chang, H.Y.; Shen, C.K.J. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J. Neurochem. 2008, 105, 797–806. [Google Scholar] [CrossRef]
- Liu-Yesucevitz, L.; Bassell, G.J.; Gitler, A.D.; Hart, A.C.; Klann, E.; Richter, J.D.; Warren, S.T.; Wolozin, B. Local RNA translation at the synapse and in disease. J. Neurosci. 2011, 31, 16086–16093. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, S.; Takeda, T.; Shibata, N.; Kobayashi, M. Alterations in subcellular localization of TDP-43 immunoreactivity in the anterior horns in sporadic amyotrophic lateral sclerosis. Neurosci. Lett. 2010, 478, 72–76. [Google Scholar] [CrossRef]
- Ofer, N.; Berger, D.R.; Kasthuri, N.; Lichtman, J.W.; Yuste, R. Ultrastructural analysis of dendritic spine necks reveals a continuum of spine morphologies. Dev. Neurobiol. 2021. [Google Scholar] [CrossRef]
- Holtmaat, A.J.; Trachtenberg, J.T.; Wilbrecht, L.; Shepherd, G.M.; Zhang, X.; Knott, G.W.; Svoboda, K. Transient and persistent dendritic spines in the neocortex in vivo. Neuron 2005, 45, 279–291. [Google Scholar] [CrossRef] [Green Version]
- Nakahata, Y.; Yasuda, R. Plasticity of Spine Structure: Local Signaling, Translation and Cytoskeletal Reorganization. Front. Synaptic Neurosci. 2018, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, R.K.; Mangelsdorf, M.; Panwar, A.; Butler, T.J.; Noakes, P.G.; Wallace, R.H. Identification of RNA bound to the TDP-43 ribonucleoprotein complex in the adult mouse brain. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Sephton, C.F.; Cenik, C.; Kucukural, A.; Dammer, E.B.; Cenik, B.; Han, Y.; Dewey, C.M.; Roth, F.P.; Herz, J.; Peng, J.; et al. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J. Biol. Chem. 2011, 286, 1204–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geevasinga, N.; Menon, P.; Özdinler, P.H.; Kiernan, M.C.; Vucic, S. Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nat. Rev. Neurol. 2016, 12, 651–661. [Google Scholar] [CrossRef]
- Vucic, S.; Kiernan, M.C. Axonal excitability properties in amyotrophic lateral sclerosis. Clin. Neurophysiol. 2006, 117, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Saba, L.; Viscomi, M.T.; Martini, A.; Caioli, S.; Mercuri, N.B.; Guatteo, E.; Zona, C. Modified age-dependent expression of NaV1.6 in an ALS model correlates with motor cortex excitability alterations. Neurobiol. Dis. 2019, 130, 104532. [Google Scholar] [CrossRef] [PubMed]
- Jamann, N.; Dannehl, D.; Lehmann, N.; Wagener, R.; Thielemann, C.; Schultz, C.; Staiger, J.; Kole, M.H.; Engelhardt, M. Sensory input drives rapid homeostatic scaling of the axon initial segment in mouse barrel cortex. Nat. Commun. 2021, 12, 23. [Google Scholar] [CrossRef] [PubMed]
- Kuba, H.; Oichi, Y.; Ohmori, H. Presynaptic activity regulates Na(+) channel distribution at the axon initial segment. Nature 2010, 465, 1075–1078. [Google Scholar] [CrossRef]
- Breton, J.D.; Stuart, G.J. Loss of sensory input increases the intrinsic excitability of layer 5 pyramidal neurons in rat barrel cortex. J. Physiol. 2009, 587 Pt 21, 5107–5119. [Google Scholar] [CrossRef]
- Turrigiano, G.G.; Leslie, K.R.; Desai, N.S.; Rutherford, L.C.; Nelson, S.B. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 1998, 391, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Siddoway, B.; Hou, H.; Xia, H. Molecular mechanisms of homeostatic synaptic downscaling. Neuropharmacology 2014, 78, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Styr, B.; Slutsky, I. Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer’s disease. Nat. Neurosci. 2018, 21, 463–473. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dyer, M.S.; Woodhouse, A.; Blizzard, C.A. Cytoplasmic Human TDP-43 Mislocalization Induces Widespread Dendritic Spine Loss in Mouse Upper Motor Neurons. Brain Sci. 2021, 11, 883. https://doi.org/10.3390/brainsci11070883
Dyer MS, Woodhouse A, Blizzard CA. Cytoplasmic Human TDP-43 Mislocalization Induces Widespread Dendritic Spine Loss in Mouse Upper Motor Neurons. Brain Sciences. 2021; 11(7):883. https://doi.org/10.3390/brainsci11070883
Chicago/Turabian StyleDyer, Marcus S., Adele Woodhouse, and Catherine A. Blizzard. 2021. "Cytoplasmic Human TDP-43 Mislocalization Induces Widespread Dendritic Spine Loss in Mouse Upper Motor Neurons" Brain Sciences 11, no. 7: 883. https://doi.org/10.3390/brainsci11070883
APA StyleDyer, M. S., Woodhouse, A., & Blizzard, C. A. (2021). Cytoplasmic Human TDP-43 Mislocalization Induces Widespread Dendritic Spine Loss in Mouse Upper Motor Neurons. Brain Sciences, 11(7), 883. https://doi.org/10.3390/brainsci11070883