
Differential Epigenetic Signature of Corticospinal Motor Neurons in ALS

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Tissue Collection and Processing
2.3. Immunocytochemistry
2.4. Imaging
2.5. Quantification and Data Analysis
2.6. Statistical Analyses
3. Results
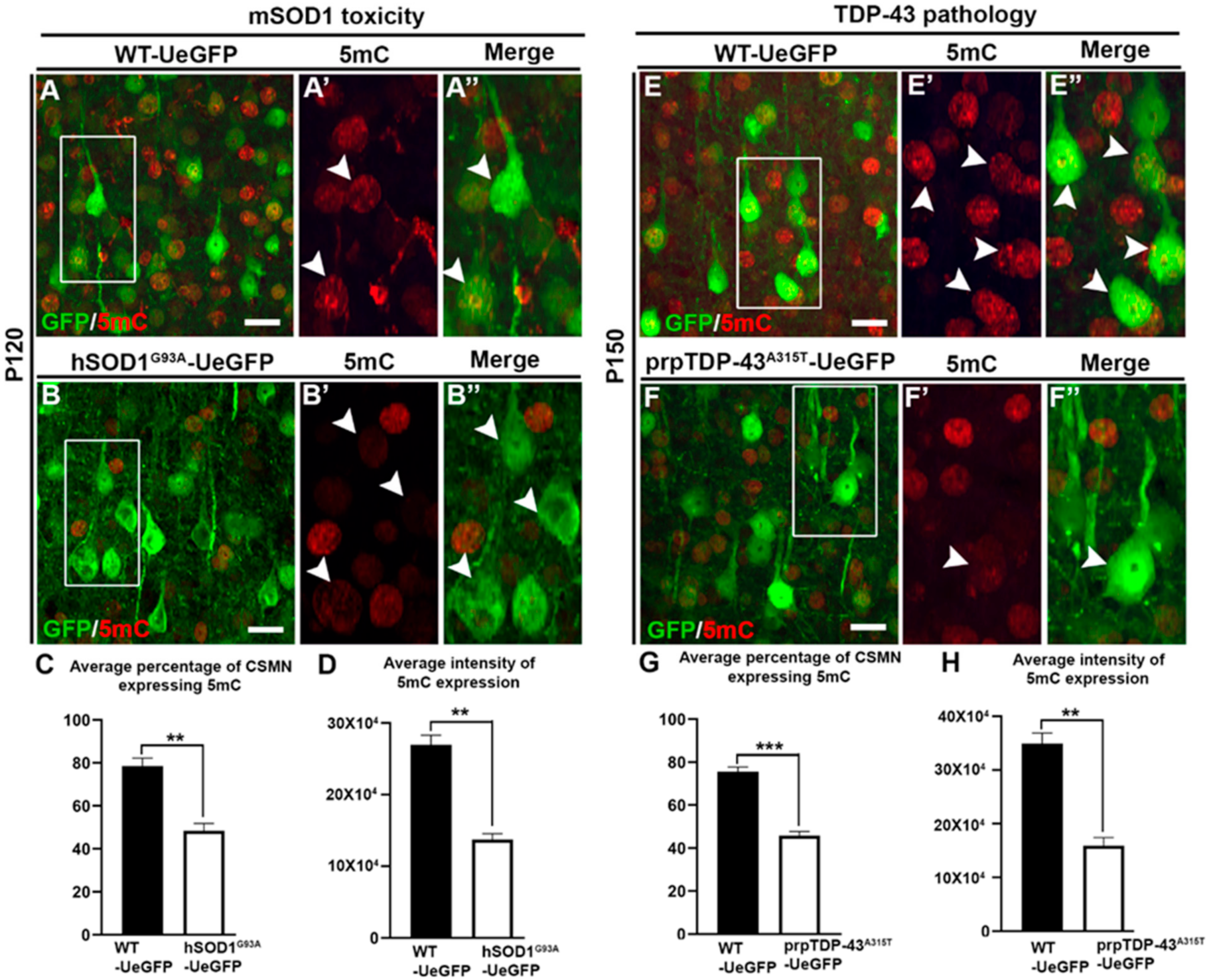
3.1. CSMN Diseased Due to mSOD1 Toxicity and TDP-43 Pathology Displayed Reduced Methylation
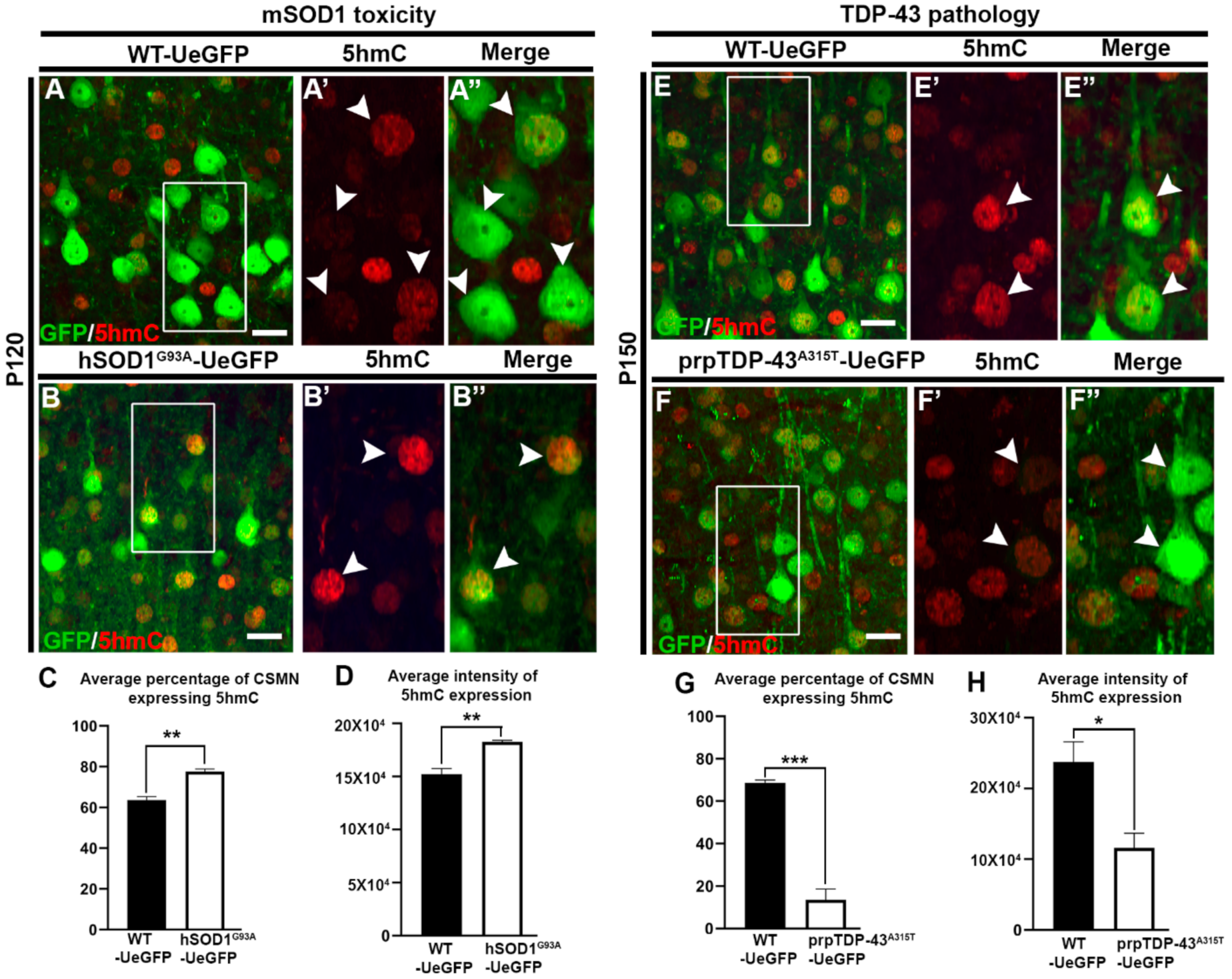
3.2. CSMN Show Increased Hydroxymethylation Due to mSOD1 Toxicity, but Decreased Hydroxymethylation Due to TDP-43 Pathology
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lemon, R.N. Descending pathways in motor control. Annu. Rev. Neurosci. 2008, 31, 195–218. [Google Scholar] [CrossRef] [PubMed]
- Fink, J.K. Progressive spastic paraparesis: Hereditary spastic paraplegia and its relation to primary and amyotrophic lateral sclerosis. Semin. Neurol. 2001, 21, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A. The Dying Forward Hypothesis of ALS: Tracing Its History. Brain Sci. 2021, 11, 300. [Google Scholar] [CrossRef] [PubMed]
- Burg, T.; Bichara, C.; Scekic-Zahirovic, J.; Fischer, M.; Stuart-Lopez, G.; Brunet, A.; Lefebvre, F.; Cordero-Erausquin, M.; Rouaux, C. Absence of Subcerebral Projection Neurons Is Beneficial in a Mouse Model of Amyotrophic Lateral Sclerosis. Ann. Neurol. 2020, 88, 688–702. [Google Scholar] [CrossRef]
- Marques, C.; Burg, T.; Scekic-Zahirovic, J.; Fischer, M.; Rouaux, C. Upper and Lower Motor Neuron Degenerations Are Somatotopically Related and Temporally Ordered in the Sod1 Mouse Model of Amyotrophic Lateral Sclerosis. Brain Sci. 2021, 11, 369. [Google Scholar] [CrossRef]
- Geevasinga, N.; Menon, P.; Ozdinler, P.H.; Kiernan, M.C.; Vucic, S. Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nat. Rev. Neurol. 2016, 12, 651–661. [Google Scholar] [CrossRef]
- Thomsen, G.M.; Gowing, G.; Latter, J.; Chen, M.; Vit, J.P.; Staggenborg, K.; Avalos, P.; Alkaslasi, M.; Ferraiuolo, L.; Likhite, S.; et al. Delayed disease onset and extended survival in the SOD1G93A rat model of amyotrophic lateral sclerosis after suppression of mutant SOD1 in the motor cortex. J. Neurosci. 2014, 34, 15587–15600. [Google Scholar] [CrossRef]
- Dervishi, I.; Gozutok, O.; Murnan, K.; Gautam, M.; Heller, D.; Bigio, E.; Ozdinler, P.H. Protein-protein interactions reveal key canonical pathways, upstream regulators, interactome domains, and novel targets in ALS. Sci. Rep. 2018, 8, 14732. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar] [CrossRef]
- Ozdinler, P.H.; Benn, S.; Yamamoto, T.H.; Guzel, M.; Brown, R.H., Jr.; Macklis, J.D. Corticospinal motor neurons and related subcerebral projection neurons undergo early and specific neurodegeneration in hSOD1G (9)(3)A transgenic ALS mice. J. Neurosci. 2011, 31, 4166–4177. [Google Scholar] [CrossRef]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Gautam, M.; Jara, J.H.; Kocak, N.; Rylaarsdam, L.E.; Kim, K.D.; Bigio, E.H.; Hande Ozdinler, P. Mitochondria, ER, and nuclear membrane defects reveal early mechanisms for upper motor neuron vulnerability with respect to TDP-43 pathology. Acta Neuropathol. 2019, 137, 47–69. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef]
- Robertson, J.; Sanelli, T.; Xiao, S.; Yang, W.; Horne, P.; Hammond, R.; Pioro, E.P.; Strong, M.J. Lack of TDP-43 abnormalities in mutant SOD1 transgenic mice shows disparity with ALS. Neurosci. Lett. 2007, 420, 128–132. [Google Scholar] [CrossRef]
- Guibert, S.; Weber, M. Functions of DNA methylation and hydroxymethylation in mammalian development. Curr. Top. Dev. Biol. 2013, 104, 47–83. [Google Scholar] [CrossRef]
- Cheng, X. Structure and function of DNA methyltransferases. Annu. Rev. Biophys. Biomol. Struct. 1995, 24, 293–318. [Google Scholar] [CrossRef]
- Guo, J.U.; Ma, D.K.; Mo, H.; Ball, M.P.; Jang, M.H.; Bonaguidi, M.A.; Balazer, J.A.; Eaves, H.L.; Xie, B.; Ford, E.; et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 2011, 14, 1345–1351. [Google Scholar] [CrossRef]
- Hashimoto, H.; Liu, Y.; Upadhyay, A.K.; Chang, Y.; Howerton, S.B.; Vertino, P.M.; Zhang, X.; Cheng, X. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 2012, 40, 4841–4849. [Google Scholar] [CrossRef]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef]
- Paez-Colasante, X.; Figueroa-Romero, C.; Sakowski, S.A.; Goutman, S.A.; Feldman, E.L. Amyotrophic lateral sclerosis: Mechanisms and therapeutics in the epigenomic era. Nat. Rev. Neurol. 2015, 11, 266–279. [Google Scholar] [CrossRef]
- Morahan, J.M.; Yu, B.; Trent, R.J.; Pamphlett, R. A genome-wide analysis of brain DNA methylation identifies new candidate genes for sporadic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 10, 418–429. [Google Scholar] [CrossRef]
- Figueroa-Romero, C.; Hur, J.; Bender, D.E.; Delaney, C.E.; Cataldo, M.D.; Smith, A.L.; Yung, R.; Ruden, D.M.; Callaghan, B.C.; Feldman, E.L. Identification of epigenetically altered genes in sporadic amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e52672. [Google Scholar] [CrossRef]
- Appleby-Mallinder, C.; Schaber, E.; Kirby, J.; Shaw, P.J.; Cooper-Knock, J.; Heath, P.R.; Highley, J.R. TDP43 proteinopathy is associated with aberrant DNA methylation in human amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2021, 47, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Esanov, R.; Belle, K.C.; van Blitterswijk, M.; Belzil, V.V.; Rademakers, R.; Dickson, D.W.; Petrucelli, L.; Boylan, K.B.; Dykxhoorn, D.M.; Wuu, J.; et al. C9orf72 promoter hypermethylation is reduced while hydroxymethylation is acquired during reprogramming of ALS patient cells. Exp. Neurol. 2016, 277, 171–177. [Google Scholar] [CrossRef]
- Belzil, V.V.; Bauer, P.O.; Gendron, T.F.; Murray, M.E.; Dickson, D.; Petrucelli, L. Characterization of DNA hypermethylation in the cerebellum of c9FTD/ALS patients. Brain Res. 2014, 1584, 15–21. [Google Scholar] [CrossRef]
- Belzil, V.V.; Bauer, P.O.; Prudencio, M.; Gendron, T.F.; Stetler, C.T.; Yan, I.K.; Pregent, L.; Daughrity, L.; Baker, M.C.; Rademakers, R.; et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013, 126, 895–905. [Google Scholar] [CrossRef]
- Xi, Z.; Zinman, L.; Moreno, D.; Schymick, J.; Liang, Y.; Sato, C.; Zheng, Y.; Ghani, M.; Dib, S.; Keith, J.; et al. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am. J. Hum. Genet. 2013, 92, 981–989. [Google Scholar] [CrossRef]
- Yasvoina, M.V.; Genc, B.; Jara, J.H.; Sheets, P.L.; Quinlan, K.A.; Milosevic, A.; Shepherd, G.M.G.; Heckman, C.J.; Özdinler, P.H. eGFP Expression under UCHL1 promoter genetically labels corticospinal motor neurons and a subpopulation of degeneration-resistant spinal motor neurons in an ALS mouse model. J. Neurosci. 2013, 33, 7890–7904. [Google Scholar] [CrossRef]
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 2011, 31, 16619–16636. [Google Scholar] [CrossRef]
- Andres-Benito, P.; Moreno, J.; Aso, E.; Povedano, M.; Ferrer, I. Amyotrophic lateral sclerosis, gene deregulation in the anterior horn of the spinal cord and frontal cortex area 8: Implications in frontotemporal lobar degeneration. Aging 2017, 9, 823–851. [Google Scholar] [CrossRef] [PubMed]
- Tremolizzo, L.; Messina, P.; Conti, E.; Sala, G.; Cecchi, M.; Airoldi, L.; Pastorelli, R.; Pupillo, E.; Bandettini Di Poggio, M.; Filosto, M.; et al. Whole-blood global DNA methylation is increased in amyotrophic lateral sclerosis independently of age of onset. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 98–105. [Google Scholar] [CrossRef]
- Ebbert, M.T.W.; Ross, C.A.; Pregent, L.J.; Lank, R.J.; Zhang, C.; Katzman, R.B.; Jansen-West, K.; Song, Y.; da Rocha, E.L.; Palmucci, C.; et al. Conserved DNA methylation combined with differential frontal cortex and cerebellar expression distinguishes C9orf72-associated and sporadic ALS, and implicates SERPINA1 in disease. Acta Neuropathol. 2017, 134, 715–728. [Google Scholar] [CrossRef]
- Van Rheenen, W.; Diekstra, F.P.; Harschnitz, O.; Westeneng, H.J.; van Eijk, K.R.; Saris, C.G.J.; Groen, E.J.N.; van Es, M.A.; Blauw, H.M.; van Vught, P.W.J.; et al. Whole blood transcriptome analysis in amyotrophic lateral sclerosis: A biomarker study. PLoS ONE 2018, 13, e0198874. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Beers, D.R.; Hooten, K.G.; Sieglaff, D.H.; Zhang, A.; Kalyana-Sundaram, S.; Traini, C.M.; Halsey, W.S.; Hughes, A.M.; Sathe, G.M.; et al. Characterization of Gene Expression Phenotype in Amyotrophic Lateral Sclerosis Monocytes. JAMA Neurol. 2017, 74, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.W.; Jeong, Y.E.; Wong, M.; Martin, L.J. DNA damage accumulates and responses are engaged in human ALS brain and spinal motor neurons and DNA repair is activatable in iPSC-derived motor neurons with SOD1 mutations. Acta Neuropathol. Commun. 2020, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Stoccoro, A.; Smith, A.R.; Mosca, L.; Marocchi, A.; Gerardi, F.; Lunetta, C.; Cereda, C.; Gagliardi, S.; Lunnon, K.; Migliore, L.; et al. Reduced mitochondrial D-loop methylation levels in sporadic amyotrophic lateral sclerosis. Clin. Epigenet. 2020, 12, 137. [Google Scholar] [CrossRef]
- Tarr, I.S.; McCann, E.P.; Benyamin, B.; Peters, T.J.; Twine, N.A.; Zhang, K.Y.; Zhao, Q.; Zhang, Z.H.; Rowe, D.B.; Nicholson, G.A.; et al. Monozygotic twins and triplets discordant for amyotrophic lateral sclerosis display differential methylation and gene expression. Sci. Rep. 2019, 9, 8254. [Google Scholar] [CrossRef]
- Xi, Z.; Rainero, I.; Rubino, E.; Pinessi, L.; Bruni, A.C.; Maletta, R.G.; Nacmias, B.; Sorbi, S.; Galimberti, D.; Surace, E.I.; et al. Hypermethylation of the CpG-island near the C9orf72 G (4)C(2)-repeat expansion in FTLD patients. Hum. Mol. Genet. 2014, 23, 5630–5637. [Google Scholar] [CrossRef]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011, 14, 1607–1616. [Google Scholar] [CrossRef]
- Szwagierczak, A.; Bultmann, S.; Schmidt, C.S.; Spada, F.; Leonhardt, H. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2010, 38, e181. [Google Scholar] [CrossRef]
- Jin, S.G.; Wu, X.; Li, A.X.; Pfeifer, G.P. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 2011, 39, 5015–5024. [Google Scholar] [CrossRef]
- Wang, T.; Pan, Q.; Lin, L.; Szulwach, K.E.; Song, C.X.; He, C.; Wu, H.; Warren, S.T.; Jin, P.; Duan, R.; et al. Genome-wide DNA hydroxymethylation changes are associated with neurodevelopmental genes in the developing human cerebellum. Hum. Mol. Genet. 2012, 21, 5500–5510. [Google Scholar] [CrossRef]
- Martinez-Iglesias, O.; Carrera, I.; Carril, J.C.; Fernandez-Novoa, L.; Cacabelos, N.; Cacabelos, R. DNA Methylation in Neurodegenerative and Cerebrovascular Disorders. Int. J. Mol. Sci. 2020, 21, 2220. [Google Scholar] [CrossRef]
- Sherwani, S.I.; Khan, H.A. Role of 5-hydroxymethylcytosine in neurodegeneration. Gene 2015, 570, 17–24. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z.; Li, L.; Xu, K.; Ma, Z.; Chow, H.M.; Herrup, K.; Li, J. Selective loss of 5hmC promotes neurodegeneration in the mouse model of Alzheimer’s disease. FASEB J. 2020, 34, 16364–16382. [Google Scholar] [CrossRef]
- Chouliaras, L.; van den Hove, D.L.; Kenis, G.; Keitel, S.; Hof, P.R.; van Os, J.; Steinbusch, H.W.; Schmitz, C.; Rutten, B.P. Age-related increase in levels of 5-hydroxymethylcytosine in mouse hippocampus is prevented by caloric restriction. Curr. Alzheimer Res. 2012, 9, 536–544. [Google Scholar] [CrossRef]
- Martin, L.J.; Chang, Q. DNA Damage Response and Repair, DNA Methylation, and Cell Death in Human Neurons and Experimental Animal Neurons Are Different. J. Neuropathol. Exp. Neurol. 2018, 77, 636–655. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozyurt, T.; Gautam, M. Differential Epigenetic Signature of Corticospinal Motor Neurons in ALS. Brain Sci. 2021, 11, 754. https://doi.org/10.3390/brainsci11060754
Ozyurt T, Gautam M. Differential Epigenetic Signature of Corticospinal Motor Neurons in ALS. Brain Sciences. 2021; 11(6):754. https://doi.org/10.3390/brainsci11060754
Chicago/Turabian StyleOzyurt, Tunch, and Mukesh Gautam. 2021. "Differential Epigenetic Signature of Corticospinal Motor Neurons in ALS" Brain Sciences 11, no. 6: 754. https://doi.org/10.3390/brainsci11060754
APA StyleOzyurt, T., & Gautam, M. (2021). Differential Epigenetic Signature of Corticospinal Motor Neurons in ALS. Brain Sciences, 11(6), 754. https://doi.org/10.3390/brainsci11060754