
Oxidative Dysregulation in Early Life Stress and Posttraumatic Stress Disorder: A Comprehensive Review




Abstract
1. Introduction
2. Oxidative Stress
2.1. Oxidative Phosphorylation
2.2. The Physiological Role of ROS/RNS
2.3. Redox Imbalance and OXS
2.4. Redox System Components
2.4.1. Nitric Oxide (NO) and the Oxidative Phosphorylation Cascade
2.4.2. Glutamate
2.4.3. Intracellular Calcium (Ca)
2.4.4. N-Acetylaspartate
2.4.5. GABA
2.4.6. The RORA System
2.4.7. 12/15-Lipoxygenase
2.4.8. GLO System
2.4.9. Klotho Gene
2.5. Measurement of OXS
3. OXS and Traumatic Stress
3.1. Animal Studies
3.1.1. OXS in Animal Models of ELS
3.1.2. OXS in Animal Models of PTSD
3.1.3. Summary and Considerations on Preclinical Animal Studies
3.2. Human Studies
3.2.1. OXS in Human ELS Studies
3.2.2. OXS in Human PTSD Studies
3.2.3. OXS-Related Genetic Findings in PTSD
3.2.4. Summary and Considerations in Human ELS and PTSD Studies

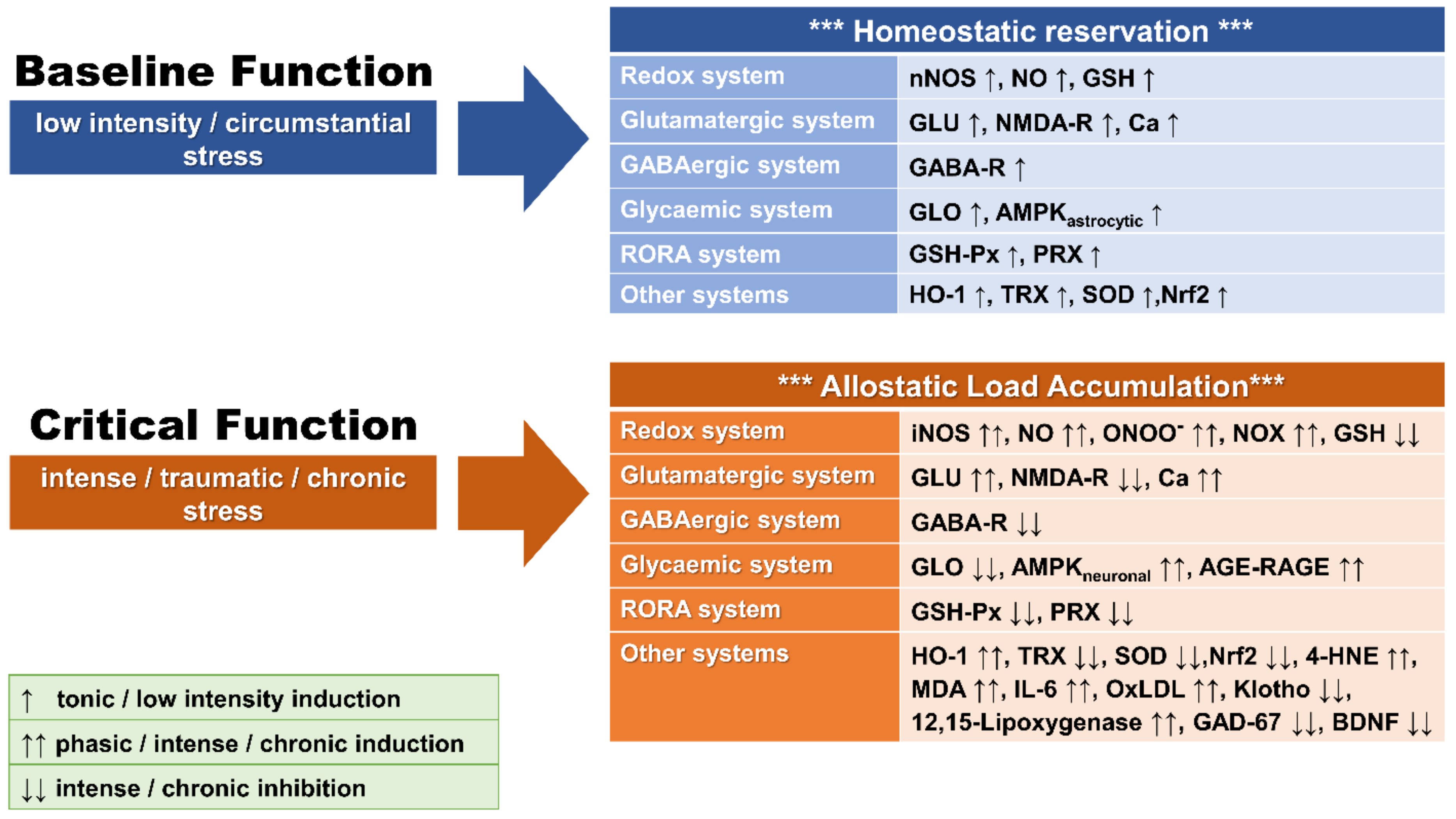{kind=link}
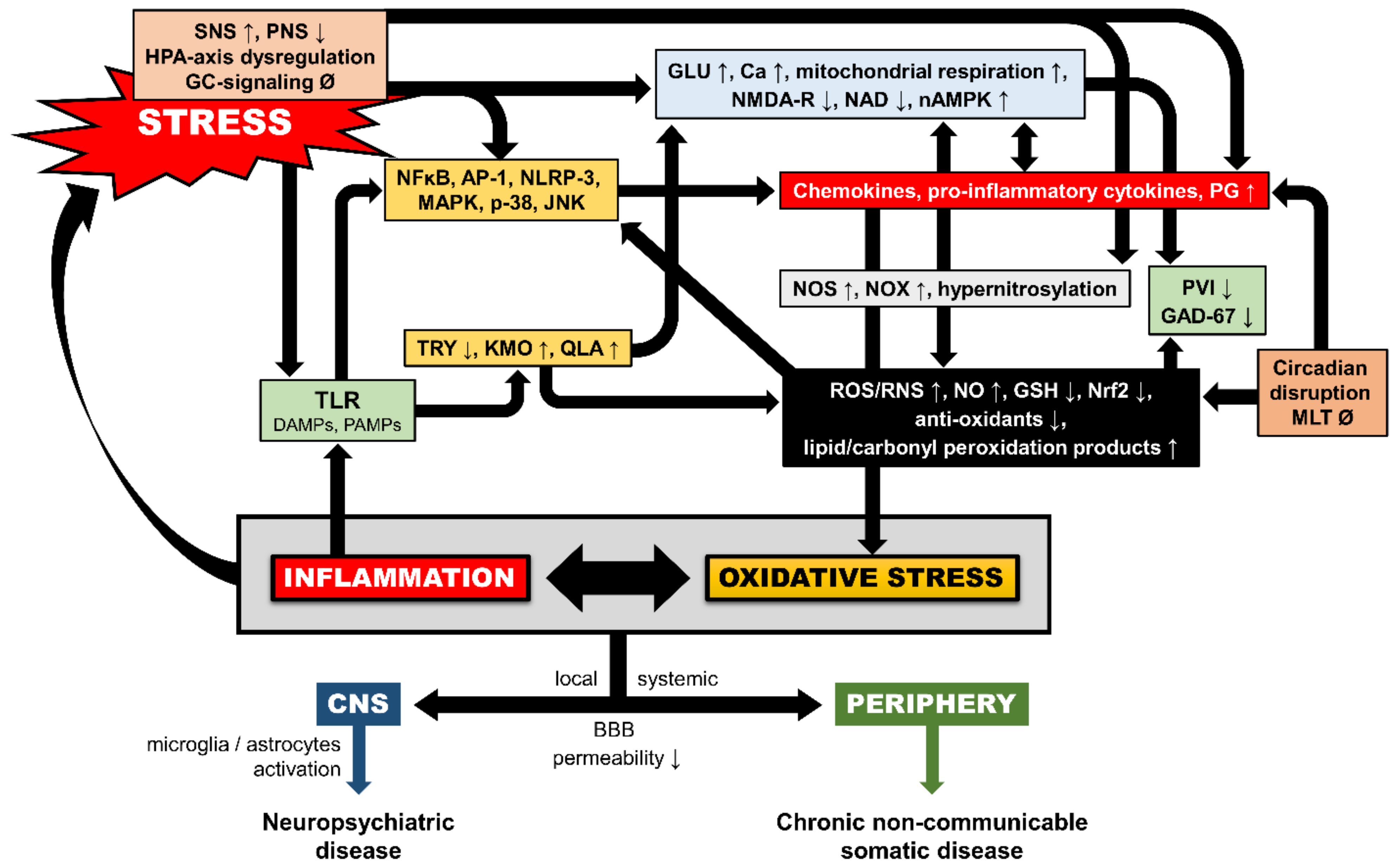{kind=link}
| Redox Index | Outcome | References | ELS/PTSD |
|---|---|---|---|
| ROS/RNS | NO ↑ | [143] | PTSD |
| ONOO− ↑ | [143] | PTSD | |
| Antioxidants levels | GSH ↑ | [167] | PTSD |
| Antioxidant activity | GSH-Px ↓ | [132] [144] | ELS PTSD |
| GSH-Px ↑ | [135] [147] | ELS PTSD | |
| GSH-Rd ↑ | [168,169] | PTSD | |
| SOD ↑ | [132] | ELS | |
| SOD Ø | [147] | PTSD | |
| SOD ↓ | [144] | PTSD | |
| PON-1 ↓ | [146,168,169] | PTSD | |
| HO-1 ↑ | [168,169] | PTSD | |
| TRAP ↓ | [132] | ELS | |
| Redox end-products | Carbonyl ↑ | [132] | ELS |
| MDA ↑ | [146] | PTSD | |
| 8-OH-DG ↑ | [133] | ELS | |
| 8-OH-DG Ø | [166] | PTSD | |
| OxLDL ↑ | [169] | PTSD | |
| Iso-Ps ↑ | [134] | ELS | |
| Thromboxane B2 Ø | [166] | PTSD | |
| Other redox-related parameters | GLU ↑ | [79] | PTSD |
| GLU ↓ | [149] | PTSD | |
| GLU Ø | [167] | PTSD | |
| NAA ↓ | [77,79] | PTSD | |
| NAA Ø | [167] | PTSD | |
| GABA ↑ | [167] | PTSD |
4. Additional Neurobiological Pathways of Trauma-Related OXS
4.1. Inflammation-Induced OXS
4.2. Glucocorticoid-Induced OXS
4.3. Epigenetic Mechanisms
4.4. Circadian Dysregulation and Melatonin
5. PTSD Comorbidities as OXS Moderators
5.1. Depression
5.2. Traumatic Brain Injury (TBI)
5.3. Other Comorbidities
6. Discussion
7. Conclusive Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chrousos, G.P. Stress and disorders of the stress system. Nat. Rev. Endocrinol. 2009, 5, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Chrousos, G.P.; Gold, P.W. The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA 1992, 267, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. Protective and damaging effects of stress mediators. N. Engl. J. Med. 1998, 338, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Agorastos, A.; Pervanidou, P.; Chrousos, G.P.; Baker, D.G. Developmental Trajectories of Early Life Stress and Trauma: A Narrative Review on Neurobiological Aspects Beyond Stress System Dysregulation. Front. Psychiatry 2019, 10, 118. [Google Scholar] [CrossRef]
- Agorastos, A.; Pervanidou, P.; Chrousos, G.P.; Kolaitis, G. Early life stress and trauma: Developmental neuroendocrine aspects of prolonged stress system dysregulation. Hormones 2018, 17, 507–520. [Google Scholar] [CrossRef]
- Frodl, T.; O’Keane, V. How does the brain deal with cumulative stress? A review with focus on developmental stress, HPA axis function and hippocampal structure in humans. Neurobiol. Dis. 2013, 52, 24–37. [Google Scholar] [CrossRef]
- Heim, C.; Nemeroff, C.B. The impact of early adverse experiences on brain systems involved in the pathophysiology of anxiety and affective disorders. Biol. Psychiatry 1999, 46, 1509–1522. [Google Scholar] [CrossRef]
- Lupien, S.J.; McEwen, B.S.; Gunnar, M.R.; Heim, C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 2009, 10, 434–445. [Google Scholar] [CrossRef]
- McEwen, B.S. Physiology and neurobiology of stress and adaptation: Central role of the brain. Physiol. Rev. 2007, 87, 873–904. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association Publishing: Washington, DC, USA, 2013. [Google Scholar]
- Heim, C. Stress, Early Life. In Encyclopedia of Behavioral Medicine; Gellman, M.D., Turner, J.R., Eds.; Springer: New York, NY, USA, 2013. [Google Scholar]
- Reynolds, R.M.; Labad, J.; Buss, C.; Ghaemmaghami, P.; Raikkonen, K. Transmitting biological effects of stress in utero: Implications for mother and offspring. Psychoneuroendocrinology 2013, 38, 1843–1849. [Google Scholar] [CrossRef]
- Danese, A. Annual Research Review: Rethinking childhood trauma-new research directions for measurement, study design and analytical strategies. J. Child. Psychol. Psychiatry 2020, 61, 236–250. [Google Scholar] [CrossRef]
- Felitti, V.J.; Anda, R.F.; Nordenberg, D.; Williamson, D.F.; Spitz, A.M.; Edwards, V.; Koss, M.P.; Marks, J.S. Relationship of childhood abuse and household dysfunction to many of the leading causes of death in adults—The adverse childhood experiences (ACE) study. Am. J. Prev. Med. 1998, 14, 245–258. [Google Scholar] [CrossRef]
- Schiavone, S.; Jaquet, V.; Trabace, L.; Krause, K.H. Severe life stress and oxidative stress in the brain: From animal models to human pathology. Antioxid. Redox Signal. 2013, 18, 1475–1490. [Google Scholar] [CrossRef]
- Yehuda, R. Post-traumatic stress disorder. N. Engl. J. Med. 2002, 346, 108–114. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Schiavone, S.; Colaianna, M.; Curtis, L. Impact of early life stress on the pathogenesis of mental disorders: Relation to brain oxidative stress. Curr. Pharm. Des. 2015, 21, 1404–1412. [Google Scholar] [CrossRef]
- Hovatta, I.; Juhila, J.; Donner, J. Oxidative stress in anxiety and comorbid disorders. Neurosci. Res. 2010, 68, 261–275. [Google Scholar] [CrossRef]
- Mhillaj, E.; Morgese, M.G.; Trabace, L. Early life and oxidative stress in psychiatric disorders: What can we learn from animal models? Curr. Pharm. Des. 2015, 21, 1396–1403. [Google Scholar] [CrossRef]
- Miller, M.W.; Lin, A.P.; Wolf, E.J.; Miller, D.R. Oxidative Stress, Inflammation, and Neuroprogression in Chronic PTSD. Harv. Rev. Psychiatry 2018, 26, 57–69. [Google Scholar] [CrossRef]
- Miller, M.W.; Sadeh, N. Traumatic stress, oxidative stress and post-traumatic stress disorder: Neurodegeneration and the accelerated-aging hypothesis. Mol. Psychiatry 2014. [Google Scholar] [CrossRef]
- Ghaemi Kerahrodi, J.; Michal, M. The fear-defense system, emotions, and oxidative stress. Redox Biol. 2020, 101588. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Algieri, C.; Tioli, G.; Lenaz, G. Molecular and Supramolecular Structure of the Mitochondrial Oxidative Phosphorylation System: Implications for Pathology. Life 2021, 11, 242. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B. Complex IV—The regulatory center of mitochondrial oxidative phosphorylation. Mitochondrion 2020. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.H. Aging: A revisited theory based on free radicals generated by NOX family NADPH oxidases. Exp. Gerontol. 2007, 42, 256–262. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Zuo, L.; Zhou, T.; Pannell, B.K.; Ziegler, A.C.; Best, T.M. Biological and physiological role of reactive oxygen species--the good, the bad and the ugly. Acta Physiol. 2015, 214, 329–348. [Google Scholar] [CrossRef]
- Bao, L.; Avshalumov, M.V.; Patel, J.C.; Lee, C.R.; Miller, E.W.; Chang, C.J.; Rice, M.E. Mitochondria are the source of hydrogen peroxide for dynamic brain-cell signaling. J. Neurosci. 2009, 29, 9002–9010. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Kamsler, A.; Segal, M. Hydrogen peroxide modulation of synaptic plasticity. J. Neurosci. 2003, 23, 269–276. [Google Scholar] [CrossRef]
- Penna, A.; Wang, D.S.; Yu, J.; Lecker, I.; Brown, P.M.; Bowie, D.; Orser, B.A. Hydrogen peroxide increases GABAA receptor-mediated tonic current in hippocampal neurons. J. Neurosci. 2014, 34, 10624–10634. [Google Scholar] [CrossRef]
- Beltran Gonzalez, A.N.; Gasulla, J.; Calvo, D.J. An intracellular redox sensor for reactive oxygen species at the M3-M4 linker of GABAA rho1 receptors. Br. J. Pharm. 2014, 171, 2291–2299. [Google Scholar] [CrossRef]
- Accardi, M.V.; Daniels, B.A.; Brown, P.M.; Fritschy, J.M.; Tyagarajan, S.K.; Bowie, D. Mitochondrial reactive oxygen species regulate the strength of inhibitory GABA-mediated synaptic transmission. Nat. Commun. 2014, 5, 3168. [Google Scholar] [CrossRef]
- Infanger, D.W.; Sharma, R.V.; Davisson, R.L. NADPH oxidases of the brain: Distribution, regulation, and function. Antioxid. Redox Signal. 2006, 8, 1583–1596. [Google Scholar] [CrossRef]
- Garthwaite, J. Concepts of neural nitric oxide-mediated transmission. Eur J. Neurosci. 2008, 27, 2783–2802. [Google Scholar] [CrossRef]
- Steinert, J.R.; Chernova, T.; Forsythe, I.D. Nitric oxide signaling in brain function, dysfunction, and dementia. Neurosci. A Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2010, 16, 435–452. [Google Scholar] [CrossRef]
- Liu, Z.; Ren, Z.; Zhang, J.; Chuang, C.C.; Kandaswamy, E.; Zhou, T.; Zuo, L. Role of ROS and Nutritional Antioxidants in Human Diseases. Front. Physiol. 2018, 9, 477. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Wang, C.H.; Wu, S.B.; Wu, Y.T.; Wei, Y.H. Oxidative stress response elicited by mitochondrial dysfunction: Implication in the pathophysiology of aging. Exp. Biol. Med. 2013, 238, 450–460. [Google Scholar] [CrossRef]
- Haines, D.D.; Juhasz, B.; Tosaki, A. Management of multicellular senescence and oxidative stress. J. Cell Mol. Med. 2013, 17, 936–957. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, Y. Chromosomal DNA fragmentation in apoptosis and necrosis induced by oxidative stress. Biochem. Pharm. 2003, 66, 1527–1535. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Martinez Leo, E.E.; Segura Campos, M.R. Systemic Oxidative Stress: A key Point in Neurodegeneration—A Review. J. Nutr. Health Aging 2019, 23, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Panahi, Y.; Javadi, B.; Sahebkar, A. The Underlying Role of Oxidative Stress in Neurodegeneration: A Mechanistic Review. CNS Neurol. Disord. Drug Targets 2018, 17, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Wadhwa, R.; Gupta, R.; Maurya, P.K. Oxidative Stress and Accelerated Aging in Neurodegenerative and Neuropsychiatric Disorder. Curr. Pharm. Des. 2018, 24, 4711–4725. [Google Scholar] [CrossRef]
- Ng, F.; Berk, M.; Dean, O.; Bush, A.I. Oxidative stress in psychiatric disorders: Evidence base and therapeutic implications. Int. J. Neuropsychopharmacol. Off. Sci. J. Coll. Int. Neuropsychopharmacol. 2008, 11, 851–876. [Google Scholar] [CrossRef]
- Clerc, P.; Rigoulet, M.; Leverve, X.; Fontaine, E. Nitric oxide increases oxidative phosphorylation efficiency. J. Bioenerg. Biomembr. 2007, 39, 158–166. [Google Scholar] [CrossRef]
- Vincent, S.R. Nitric oxide neurons and neurotransmission. Prog. Neurobiol. 2010, 90, 246–255. [Google Scholar] [CrossRef]
- Almeida, A.; Moncada, S.; Bolanos, J.P. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat. Cell Biol. 2004, 6, 45–51. [Google Scholar] [CrossRef]
- Yeo, C.T.; Stancill, J.S.; Oleson, B.J.; Schnuck, J.K.; Stafford, J.D.; Naatz, A.; Hansen, P.A.; Corbett, J.A. Regulation of ATR-dependent DNA damage response by nitric oxide. J. Biol. Chem. 2021, 100388. [Google Scholar] [CrossRef]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Ishikado, A.; Nishio, Y.; Morino, K.; Ugi, S.; Kondo, H.; Makino, T.; Kashiwagi, A.; Maegawa, H. Low concentration of 4-hydroxy hexenal increases heme oxygenase-1 expression through activation of Nrf2 and antioxidative activity in vascular endothelial cells. Biochem. Biophys. Res. Commun. 2010, 402, 99–104. [Google Scholar] [CrossRef]
- Iles, K.E.; Liu, R.M. Mechanisms of glutamate cysteine ligase (GCL) induction by 4-hydroxynonenal. Free Radic. Biol. Med. 2005, 38, 547–556. [Google Scholar] [CrossRef]
- Sedlak, T.W.; Snyder, S.H. Messenger molecules and cell death: Therapeutic implications. JAMA 2006, 295, 81–89. [Google Scholar] [CrossRef]
- Boldyrev, A.; Bulygina, E.; Makhro, A. Glutamate receptors modulate oxidative stress in neuronal cells. A mini-review. Neurotox. Res. 2004, 6, 581–587. [Google Scholar] [CrossRef]
- Jacintho, J.D.; Kovacic, P. Neurotransmission and neurotoxicity by nitric oxide, catecholamines, and glutamate: Unifying themes of reactive oxygen species and electron transfer. Curr. Med. Chem. 2003, 10, 2693–2703. [Google Scholar] [CrossRef]
- Banasr, M.; Chowdhury, G.M.; Terwilliger, R.; Newton, S.S.; Duman, R.S.; Behar, K.L.; Sanacora, G. Glial pathology in an animal model of depression: Reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol. Psychiatry 2010, 15, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Massaad, C.A.; Klann, E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox Signal. 2011, 14, 2013–2054. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pinto-Duarte, A.; Sejnowski, T.J.; Behrens, M.M. How Nox2-containing NADPH oxidase affects cortical circuits in the NMDA receptor antagonist model of schizophrenia. Antioxid. Redox Signal. 2013, 18, 1444–1462. [Google Scholar] [CrossRef] [PubMed]
- Smaili, S.; Hirata, H.; Ureshino, R.; Monteforte, P.T.; Morales, A.P.; Muler, M.L.; Terashima, J.; Oseki, K.; Rosenstock, T.R.; Lopes, G.S.; et al. Calcium and cell death signaling in neurodegeneration and aging. An. Acad. Bras. Cienc. 2009, 81, 467–475. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003, 26, 81–89. [Google Scholar] [CrossRef]
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Varnai, P.; Hajnoczky, G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol. Cell 2016, 63, 240–248. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L.; Snyder, S.H. Molecular mechanisms of nitric oxide actions in the brain. Ann. N. Y. Acad. Sci. 1994, 738, 76–85. [Google Scholar] [CrossRef]
- Matthews, R.P.; Guthrie, C.R.; Wailes, L.M.; Zhao, X.; Means, A.R.; McKnight, G.S. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol. Cell Biol. 1994, 14, 6107–6116. [Google Scholar] [CrossRef]
- Bodhinathan, K.; Kumar, A.; Foster, T.C. Intracellular redox state alters NMDA receptor response during aging through Ca2+/calmodulin-dependent protein kinase II. J. Neurosci. 2010, 30, 1914–1924. [Google Scholar] [CrossRef]
- Ebisu, T.; Rooney, W.D.; Graham, S.H.; Weiner, M.W.; Maudsley, A.A. N-acetylaspartate as an in vivo marker of neuronal viability in kainate-induced status epilepticus: 1H magnetic resonance spectroscopic imaging. J. Cereb. Blood Flow Metab. 1994, 14, 373–382. [Google Scholar] [CrossRef]
- Schuff, N.; Meyerhoff, D.J.; Mueller, S.; Chao, L.; Sacrey, D.T.; Laxer, K.; Weiner, M.W. N-Acetylaspartate as a Marker of Neuronal Injury in Neurodegenerative Disease; Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 2006; Volume 576, pp. 241–262. [Google Scholar] [CrossRef]
- Grachev, I.D.; Kumar, R.; Ramachandran, T.S.; Szeverenyi, N.M. Cognitive interference is associated with neuronal marker N-acetyl aspartate in the anterior cingulate cortex: An in vivo (1)H-MRS study of the Stroop Color-Word task. Mol. Psychiatry 2001, 6, 529–539. [Google Scholar] [CrossRef]
- Schuff, N.; Neylan, T.C.; Fox-Bosetti, S.; Lenoci, M.; Samuelson, K.W.; Studholme, C.; Kornak, J.; Marmar, C.R.; Weiner, M.W. Abnormal N-acetylaspartate in hippocampus and anterior cingulate in posttraumatic stress disorder. Psychiatry Res. 2008, 162, 147–157. [Google Scholar] [CrossRef]
- Clark, J.F.; Doepke, A.; Filosa, J.A.; Wardle, R.L.; Lu, A.; Meeker, T.J.; Pyne-Geithman, G.J. N-acetylaspartate as a reservoir for glutamate. Med. Hypotheses 2006, 67, 506–512. [Google Scholar] [CrossRef]
- Rosso, I.M.; Crowley, D.J.; Silveri, M.M.; Rauch, S.L.; Jensen, J.E. Hippocampus Glutamate and N-Acetyl Aspartate Markers of Excitotoxic Neuronal Compromise in Posttraumatic Stress Disorder. Neuropsychopharmacology 2017, 42, 1698–1705. [Google Scholar] [CrossRef]
- Avoli, M.; Krnjevic, K. The Long and Winding Road to Gamma-Amino-Butyric Acid as Neurotransmitter. Can. J. Neurol. Sci. 2016, 43, 219–226. [Google Scholar] [CrossRef]
- Kann, O.; Huchzermeyer, C.; Kovacs, R.; Wirtz, S.; Schuelke, M. Gamma oscillations in the hippocampus require high complex I gene expression and strong functional performance of mitochondria. Brain 2011, 134, 345–358. [Google Scholar] [CrossRef]
- Calvo, D.J.; Beltran Gonzalez, A.N. Dynamic Regulation of the GABAA Receptor Function by Redox Mechanisms. Mol. Pharm. 2016, 90, 326–333. [Google Scholar] [CrossRef]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef]
- Dafre, A.L.; Rosa, J.M.; Rodrigues, A.L.S.; Cunha, M.P. Multiple cellular targets involved in the antidepressant-like effect of glutathione. Chem. Biol. Interact. 2020, 328, 109195. [Google Scholar] [CrossRef]
- Jetten, A.M.; Kurebayashi, S.; Ueda, E. The ROR nuclear orphan receptor subfamily: Critical regulators of multiple biological processes. Prog. Nucleic Acid Res. Mol. Biol. 2001, 69, 205–247. [Google Scholar] [CrossRef]
- Miller, M.W.; Wolf, E.J.; Logue, M.W.; Baldwin, C.T. The retinoid-related orphan receptor alpha (RORA) gene and fear-related psychopathology. J. Affect. Disord. 2013, 151, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Boukhtouche, F.; Vodjdani, G.; Jarvis, C.I.; Bakouche, J.; Staels, B.; Mallet, J.; Mariani, J.; Lemaigre-Dubreuil, Y.; Brugg, B. Human retinoic acid receptor-related orphan receptor alpha1 overexpression protects neurones against oxidative stress-induced apoptosis. J. Neurochem. 2006, 96, 1778–1789. [Google Scholar] [CrossRef] [PubMed]
- Pallast, S.; Arai, K.; Wang, X.; Lo, E.H.; van Leyen, K. 12/15-Lipoxygenase targets neuronal mitochondria under oxidative stress. J. Neurochem. 2009, 111, 882–889. [Google Scholar] [CrossRef]
- Maessen, D.E.; Stehouwer, C.D.; Schalkwijk, C.G. The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin. Sci. 2015, 128, 839–861. [Google Scholar] [CrossRef] [PubMed]
- Sousa Silva, M.; Gomes, R.A.; Ferreira, A.E.; Ponces Freire, A.; Cordeiro, C. The glyoxalase pathway: The first hundred years... and beyond. Biochem. J. 2013, 453, 1–15. [Google Scholar] [CrossRef]
- Sharma, C.; Kaur, A.; Thind, S.S.; Singh, B.; Raina, S. Advanced glycation End-products (AGEs): An emerging concern for processed food industries. J. Food Sci. Technol. 2015, 52, 7561–7576. [Google Scholar] [CrossRef]
- Shen, C.; Ma, Y.; Zeng, Z.; Yin, Q.; Hong, Y.; Hou, X.; Liu, X. RAGE-Specific Inhibitor FPS-ZM1 Attenuates AGEs-Induced Neuroinflammation and Oxidative Stress in Rat Primary Microglia. Neurochem. Res. 2017, 42, 2902–2911. [Google Scholar] [CrossRef]
- Allaman, I.; Belanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef]
- De Bari, L.; Scire, A.; Minnelli, C.; Cianfruglia, L.; Kalapos, M.P.; Armeni, T. Interplay among Oxidative Stress, Methylglyoxal Pathway and S-Glutathionylation. Antioxidants 2020, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Gurnani, P.; Nandi, A.; Kurosu, H.; Miyoshi, M.; Ogawa, Y.; Castrillon, D.H.; Rosenblatt, K.P.; et al. Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 2005, 280, 38029–38034. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M. Klotho as a regulator of oxidative stress and senescence. Biol. Chem. 2008, 389, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.J.; Morrison, F.G.; Sullivan, D.R.; Logue, M.W.; Guetta, R.E.; Stone, A.; Schichman, S.A.; McGlinchey, R.E.; Milberg, W.P.; Miller, M.W. The goddess who spins the thread of life: Klotho, psychiatric stress, and accelerated aging. Brain Behav. Immun. 2019, 80, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxid Med. Cell Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef]
- Menzel, A.; Samouda, H.; Dohet, F.; Loap, S.; Ellulu, M.S.; Bohn, T. Common and Novel Markers for Measuring Inflammation and Oxidative Stress Ex Vivo in Research and Clinical Practice-Which to Use Regarding Disease Outcomes? Antioxidants 2021, 10, 414. [Google Scholar] [CrossRef]
- Ferrucci, L.; Gonzalez-Freire, M.; Fabbri, E.; Simonsick, E.; Tanaka, T.; Moore, Z.; Salimi, S.; Sierra, F.; de Cabo, R. Measuring biological aging in humans: A quest. Aging Cell 2020, 19, e13080. [Google Scholar] [CrossRef]
- Price, L.H.; Kao, H.T.; Burgers, D.E.; Carpenter, L.L.; Tyrka, A.R. Telomeres and early-life stress: An overview. Biol. Psychiatry 2013, 73, 15–23. [Google Scholar] [CrossRef]
- Ridout, K.K.; Khan, M.; Ridout, S.J. Adverse Childhood Experiences Run Deep: Toxic Early Life Stress, Telomeres, and Mitochondrial DNA Copy Number, the Biological Markers of Cumulative Stress. Bioessays 2018, 40, e1800077. [Google Scholar] [CrossRef]
- Grigoruta, M.; Chavez-Solano, M.; Varela-Ramirez, A.; Sierra-Fonseca, J.A.; Orozco-Lucero, E.; Hamdan, J.N.; Gosselink, K.L.; Martinez-Martinez, A. Maternal separation induces retinal and peripheral blood mononuclear cell alterations across the lifespan of female rats. Brain Res. 2020, 1749, 147117. [Google Scholar] [CrossRef]
- Malcon, L.M.C.; Wearick-Silva, L.E.; Zaparte, A.; Orso, R.; Luft, C.; Tractenberg, S.G.; Donadio, M.V.F.; de Oliveira, J.R.; Grassi-Oliveira, R. Maternal separation induces long-term oxidative stress alterations and increases anxiety-like behavior of male Balb/cJ mice. Exp. Brain Res. 2020, 238, 2097–2107. [Google Scholar] [CrossRef]
- Sahafi, E.; Peeri, M.; Hosseini, M.J.; Azarbyjani, M.A. Cardiac oxidative stress following maternal separation stress was mitigated following adolescent voluntary exercise in adult male rat. Physiol. Behav. 2018, 183, 39–45. [Google Scholar] [CrossRef]
- Ho, D.H.; Burch, M.L.; Musall, B.; Musall, J.B.; Hyndman, K.A.; Pollock, J.S. Early life stress in male mice induces superoxide production and endothelial dysfunction in adulthood. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1267–H1274. [Google Scholar] [CrossRef]
- Khodamoradi, K.; Amini-Khoei, H.; Khosravizadeh, Z.; Hosseini, S.R.; Dehpour, A.R.; Hassanzadeh, G. Oxidative stress, inflammatory reactions and apoptosis mediated the negative effect of chronic stress induced by maternal separation on the reproductive system in male mice. Reprod. Biol. 2019, 19, 340–348. [Google Scholar] [CrossRef]
- Ghatebi, M.; Zavareh, S.; Lashkarbolouki, T.; Elahdadi Salmani, M. Implications from early life stress on the development of mouse ovarian follicles: Focus on oxidative stress. J. Obs. Gynaecol. Res. 2019, 45, 1506–1514. [Google Scholar] [CrossRef]
- Stevens, H.E.; Su, T.; Yanagawa, Y.; Vaccarino, F.M. Prenatal stress delays inhibitory neuron progenitor migration in the developing neocortex. Psychoneuroendocrinology 2013, 38, 509–521. [Google Scholar] [CrossRef]
- Brenhouse, H.C.; Andersen, S.L. Nonsteroidal anti-inflammatory treatment prevents delayed effects of early life stress in rats. Biol. Psychiatry 2011, 70, 434–440. [Google Scholar] [CrossRef]
- Harte, M.K.; Powell, S.B.; Swerdlow, N.R.; Geyer, M.A.; Reynolds, G.P. Deficits in parvalbumin and calbindin immunoreactive cells in the hippocampus of isolation reared rats. J. Neural. Transm. 2007, 114, 893–898. [Google Scholar] [CrossRef]
- Shao, Y.; Yan, G.; Xuan, Y.; Peng, H.; Huang, Q.J.; Wu, R.; Xu, H. Chronic social isolation decreases glutamate and glutamine levels and induces oxidative stress in the rat hippocampus. Behav. Brain Res. 2015, 282, 201–208. [Google Scholar] [CrossRef]
- Reus, G.Z.; Fernandes, G.C.; de Moura, A.B.; Silva, R.H.; Darabas, A.C.; de Souza, T.G.; Abelaira, H.M.; Carneiro, C.; Wendhausen, D.; Michels, M.; et al. Early life experience contributes to the developmental programming of depressive-like behaviour, neuroinflammation and oxidative stress. J. Psychiatr. Res. 2017, 95, 196–207. [Google Scholar] [CrossRef]
- Marasco, V.; Spencer, K.A.; Robinson, J.; Herzyk, P.; Costantini, D. Developmental post-natal stress can alter the effects of pre-natal stress on the adult redox balance. Gen. Comp. Endocrinol. 2013, 191, 239–246. [Google Scholar] [CrossRef]
- Mejia-Carmona, G.E.; Gosselink, K.L.; Perez-Ishiwara, G.; Martinez-Martinez, A. Oxidant/antioxidant effects of chronic exposure to predator odor in prefrontal cortex, amygdala, and hypothalamus. Mol. Cell Biochem. 2015, 406, 121–129. [Google Scholar] [CrossRef]
- Campos, A.C.; Piorino, E.M.; Ferreira, F.R.; Guimaraes, F.S. Increased nitric oxide-mediated neurotransmission in the medial prefrontal cortex is associated with the long lasting anxiogenic-like effect of predator exposure. Behav. Brain Res. 2013, 256, 391–397. [Google Scholar] [CrossRef]
- Wilson, C.B.; McLaughlin, L.D.; Nair, A.; Ebenezer, P.J.; Dange, R.; Francis, J. Inflammation and oxidative stress are elevated in the brain, blood, and adrenal glands during the progression of post-traumatic stress disorder in a predator exposure animal model. PLoS ONE 2013, 8, e76146. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Duan, F.; Wu, J.; Min, Q.; Huang, Q.; Luo, M.; He, Z. Effect of cyclooxygenase2 inhibition on the development of posttraumatic stress disorder in rats. Mol. Med. Rep. 2018, 17, 4925–4932. [Google Scholar] [CrossRef]
- Petrovic, R.; Puskas, L.; Jevtic Dozudic, G.; Stojkovic, T.; Velimirovic, M.; Nikolic, T.; Zivkovic, M.; Djorovic, D.J.; Nenadovic, M.; Petronijevic, N. NADPH oxidase and redox status in amygdala, hippocampus and cortex of male Wistar rats in an animal model of post-traumatic stress disorder. Stress 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xiao, B.; Han, F.; Shi, Y. Metformin Alleviated the Neuronal Oxidative Stress in Hippocampus of Rats under Single Prolonged Stress. J. Mol. Neurosci. 2017, 63, 28–35. [Google Scholar] [CrossRef]
- Peng, Z.; Wang, H.; Zhang, R.; Chen, Y.; Xue, F.; Nie, H.; Chen, Y.; Wu, D.; Wang, Y.; Wang, H.; et al. Gastrodin ameliorates anxiety-like behaviors and inhibits IL-1beta level and p38 MAPK phosphorylation of hippocampus in the rat model of posttraumatic stress disorder. Physiol. Res. 2013, 62, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.R.; Zhang, H.; Zhao, H.T.; Ji, M.H.; Li, H.H.; Wu, J.; Li, K.Y.; Yang, J.J. Amelioration of oxidative stress-induced phenotype loss of parvalbumin interneurons might contribute to the beneficial effects of environmental enrichment in a rat model of post-traumatic stress disorder. Behav. Brain Res. 2016, 312, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Harvey, B.H.; Bothma, T.; Nel, A.; Wegener, G.; Stein, D.J. Involvement of the NMDA receptor, NO-cyclic GMP and nuclear factor K-beta in an animal model of repeated trauma. Hum. Psychopharmacol. 2005, 20, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Harvey, B.H.; Oosthuizen, F.; Brand, L.; Wegener, G.; Stein, D.J. Stress-restress evokes sustained iNOS activity and altered GABA levels and NMDA receptors in rat hippocampus. Psychopharmacology 2004, 175, 494–502. [Google Scholar] [CrossRef]
- Ebenezer, P.J.; Wilson, C.B.; Wilson, L.D.; Nair, A.R.; J, F. The Anti-Inflammatory Effects of Blueberries in an Animal Model of Post-Traumatic Stress Disorder (PTSD). PLoS ONE 2016, 11, e0160923. [Google Scholar] [CrossRef]
- Gautam, A.; D’Arpa, P.; Donohue, D.E.; Muhie, S.; Chakraborty, N.; Luke, B.T.; Grapov, D.; Carroll, E.E.; Meyerhoff, J.L.; Hammamieh, R.; et al. Acute and chronic plasma metabolomic and liver transcriptomic stress effects in a mouse model with features of post-traumatic stress disorder. PLoS ONE 2015, 10, e0117092. [Google Scholar] [CrossRef]
- Daskalakis, N.P.; Cohen, H.; Cai, G.; Buxbaum, J.D.; Yehuda, R. Expression profiling associates blood and brain glucocorticoid receptor signaling with trauma-related individual differences in both sexes. Proc. Natl. Acad. Sci. USA 2014, 111, 13529–13534. [Google Scholar] [CrossRef]
- Oosthuizen, F.; Wegener, G.; Harvey, B.H. Nitric oxide as inflammatory mediator in post-traumatic stress disorder (PTSD): Evidence from an animal model. Neuropsychiatr. Dis. Treat. 2005, 1, 109–123. [Google Scholar] [CrossRef]
- Uysal, N.; Gonenc, S.; Acikgoz, O.; Pekcetin, C.; Kayatekin, B.M.; Sonmez, A.; Semin, I. Age-dependent effects of maternal deprivation on oxidative stress in infant rat brain. Neurosci. Lett. 2005, 384, 98–101. [Google Scholar] [CrossRef]
- Pajovic, S.B.; Pejic, S.; Stojiljkovic, V.; Gavrilovic, L.; Dronjak, S.; Kanazir, D.T. Alterations in hippocampal antioxidant enzyme activities and sympatho-adrenomedullary system of rats in response to different stress models. Physiol. Res. 2006, 55, 453–460. [Google Scholar]
- Djordjevic, J.; Djordjevic, A.; Adzic, M.; Radojcic, M.B. Chronic social isolation compromises the activity of both glutathione peroxidase and catalase in hippocampus of male wistar rats. Cell. Mol. Neurobiol. 2010, 30, 693–700. [Google Scholar] [CrossRef]
- Do Prado, C.H.; Grassi-Oliveira, R.; Wieck, A.; Zaparte, A.; Filho, L.D.; da Silva Morrone, M.; Moreira, J.C.; Bauer, M.E. The impact of childhood maltreatment on redox state: Relationship with oxidative damage and antioxidant defenses in adolescents with no psychiatric disorder. Neurosci. Lett. 2016, 617, 173–177. [Google Scholar] [CrossRef]
- Fanning, J.R.; Lee, R.; Gozal, D.; Coussons-Read, M.; Coccaro, E.F. Childhood trauma and parental style: Relationship with markers of inflammation, oxidative stress, and aggression in healthy and personality disordered subjects. Biol. Psychol. 2015, 112, 56–65. [Google Scholar] [CrossRef]
- Horn, S.R.; Leve, L.D.; Levitt, P.; Fisher, P.A. Childhood adversity, mental health, and oxidative stress: A pilot study. PLoS ONE 2019, 14, e0215085. [Google Scholar] [CrossRef] [PubMed]
- Alameda, L.; Fournier, M.; Khadimallah, I.; Griffa, A.; Cleusix, M.; Jenni, R.; Ferrari, C.; Klauser, P.; Baumann, P.S.; Cuenod, M.; et al. Redox dysregulation as a link between childhood trauma and psychopathological and neurocognitive profile in patients with early psychosis. Proc. Natl. Acad. Sci. USA 2018, 115, 12495–12500. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, B.M.; Carvalho, C.M.; Moretti, P.N.; Mello, M.F.; Belangero, S.I. Stress-related telomere length in children: A systematic review. J. Psychiatr. Res. 2017, 92, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Tyrka, A.R.; Parade, S.H.; Price, L.H.; Kao, H.T.; Porton, B.; Philip, N.S.; Welch, E.S.; Carpenter, L.L. Alterations of Mitochondrial DNA Copy Number and Telomere Length With Early Adversity and Psychopathology. Biol. Psychiatry 2016, 79, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.; Hovatta, I.; Frissa, S.; Goodwin, L.; Hotopf, M.; Hatch, S.L.; Breen, G.; Powell, T.R. Assessing the contributions of childhood maltreatment subtypes and depression case-control status on telomere length reveals a specific role of physical neglect. J. Affect. Disord. 2017, 213, 16–22. [Google Scholar] [CrossRef]
- Ridout, K.K.; Parade, S.H.; Kao, H.T.; Magnan, S.; Seifer, R.; Porton, B.; Price, L.H.; Tyrka, A.R. Childhood maltreatment, behavioral adjustment, and molecular markers of cellular aging in preschool-aged children: A cohort study. Psychoneuroendocrinology 2019, 107, 261–269. [Google Scholar] [CrossRef]
- Savolainen, K.; Eriksson, J.G.; Kananen, L.; Kajantie, E.; Pesonen, A.K.; Heinonen, K.; Raikkonen, K. Associations between early life stress, self-reported traumatic experiences across the lifespan and leukocyte telomere length in elderly adults. Biol. Psychol. 2014, 97, 35–42. [Google Scholar] [CrossRef]
- Xavier, G.; Spindola, L.M.; Ota, V.K.; Carvalho, C.M.; Maurya, P.K.; Tempaku, P.F.; Moretti, P.N.; Mazotti, D.R.; Sato, J.R.; Brietzke, E.; et al. Effect of male-specific childhood trauma on telomere length. J. Psychiatr. Res. 2018, 107, 104–109. [Google Scholar] [CrossRef]
- Beijers, R.; Hartman, S.; Shalev, I.; Hastings, W.; Mattern, B.C.; de Weerth, C.; Belsky, J. Testing three hypotheses about effects of sensitive-insensitive parenting on telomeres. Dev. Psychol. 2020, 56, 237–250. [Google Scholar] [CrossRef]
- Pall, M.L.; Satterlee, J.D. Elevated nitric oxide/peroxynitrite mechanism for the common etiology of multiple chemical sensitivity, chronic fatigue syndrome, and posttraumatic stress disorder. Ann. N. Y. Acad. Sci. 2001, 933, 323–329. [Google Scholar] [CrossRef]
- Borovac Stefanovic, L.; Kalinic, D.; Mimica, N.; Beer Ljubic, B.; Aladrovic, J.; Mandelsamen Perica, M.; Curic, M.; Grosic, P.F.; Delas, I. Oxidative status and the severity of clinical symptoms in patients with post-traumatic stress disorder. Ann. Clin. Biochem. 2015, 52, 95–104. [Google Scholar] [CrossRef]
- Konjevod, M.; Nedic Erjavec, G.; Nikolac Perkovic, M.; Saiz, J.; Tudor, L.; Uzun, S.; Kozumplik, O.; Svob Strac, D.; Zarkovic, N.; Pivac, N. Metabolomics in posttraumatic stress disorder: Untargeted metabolomic analysis of plasma samples from Croatian war veterans. Free Radic. Biol. Med. 2020. [Google Scholar] [CrossRef]
- Atli, A.; Bulut, M.; Bez, Y.; Kaplan, I.; Ozdemir, P.G.; Uysal, C.; Selcuk, H.; Sir, A. Altered lipid peroxidation markers are related to post-traumatic stress disorder (PTSD) and not trauma itself in earthquake survivors. Eur. Arch. Psychiatry Clin. Neurosci. 2016, 266, 329–336. [Google Scholar] [CrossRef]
- Tezcan, E.; Atmaca, M.; Kuloglu, M.; Ustundag, B. Free radicals in patients with post-traumatic stress disorder. Eur. Arch. Psychiatry Clin. Neurosci. 2003, 253, 89–91. [Google Scholar] [CrossRef]
- Averill, L.A.; Purohit, P.; Averill, C.L.; Boesl, M.A.; Krystal, J.H.; Abdallah, C.G. Glutamate dysregulation and glutamatergic therapeutics for PTSD: Evidence from human studies. Neurosci. Lett. 2017, 649, 147–155. [Google Scholar] [CrossRef]
- Harnett, N.G.; Wood, K.H.; Ference, E.W., 3rd; Reid, M.A.; Lahti, A.C.; Knight, A.J.; Knight, D.C. Glutamate/glutamine concentrations in the dorsal anterior cingulate vary with Post-Traumatic Stress Disorder symptoms. J. Psychiatr. Res. 2017, 91, 169–176. [Google Scholar] [CrossRef]
- Ousdal, O.T.; Milde, A.M.; Craven, A.R.; Ersland, L.; Endestad, T.; Melinder, A.; Huys, Q.J.; Hugdahl, K. Prefrontal glutamate levels predict altered amygdala-prefrontal connectivity in traumatized youths. Psychol. Med. 2019, 49, 1822–1830. [Google Scholar] [CrossRef]
- Miller, M.W.; Wolf, E.J.; Sadeh, N.; Logue, M.; Spielberg, J.M.; Hayes, J.P.; Sperbeck, E.; Schichman, S.A.; Stone, A.; Carter, W.C.; et al. A novel locus in the oxidative stress-related gene ALOX12 moderates the association between PTSD and thickness of the prefrontal cortex. Psychoneuroendocrinology 2015, 62, 359–365. [Google Scholar] [CrossRef]
- Glatt, S.J.; Tylee, D.S.; Chandler, S.D.; Pazol, J.; Nievergelt, C.M.; Woelk, C.H.; Baker, D.G.; Lohr, J.B.; Kremen, W.S.; Litz, B.T.; et al. Blood-based gene-expression predictors of PTSD risk and resilience among deployed marines: A pilot study. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2013, 162B, 313–326. [Google Scholar] [CrossRef]
- Tylee, D.S.; Chandler, S.D.; Nievergelt, C.M.; Liu, X.; Pazol, J.; Woelk, C.H.; Lohr, J.B.; Kremen, W.S.; Baker, D.G.; Glatt, S.J.; et al. Blood-based gene-expression biomarkers of post-traumatic stress disorder among deployed marines: A pilot study. Psychoneuroendocrinology 2015, 51, 472–494. [Google Scholar] [CrossRef]
- Bruenig, D.; Morris, C.P.; Mehta, D.; Harvey, W.; Lawford, B.; Young, R.M.; Voisey, J. Nitric oxide pathway genes (NOS1AP and NOS1) are involved in PTSD severity, depression, anxiety, stress and resilience. Gene 2017, 625, 42–48. [Google Scholar] [CrossRef]
- Logue, M.W.; Baldwin, C.; Guffanti, G.; Melista, E.; Wolf, E.J.; Reardon, A.F.; Uddin, M.; Wildman, D.; Galea, S.; Koenen, K.C.; et al. A genome-wide association study of post-traumatic stress disorder identifies the retinoid-related orphan receptor alpha (RORA) gene as a significant risk locus. Mol. Psychiatry 2013, 18, 937–942. [Google Scholar] [CrossRef]
- Lowe, S.R.; Meyers, J.L.; Galea, S.; Aiello, A.E.; Uddin, M.; Wildman, D.E.; Koenen, K.C. RORA and posttraumatic stress trajectories: Main effects and interactions with childhood physical abuse history. Brain Behav. 2015, 5, e00323. [Google Scholar] [CrossRef]
- Li, X.; Wang, J.; Zhou, J.; Huang, P.; Li, J. The association between post-traumatic stress disorder and shorter telomere length: A systematic review and meta-analysis. J. Affect. Disord. 2017, 218, 322–326. [Google Scholar] [CrossRef]
- Kang, J.I.; Mueller, S.G.; Wu, G.W.Y.; Lin, J.; Ng, P.; Yehuda, R.; Flory, J.D.; Abu-Amara, D.; Reus, V.I.; Gautam, A.; et al. Effect of Combat Exposure and Posttraumatic Stress Disorder on Telomere Length and Amygdala Volume. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2020, 5, 678–687. [Google Scholar] [CrossRef]
- Avetyan, D.; Zakharyan, R.; Petrek, M.; Arakelyan, A. Telomere shortening in blood leukocytes of patients with posttraumatic stress disorder. J. Psychiatr. Res. 2019, 111, 83–88. [Google Scholar] [CrossRef]
- Zieker, J.; Zieker, D.; Jatzko, A.; Dietzsch, J.; Nieselt, K.; Schmitt, A.; Bertsch, T.; Fassbender, K.; Spanagel, R.; Northoff, H.; et al. Differential gene expression in peripheral blood of patients suffering from post-traumatic stress disorder. Mol. Psychiatry 2007, 12, 116–118. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.Y.; Kim, S.J.; Choi, J.R.; Lee, S.T.; Kim, J.; Hwang, I.S.; Chung, H.G.; Choi, J.H.; Kim, H.W.; Kim, S.H.; et al. The effect of trauma and PTSD on telomere length: An exploratory study in people exposed to combat trauma. Sci. Rep. 2017, 7, 4375. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.Y.; Levin, Y.; Uziel, O.; Abumock, H.; Solomon, Z. Traumatic stress and cellular senescence: The role of war-captivity and homecoming stressors in later life telomere length. J. Affect. Disord. 2018, 238, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.L.; Koenen, K.C.; Chen, Q.; Gilsanz, P.; Mason, S.M.; Prescott, J.; Ratanatharathorn, A.; Rimm, E.B.; Sumner, J.A.; Winning, A.; et al. Posttraumatic stress disorder and accelerated aging: PTSD and leukocyte telomere length in a sample of civilian women. Depress. Anxiety 2017, 34, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.B.; Leite-Morris, K.A.; Wang, L.; Rumbika, K.K.; Heinrichs, S.C.; Zeng, X.; Wu, L.; Arena, D.T.; Teng, Y.D. Pathophysiological Bases of Comorbidity: Traumatic Brain Injury and Post-Traumatic Stress Disorder. J. Neurotrauma 2018, 35, 210–225. [Google Scholar] [CrossRef]
- Loignon, A.; Ouellet, M.C.; Belleville, G. A Systematic Review and Meta-analysis on PTSD Following TBI Among Military/Veteran and Civilian Populations. J. Head Trauma Rehabil. 2020, 35, E21–E35. [Google Scholar] [CrossRef]
- Ceprnja, M.; Derek, L.; Unic, A.; Blazev, M.; Fistonic, M.; Kozaric-Kovacic, D.; Franic, M.; Romic, Z. Oxidative stress markers in patients with post-traumatic stress disorder. Coll. Antropol. 2011, 35, 1155–1160. [Google Scholar]
- Michels, L.; Schulte-Vels, T.; Schick, M.; O’Gorman, R.L.; Zeffiro, T.; Hasler, G.; Mueller-Pfeiffer, C. Prefrontal GABA and glutathione imbalance in posttraumatic stress disorder: Preliminary findings. Psychiatry Res. 2014, 224, 288–295. [Google Scholar] [CrossRef]
- Oglodek, E.A. The role of PON-1, GR, IL-18, and OxLDL in depression with and without posttraumatic stress disorder. Pharmacol. Rep. Pr. 2017, 69, 837–845. [Google Scholar] [CrossRef]
- Oglodek, E.A. Changes in the concentrations of inflammatory and oxidative status biomediators (MIP-1 alpha, PMN elastase, MDA, and IL-12) in depressed patients with and without posttraumatic stress disorder. Pharmacol. Rep. Pr. 2018, 70, 110–118. [Google Scholar] [CrossRef]
- Czarny, P.; Wigner, P.; Galecki, P.; Sliwinski, T. The interplay between inflammation, oxidative stress, DNA damage, DNA repair and mitochondrial dysfunction in depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 80, 309–321. [Google Scholar] [CrossRef]
- Iwata, M.; Ota, K.T.; Duman, R.S. The inflammasome: Pathways linking psychological stress, depression, and systemic illnesses. Brain Behav. Immun. 2013, 31, 105–114. [Google Scholar] [CrossRef]
- Baumeister, D.; Akhtar, R.; Ciufolini, S.; Pariante, C.M.; Mondelli, V. Childhood trauma and adulthood inflammation: A meta-analysis of peripheral C-reactive protein, interleukin-6 and tumour necrosis factor-alpha. Mol. Psychiatry 2016, 21, 642–649. [Google Scholar] [CrossRef]
- Danese, A.; Caspi, A.; Williams, B.; Ambler, A.; Sugden, K.; Mika, J.; Werts, H.; Freeman, J.; Pariante, C.M.; Moffitt, T.E.; et al. Biological embedding of stress through inflammation processes in childhood. Mol. Psychiatry 2011, 16, 244–246. [Google Scholar] [CrossRef]
- Danese, A.; Pariante, C.M.; Caspi, A.; Taylor, A.; Poulton, R. Childhood maltreatment predicts adult inflammation in a life-course study. Proc. Natl. Acad. Sci. USA 2007, 104, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Danese, A.; Lewis, S.J. Psychoneuroimmunology of Early-Life Stress: The Hidden Wounds of Childhood Trauma? Neuropsychopharmacology 2017, 42, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Coelho, R.; Viola, T.W.; Walss-Bass, C.; Brietzke, E.; Grassi-Oliveira, R. Childhood maltreatment and inflammatory markers: A systematic review. Acta Psychiatr. Scand. 2014, 129, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.D.; Lee, S.; Yoon, S. Inflammation in Post-Traumatic Stress Disorder (PTSD): A Review of Potential Correlates of PTSD with a Neurological Perspective. Antioxidants 2020, 9, 107. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Jiang, W. Immune biomarkers alterations in post-traumatic stress disorder: A systematic review and meta-analysis. J. Affect. Disord. 2020, 268, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Speer, K.; Upton, D.; Semple, S.; McKune, A. Systemic low-grade inflammation in post-traumatic stress disorder: A systematic review. J. Inflamm. Res. 2018, 11, 111–121. [Google Scholar] [CrossRef]
- Wahl, S.M.; McCartney-Francis, N.; Chan, J.; Dionne, R.; Ta, L.; Orenstein, J.M. Nitric oxide in experimental joint inflammation. Benefit or detriment? Cells Tissues Organs 2003, 174, 26–33. [Google Scholar] [CrossRef]
- Wu, F.; Tyml, K.; Wilson, J.X. iNOS expression requires NADPH oxidase-dependent redox signaling in microvascular endothelial cells. J. Cell Physiol. 2008, 217, 207–214. [Google Scholar] [CrossRef]
- Brown, G.C. Mechanisms of inflammatory neurodegeneration: INOS and NADPH oxidase. Biochem. Soc. Trans. 2007, 35, 1119–1121. [Google Scholar] [CrossRef]
- Krakauer, T. Inflammasomes, Autophagy, and Cell Death: The Trinity of Innate Host Defense against Intracellular Bacteria. Mediat. Inflamm. 2019, 2019, 2471215. [Google Scholar] [CrossRef]
- Gill, R.; Tsung, A.; Billiar, T. Linking oxidative stress to inflammation: Toll-like receptors. Free Radic. Biol. Med. 2010, 48, 1121–1132. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, D.; Song, P.; Zou, M.H. Tryptophan-kynurenine pathway is dysregulated in inflammation, and immune activation. Front. Biosci. 2015, 20, 1116–1143. [Google Scholar] [CrossRef]
- Garrison, A.M.; Parrott, J.M.; Tunon, A.; Delgado, J.; Redus, L.; O’Connor, J.C. Kynurenine pathway metabolic balance influences microglia activity: Targeting kynurenine monooxygenase to dampen neuroinflammation. Psychoneuroendocrinology 2018, 94, 1–10. [Google Scholar] [CrossRef]
- Braidy, N.; Grant, R.; Adams, S.; Brew, B.J.; Guillemin, G.J. Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox. Res. 2009, 16, 77–86. [Google Scholar] [CrossRef]
- Steullet, P.; Cabungcal, J.H.; Monin, A.; Dwir, D.; O’Donnell, P.; Cuenod, M.; Do, K.Q. Redox dysregulation, neuroinflammation, and NMDA receptor hypofunction: A "central hub" in schizophrenia pathophysiology? Schizophr. Res. 2016, 176, 41–51. [Google Scholar] [CrossRef]
- Speer, K.E.; Semple, S.; Naumovski, N.; D’Cunha, N.M.; McKune, A.J. HPA axis function and diurnal cortisol in post-traumatic stress disorder: A systematic review. Neurobiol. Stress 2019, 11, 100180. [Google Scholar] [CrossRef]
- Dunlop, B.W.; Wong, A. The hypothalamic-pituitary-adrenal axis in PTSD: Pathophysiology and treatment interventions. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 89, 361–379. [Google Scholar] [CrossRef]
- Sapolsky, R.M.; Romero, L.M.; Munck, A.U. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr. Rev. 2000, 21, 55–89. [Google Scholar] [CrossRef]
- Huo, Y.; Rangarajan, P.; Ling, E.A.; Dheen, S.T. Dexamethasone inhibits the Nox-dependent ROS production via suppression of MKP-1-dependent MAPK pathways in activated microglia. BMC Neurosci. 2011, 12, 49. [Google Scholar] [CrossRef]
- Long, F.; Wang, Y.X.; Liu, L.; Zhou, J.; Cui, R.Y.; Jiang, C.L. Rapid nongenomic inhibitory effects of glucocorticoids on phagocytosis and superoxide anion production by macrophages. Steroids 2005, 70, 55–61. [Google Scholar] [CrossRef]
- MacPherson, A.; Dinkel, K.; Sapolsky, R. Glucocorticoids worsen excitotoxin-induced expression of pro-inflammatory cytokines in hippocampal cultures. Exp. Neurol. 2005, 194, 376–383. [Google Scholar] [CrossRef]
- Frank, M.G.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Stress-induced neuroinflammatory priming: A liability factor in the etiology of psychiatric disorders. Neurobiol. Stress 2016, 4, 62–70. [Google Scholar] [CrossRef]
- Joergensen, A.; Broedbaek, K.; Weimann, A.; Semba, R.D.; Ferrucci, L.; Joergensen, M.B.; Poulsen, H.E. Association between urinary excretion of cortisol and markers of oxidatively damaged DNA and RNA in humans. PLoS ONE 2011, 6, e20795. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, L.J.; Sapolsky, R.M. Glucocorticoids may enhance oxygen radical-mediated neurotoxicity. Neurotoxicology 1996, 17, 873–882. [Google Scholar] [PubMed]
- Sato, H.; Takahashi, T.; Sumitani, K.; Takatsu, H.; Urano, S. Glucocorticoid Generates ROS to Induce Oxidative Injury in the Hippocampus, Leading to Impairment of Cognitive Function of Rats. J. Clin. Biochem. Nutr. 2010, 47, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Costantini, D.; Marasco, V.; Moller, A.P. A meta-analysis of glucocorticoids as modulators of oxidative stress in vertebrates. J. Comp. Physiol. B 2011, 181, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Logue, M.W.; Smith, A.K.; Baldwin, C.; Wolf, E.J.; Guffanti, G.; Ratanatharathorn, A.; Stone, A.; Schichman, S.A.; Humphries, D.; Binder, E.B.; et al. An analysis of gene expression in PTSD implicates genes involved in the glucocorticoid receptor pathway and neural responses to stress. Psychoneuroendocrinology 2015, 57, 1–13. [Google Scholar] [CrossRef]
- Fuchikami, M.; Yamamoto, S.; Morinobu, S.; Takei, S.; Yamawaki, S. Epigenetic regulation of BDNF gene in response to stress. Psychiatry Investig. 2010, 7, 251–256. [Google Scholar] [CrossRef]
- Rodrigues, G.M., Jr.; Toffoli, L.V.; Manfredo, M.H.; Francis-Oliveira, J.; Silva, A.S.; Raquel, H.A.; Martins-Pinge, M.C.; Moreira, E.G.; Fernandes, K.B.; Pelosi, G.G.; et al. Acute stress affects the global DNA methylation profile in rat brain: Modulation by physical exercise. Behav. Brain Res. 2015, 279, 123–128. [Google Scholar] [CrossRef]
- Li, S.; Papale, L.A.; Zhang, Q.; Madrid, A.; Chen, L.; Chopra, P.; Keles, S.; Jin, P.; Alisch, R.S. Genome-wide alterations in hippocampal 5-hydroxymethylcytosine links plasticity genes to acute stress. Neurobiol. Dis. 2016, 86, 99–108. [Google Scholar] [CrossRef][Green Version]
- Li, S.; Papale, L.A.; Kintner, D.B.; Sabat, G.; Barrett-Wilt, G.A.; Cengiz, P.; Alisch, R.S. Hippocampal increase of 5-hmC in the glucocorticoid receptor gene following acute stress. Behav. Brain Res. 2015, 286, 236–240. [Google Scholar] [CrossRef]
- Nasca, C.; Zelli, D.; Bigio, B.; Piccinin, S.; Scaccianoce, S.; Nistico, R.; McEwen, B.S. Stress dynamically regulates behavior and glutamatergic gene expression in hippocampus by opening a window of epigenetic plasticity. Proc. Natl. Acad. Sci. USA 2015, 112, 14960–14965. [Google Scholar] [CrossRef]
- Yehuda, R.; Daskalakis, N.P.; Bierer, L.M.; Bader, H.N.; Klengel, T.; Holsboer, F.; Binder, E.B. Holocaust Exposure Induced Intergenerational Effects on FKBP5 Methylation. Biol. Psychiatry 2016, 80, 372–380. [Google Scholar] [CrossRef]
- Yehuda, R.; Flory, J.D.; Bierer, L.M.; Henn-Haase, C.; Lehrner, A.; Desarnaud, F.; Makotkine, I.; Daskalakis, N.P.; Marmar, C.R.; Meaney, M.J. Lower methylation of glucocorticoid receptor gene promoter 1F in peripheral blood of veterans with posttraumatic stress disorder. Biol. Psychiatry 2015, 77, 356–364. [Google Scholar] [CrossRef]
- Yehuda, R.; Daskalakis, N.P.; Lehrner, A.; Desarnaud, F.; Bader, H.N.; Makotkine, I.; Flory, J.D.; Bierer, L.M.; Meaney, M.J. Influences of maternal and paternal PTSD on epigenetic regulation of the glucocorticoid receptor gene in Holocaust survivor offspring. Am. J. Psychiatry 2014, 171, 872–880. [Google Scholar] [CrossRef]
- Blouin, A.M.; Sillivan, S.E.; Joseph, N.F.; Miller, C.A. The potential of epigenetics in stress-enhanced fear learning models of PTSD. Learn. Mem. 2016, 23, 576–586. [Google Scholar] [CrossRef]
- Wolf, E.J.; Logue, M.W.; Zhao, X.; Daskalakis, N.P.; Morrison, F.G.; Escarfulleri, S.; Stone, A.; Schichman, S.A.; McGlinchey, R.E.; Milberg, W.P.; et al. PTSD and the klotho longevity gene: Evaluation of longitudinal effects on inflammation via DNA methylation. Psychoneuroendocrinology 2020, 117, 104656. [Google Scholar] [CrossRef]
- Spoormaker, V.I.; Montgomery, P. Disturbed sleep in post-traumatic stress disorder: Secondary symptom or core feature? Sleep Med. Rev. 2008, 12, 169–184. [Google Scholar] [CrossRef]
- Germain, A. Sleep disturbances as the hallmark of PTSD: Where are we now? Am. J. Psychiatry 2013, 170, 372–382. [Google Scholar] [CrossRef]
- Agorastos, A.; Kellner, M.; Baker, D.G.; Otte, C. When time stands still. An integrative review on the role of chronodisruption in PTSD. Curr. Opin. Psychiatry 2014, 27, 385–392. [Google Scholar] [CrossRef]
- Agorastos, A.; Olff, M. Traumatic stress and the circadian system: Neurobiology, timing and treatment of posttraumatic chronodisruption. Eur J. Psychotraumatol. 2020, 11, 1833644. [Google Scholar] [CrossRef]
- Agorastos, A.; Nicolaides, N.C.; Bozikas, V.P.; Chrousos, G.P.; Pervanidou, P. Multilevel Interactions of Stress and Circadian System: Implications for Traumatic Stress. Front. Psychiatry 2019, 10, 1003. [Google Scholar] [CrossRef]
- Patel, S.A.; Velingkaar, N.S.; Kondratov, R.V. Transcriptional control of antioxidant defense by the circadian clock. Antioxid. Redox Signal. 2014, 20, 2997–3006. [Google Scholar] [CrossRef]
- Hardeland, R.; Coto-Montes, A.; Poeggeler, B. Circadian rhythms, oxidative stress, and antioxidative defense mechanisms. Chronobiol. Int. 2003, 20, 921–962. [Google Scholar] [CrossRef]
- Alzoubi, K.H.; Khabour, O.F.; Salah, H.A.; Abu Rashid, B.E. The combined effect of sleep deprivation and Western diet on spatial learning and memory: Role of BDNF and oxidative stress. J. Mol. Neurosci. 2013, 50, 124–133. [Google Scholar] [CrossRef]
- Villafuerte, G.; Miguel-Puga, A.; Rodriguez, E.M.; Machado, S.; Manjarrez, E.; Arias-Carrion, O. Sleep deprivation and oxidative stress in animal models: A systematic review. Oxid. Med. Cell. Longev. 2015, 2015, 234952. [Google Scholar] [CrossRef]
- Gulec, M.; Ozkol, H.; Selvi, Y.; Tuluce, Y.; Aydin, A.; Besiroglu, L.; Ozdemir, P.G. Oxidative stress in patients with primary insomnia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 37, 247–251. [Google Scholar] [CrossRef]
- Atrooz, F.; Salim, S. Sleep deprivation, oxidative stress and inflammation. Adv. Protein Chem. Struct. Biol. 2020, 119, 309–336. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Stubbs, B.; Kohler, C.A.; Walder, K.; Slyepchenko, A.; Berk, M.; Carvalho, A.F. The putative role of oxidative stress and inflammation in the pathophysiology of sleep dysfunction across neuropsychiatric disorders: Focus on chronic fatigue syndrome, bipolar disorder and multiple sclerosis. Sleep Med. Rev. 2018, 41, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Manchester, L.C.; Coto-Montes, A.; Boga, J.A.; Andersen, L.P.; Zhou, Z.; Galano, A.; Vriend, J.; Tan, D.X.; Reiter, R.J. Melatonin: An ancient molecule that makes oxygen metabolically tolerable. J. Pineal Res. 2015, 59, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Esteban-Zubero, E.; Zhou, Z.; Reiter, R.J. Melatonin as a Potent and Inducible Endogenous Antioxidant: Synthesis and Metabolism. Molecules 2015, 20, 18886–18906. [Google Scholar] [CrossRef]
- Garcia, J.J.; Lopez-Pingarron, L.; Almeida-Souza, P.; Tres, A.; Escudero, P.; Garcia-Gil, F.A.; Tan, D.X.; Reiter, R.J.; Ramirez, J.M.; Bernal-Perez, M. Protective effects of melatonin in reducing oxidative stress and in preserving the fluidity of biological membranes: A review. J. Pineal Res. 2014, 56, 225–237. [Google Scholar] [CrossRef]
- Galano, A.; Medina, M.E.; Tan, D.X.; Reiter, R.J. Melatonin and its metabolites as copper chelating agents and their role in inhibiting oxidative stress: A physicochemical analysis. J. Pineal Res. 2015, 58, 107–116. [Google Scholar] [CrossRef]
- Galano, A.; Tan, D.X.; Reiter, R.J. Melatonin as a natural ally against oxidative stress: A physicochemical examination. J. Pineal Res. 2011, 51, 1–16. [Google Scholar] [CrossRef]
- Hardeland, R.; Cardinali, D.P.; Srinivasan, V.; Spence, D.W.; Brown, G.M.; Pandi-Perumal, S.R. Melatonin--a pleiotropic, orchestrating regulator molecule. Prog. Neurobiol. 2011, 93, 350–384. [Google Scholar] [CrossRef]
- Hardeland, R.; Cardinali, D.P.; Brown, G.M.; Pandi-Perumal, S.R. Melatonin and brain inflammaging. Prog. Neurobiol. 2015. [Google Scholar] [CrossRef]
- Korkmaz, A.; Rosales-Corral, S.; Reiter, R.J. Gene regulation by melatonin linked to epigenetic phenomena. Gene 2012, 503, 1–11. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Galano, A. Melatonin: Exceeding expectations. Physiology 2014, 29, 325–333. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Kim, S.J.; Cruz, M.H. Delivery of pineal melatonin to the brain and SCN: Role of canaliculi, cerebrospinal fluid, tanycytes and Virchow-Robin perivascular spaces. Brain Struct. Funct. 2014, 219, 1873–1887. [Google Scholar] [CrossRef]
- McFarlane, A.C.; Barton, C.A.; Briggs, N.; Kennaway, D.J. The relationship between urinary melatonin metabolite excretion and posttraumatic symptoms following traumatic injury. J. Affect. Disord. 2010, 127, 365–369. [Google Scholar] [CrossRef]
- Ali, T.; Badshah, H.; Kim, T.H.; Kim, M.O. Melatonin attenuates D-galactose-induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NF-K B/JNK signaling pathway in aging mouse model. J. Pineal Res. 2015, 58, 71–85. [Google Scholar] [CrossRef]
- Sainz, R.M.; Mayo, J.C.; Reiter, R.J.; Antolin, I.; Esteban, M.M.; Rodriguez, C. Melatonin regulates glucocorticoid receptor: An answer to its antiapoptotic action in thymus. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1999, 13, 1547–1556. [Google Scholar] [CrossRef]
- Quiros, I.; Mayo, J.C.; Garcia-Suarez, O.; Hevia, D.; Martin, V.; Rodriguez, C.; Sainz, R.M. Melatonin prevents glucocorticoid inhibition of cell proliferation and toxicity in hippocampal cells by reducing glucocorticoid receptor nuclear translocation. J. Steroid Biochem. Mol. Biol. 2008, 110, 116–124. [Google Scholar] [CrossRef]
- Yadav, S.K.; Haldar, C. Experimentally induced stress, oxidative load and changes in immunity in a tropical wild bird, Perdicula asiatica: Involvement of melatonin and glucocorticoid receptors. Zoology 2014, 117, 261–268. [Google Scholar] [CrossRef]
- Ekthuwapranee, K.; Sotthibundhu, A.; Tocharus, C.; Govitrapong, P. Melatonin ameliorates dexamethasone-induced inhibitory effects on the proliferation of cultured progenitor cells obtained from adult rat hippocampus. J. Steroid Biochem. Mol. Biol. 2015, 145, 38–48. [Google Scholar] [CrossRef]
- Tongjaroenbuangam, W.; Ruksee, N.; Mahanam, T.; Govitrapong, P. Melatonin attenuates dexamethasone-induced spatial memory impairment and dexamethasone-induced reduction of synaptic protein expressions in the mouse brain. Neurochem. Int. 2013, 63, 482–491. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, H.Q.; Liang, X.Y.; Zhang, H.F.; Zhang, T.; Liu, F.E. Melatonin ameliorates cognitive impairment induced by sleep deprivation in rats: Role of oxidative stress, BDNF and CaMKII. Behav. Brain Res. 2013, 256, 72–81. [Google Scholar] [CrossRef]
- Ballenger, J.C.; Davidson, J.R.; Lecrubier, Y.; Nutt, D.J.; Foa, E.B.; Kessler, R.C.; McFarlane, A.C.; Shalev, A.Y. Consensus statement on posttraumatic stress disorder from the International Consensus Group on Depression and Anxiety. J. Clin. Psychiatry 2000, 61 (Suppl. 5), 60–66. [Google Scholar]
- Maul, S.; Giegling, I.; Fabbri, C.; Corponi, F.; Serretti, A.; Rujescu, D. Genetics of resilience: Implications from genome-wide association studies and candidate genes of the stress response system in posttraumatic stress disorder and depression. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2020, 183, 77–94. [Google Scholar] [CrossRef]
- Rohleder, N.; Wolf, J.M.; Wolf, O.T. Glucocorticoid sensitivity of cognitive and inflammatory processes in depression and posttraumatic stress disorder. Neurosci. Biobehav. Rev. 2010, 35, 104–114. [Google Scholar] [CrossRef]
- Black, C.N.; Bot, M.; Scheffer, P.G.; Cuijpers, P.; Penninx, B.W. Is depression associated with increased oxidative stress? A systematic review and meta-analysis. Psychoneuroendocrinology 2015, 51, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Kubera, M.; Obuchowiczwa, E.; Goehler, L.; Brzeszcz, J. Depression’s multiple comorbidities explained by (neuro)inflammatory and oxidative & nitrosative stress pathways. Neuro Endocrinol. Lett. 2011, 32, 7–24. [Google Scholar] [PubMed]
- Black, C.N.; Bot, M.; Revesz, D.; Scheffer, P.G.; Penninx, B. The association between three major physiological stress systems and oxidative DNA and lipid damage. Psychoneuroendocrinology 2017, 80, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Bahraini, N.H.; Breshears, R.E.; Hernandez, T.D.; Schneider, A.L.; Forster, J.E.; Brenner, L.A. Traumatic brain injury and posttraumatic stress disorder. Psychiatr. Clin. N. Am. 2014, 37, 55–75. [Google Scholar] [CrossRef]
- Ragsdale, K.A.; Neer, S.M.; Beidel, D.C.; Frueh, B.C.; Stout, J.W. Posttraumatic stress disorder in OEF/OIF veterans with and without traumatic brain injury. J. Anxiety Disord. 2013, 27, 420–426. [Google Scholar] [CrossRef]
- Prasad, K.N.; Bondy, S.C. Common biochemical defects linkage between post-traumatic stress disorders, mild traumatic brain injury (TBI) and penetrating TBI. Brain Res. 2015, 1599C, 103–114. [Google Scholar] [CrossRef]
- Spielberg, J.M.; McGlinchey, R.E.; Milberg, W.P.; Salat, D.H. Brain Network Disturbance Related to Posttraumatic Stress and Traumatic Brain Injury in Veterans. Biol. Psychiatry 2015. [Google Scholar] [CrossRef]
- Williamson, J.B.; Heilman, K.M.; Porges, E.C.; Lamb, D.G.; Porges, S.W. A possible mechanism for PTSD symptoms in patients with traumatic brain injury: Central autonomic network disruption. Front. Neuroeng. 2013, 6, 13. [Google Scholar] [CrossRef]
- Hoge, C.W.; McGurk, D.; Thomas, J.L.; Cox, A.L.; Engel, C.C.; Castro, C.A. Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N. Engl. J. Med. 2008, 358, 453–463. [Google Scholar] [CrossRef]
- Schneiderman, A.I.; Braver, E.R.; Kang, H.K. Understanding sequelae of injury mechanisms and mild traumatic brain injury incurred during the conflicts in Iraq and Afghanistan: Persistent postconcussive symptoms and posttraumatic stress disorder. Am. J. Epidemiol. 2008, 167, 1446–1452. [Google Scholar] [CrossRef]
- Carlson, K.F.; Kehle, S.M.; Meis, L.A.; Greer, N.; Macdonald, R.; Rutks, I.; Sayer, N.A.; Dobscha, S.K.; Wilt, T.J. Prevalence, assessment, and treatment of mild traumatic brain injury and posttraumatic stress disorder: A systematic review of the evidence. J. Head Trauma Rehabil. 2011, 26, 103–115. [Google Scholar] [CrossRef]
- Tanev, K.S.; Pentel, K.Z.; Kredlow, M.A.; Charney, M.E. PTSD and TBI co-morbidity: Scope, clinical presentation and treatment options. Brain Inj. 2014, 28, 261–270. [Google Scholar] [CrossRef]
- Yurgil, K.A.; Barkauskas, D.A.; Vasterling, J.J.; Nievergelt, C.M.; Larson, G.E.; Schork, N.J.; Litz, B.T.; Nash, W.P.; Baker, D.G.; Marine Resiliency Study, T. Association between traumatic brain injury and risk of posttraumatic stress disorder in active-duty Marines. JAMA Psychiatry 2014, 71, 149–157. [Google Scholar] [CrossRef]
- Lozano, D.; Gonzales-Portillo, G.S.; Acosta, S.; de la Pena, I.; Tajiri, N.; Kaneko, Y.; Borlongan, C.V. Neuroinflammatory responses to traumatic brain injury: Etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr. Dis. Treat. 2015, 11, 97–106. [Google Scholar] [CrossRef]
- Rodriguez-Rodriguez, A.; Egea-Guerrero, J.J.; Murillo-Cabezas, F.; Carrillo-Vico, A. Oxidative stress in traumatic brain injury. Curr. Med. Chem. 2014, 21, 1201–1211. [Google Scholar] [CrossRef]
- Cornelius, C.; Crupi, R.; Calabrese, V.; Graziano, A.; Milone, P.; Pennisi, G.; Radak, Z.; Calabrese, E.J.; Cuzzocrea, S. Traumatic brain injury: Oxidative stress and neuroprotection. Antioxid. Redox Signal. 2013, 19, 836–853. [Google Scholar] [CrossRef]
- Haghighi, F.; Ge, Y.; Chen, S.; Xin, Y.; Umali, M.U.; De Gasperi, R.; Gama Sosa, M.A.; Ahlers, S.T.; Elder, G.A. Neuronal DNA Methylation Profiling of Blast-Related Traumatic Brain Injury. J. Neurotrauma 2015. [Google Scholar] [CrossRef]
- Dansie, E.J.; Heppner, P.; Furberg, H.; Goldberg, J.; Buchwald, D.; Afari, N. The Comorbidity of Self-Reported Chronic Fatigue Syndrome, Post-Traumatic Stress Disorder, and Traumatic Symptoms. Psychosomatics 2012, 53, 250–257. [Google Scholar] [CrossRef][Green Version]
- Eglinton, R.; Chung, M.C. The relationship between posttraumatic stress disorder, illness cognitions, defence styles, fatigue severity and psychological well-being in chronic fatigue syndrome. Psychiatry Res. 2011, 188, 245–252. [Google Scholar] [CrossRef]
- Heim, C.; Nater, U.M.; Maloney, E.; Boneva, R.; Jones, J.F.; Reeves, W.C. Childhood Trauma and Risk for Chronic Fatigue Syndrome Association With Neuroendocrine Dysfunction. Arch. Gen. Psychiatry 2009, 66, 72–80. [Google Scholar] [CrossRef]
- Afari, N.; Ahumada, S.M.; Wright, L.J.; Mostoufi, S.; Golnari, G.; Reis, V.; Cuneo, J.G. Psychological trauma and functional somatic syndromes: A systematic review and meta-analysis. Psychosom. Med. 2014, 76, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Dell’Osso, L.; Carmassi, C.; Consoli, G.; Conversano, C.; Ramacciotti, C.E.; Musetti, L.; Massimetti, E.; Pergentini, I.; Corsi, M.; Ciapparelli, A.; et al. Lifetime post-traumatic stress symptoms are related to the health-related quality of life and severity of pain/fatigue in patients with fibromyalgia. Clin. Exp. Rheumatol. 2011, 29, S73–S78. [Google Scholar] [PubMed]
- Galek, A.; Erbsloh-Moller, B.; Kollner, V.; Kuhn-Becker, H.; Langhorst, J.; Petermann, F.; Prothmann, U.; Winkelmann, A.; Hauser, W. Mental disorders in patients with fibromyalgia syndrome. Screening in centres of different medical specialties. Schmerz 2013, 27, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Haviland, M.G.; Morton, K.R.; Oda, K.; Fraser, G.E. Traumatic experiences, major life stressors, and self-reporting a physician-given fibromyalgia diagnosis. Psychiatry Res. 2010, 177, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Hauser, W.; Galek, A.; Erbsloh-Moller, B.; Kollner, V.; Kuhn-Becker, H.; Langhorst, J.; Petermann, F.; Prothmann, U.; Winkelmann, A.; Schmutzer, G.; et al. Posttraumatic stress disorder in fibromyalgia syndrome: Prevalence, temporal relationship between posttraumatic stress and fibromyalgia symptoms, and impact on clinical outcome. Pain 2013, 154, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Ng, Q.X.; Soh, A.Y.S.; Loke, W.; Venkatanarayanan, N.; Lim, D.Y.; Yeo, W.S. Systematic review with meta-analysis: The association between post-traumatic stress disorder and irritable bowel syndrome. J. Gastroenterol. Hepatol. 2019, 34, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Boscarino, J.A.; Forsberg, C.W.; Goldberg, J. A twin study of the association between PTSD symptoms and rheumatoid arthritis. Psychosom. Med. 2010, 72, 481–486. [Google Scholar] [CrossRef]
- Nater, U.M.; Youngblood, L.S.; Jones, J.F.; Unger, E.R.; Miller, A.H.; Reeves, W.C.; Heim, C. Alterations in diurnal salivary cortisol rhythm in a population-based sample of cases with chronic fatigue syndrome. Psychosom. Med. 2008, 70, 298–305. [Google Scholar] [CrossRef]
- Adler, G.K.; Manfredsdottir, V.F.; Creskoff, K.W. Neuroendocrine abnormalities in fibromyalgia. Curr. Pain Headache Rep. 2002, 6, 289–298. [Google Scholar] [CrossRef]
- Nijhof, S.L.; Rutten, J.M.; Uiterwaal, C.S.; Bleijenberg, G.; Kimpen, J.L.; Putte, E.M. The role of hypocortisolism in chronic fatigue syndrome. Psychoneuroendocrinology 2014, 42, 199–206. [Google Scholar] [CrossRef]
- Parker, A.J.; Wessely, S.; Cleare, A.J. The neuroendocrinology of chronic fatigue syndrome and fibromyalgia. Psychol. Med. 2001, 31, 1331–1345. [Google Scholar] [CrossRef]
- Tanriverdi, F.; Karaca, Z.; Unluhizarci, K.; Kelestimur, F. The hypothalamo-pituitary-adrenal axis in chronic fatigue syndrome and fibromyalgia syndrome. Stress 2007, 10, 13–25. [Google Scholar] [CrossRef]
- Bjorklund, G.; Dadar, M.; Pivina, L.; Dosa, M.D.; Semenova, Y.; Maes, M. Environmental, Neuro-immune, and Neuro-oxidative Stress Interactions in Chronic Fatigue Syndrome. Mol. Neurobiol. 2020, 57, 4598–4607. [Google Scholar] [CrossRef]
- Cordero, M.D.; de Miguel, M.; Carmona-Lopez, I.; Bonal, P.; Campa, F.; Moreno-Fernandez, A.M. Oxidative stress and mitochondrial dysfunction in fibromyalgia. Neuro Endocrinol. Lett. 2010, 31, 169–173. [Google Scholar]
- Fatima, G.; Das, S.K.; Mahdi, A.A. Oxidative stress and antioxidative parameters and metal ion content in patients with fibromyalgia syndrome: Implications in the pathogenesis of the disease. Clin. Exp. Rheumatol. 2013, 31, S128–S133. [Google Scholar]
- Maes, M. Inflammatory and oxidative and nitrosative stress cascades as new drug targets in myalgic encephalomyelitis and chronic fatigue syndrome. Mod. Trends Pharm. 2013, 28, 162–174. [Google Scholar] [CrossRef]
- Maes, M.; Twisk, F.N. Why myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) may kill you: Disorders in the inflammatory and oxidative and nitrosative stress (IO&NS) pathways may explain cardiovascular disorders in ME/CFS. Neuro Endocrinol. Lett. 2009, 30, 677–693. [Google Scholar]
- Phull, A.R.; Nasir, B.; Haq, I.U.; Kim, S.J. Oxidative stress, consequences and ROS mediated cellular signaling in rheumatoid arthritis. Chem. Biol. Interact. 2018, 281, 121–136. [Google Scholar] [CrossRef]
- Quinonez-Flores, C.M.; Gonzalez-Chavez, S.A.; Del Rio Najera, D.; Pacheco-Tena, C. Oxidative Stress Relevance in the Pathogenesis of the Rheumatoid Arthritis: A Systematic Review. Biomed. Res. Int. 2016, 2016, 6097417. [Google Scholar] [CrossRef]
- Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci. 2015, 18, 199–209. [Google Scholar] [CrossRef]
- Seyedsadjadi, N.; Grant, R. The Potential Benefit of Monitoring Oxidative Stress and Inflammation in the Prevention of Non-Communicable Diseases (NCDs). Antioxidants 2020, 10, 15. [Google Scholar] [CrossRef]
- Fu, S.S.; McFall, M.; Saxon, A.J.; Beckham, J.C.; Carmody, T.P.; Baker, D.G.; Joseph, A.M. Post-traumatic stress disorder and smoking: A systematic review. Nicotine Tob. Res. 2007, 9, 1071–1084. [Google Scholar] [CrossRef]
- Fukuda, S.; Morimoto, K.; Mure, K.; Maruyama, S. Posttraumatic stress and change in lifestyle among the Hanshin-Awaji earthquake victims. Prev. Med. 1999, 29, 147–151. [Google Scholar] [CrossRef]
- Csiszar, A.; Podlutsky, A.; Wolin, M.S.; Losonczy, G.; Pacher, P.; Ungvari, Z. Oxidative stress and accelerated vascular aging: Implications for cigarette smoking. Front. Biosci. 2009, 14, 3128–3144. [Google Scholar] [CrossRef]
- Ellegaard, P.K.; Poulsen, H.E. Tobacco smoking and oxidative stress to DNA: A meta-analysis of studies using chromatographic and immunological methods. Scand. J. Clin. Lab. Invest. 2016, 76, 151–158. [Google Scholar] [CrossRef]
- Johannsen, A.; Susin, C.; Gustafsson, A. Smoking and inflammation: Evidence for a synergistic role in chronic disease. Periodontology 2000 2014, 64, 111–126. [Google Scholar] [CrossRef]
- Ruiz-Nunez, B.; Pruimboom, L.; Dijck-Brouwer, D.A.; Muskiet, F.A. Lifestyle and nutritional imbalances associated with Western diseases: Causes and consequences of chronic systemic low-grade inflammation in an evolutionary context. J. Nutr. Biochem. 2013, 24, 1183–1201. [Google Scholar] [CrossRef]
- Perkins, D.O.; Jeffries, C.D.; Do, K.Q. Potential Roles of Redox Dysregulation in the Development of Schizophrenia. Biol. Psychiatry 2020, 88, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Cabungcal, J.H.; Steullet, P.; Kraftsik, R.; Cuenod, M.; Do, K.Q. A developmental redox dysregulation leads to spatio-temporal deficit of parvalbumin neuron circuitry in a schizophrenia mouse model. Schizophr. Res. 2019, 213, 96–106. [Google Scholar] [CrossRef]


| Term | Definition |
|---|---|
| Traumatic Event | An event that threatens actual or perceived injury, death, or the physical integrity of self or others and also causes horror, terror, or helplessness at the time it occurs and overwhelms a person’s ability to cope (e.g., physical/sexual abuse, medical trauma, motor vehicle accident, acts of terrorism, war experiences, natural and human-made disasters, witnessed homicides/suicides) [10]. |
| Early Life Stress (ELS) | A broad spectrum of adverse and stressful experiences (e.g., maltreatment, neglect, parental separation, parental loss, extreme poverty, starvation, domestic/community/school violence, medical trauma/illness, war and disaster experiences, etc.) during the first months of life, early and late childhood, and adolescence [11], while the term has been recently extended by some authors and includes also prenatal life events [12]. |
| Childhood Trauma/ Maltreatment (CT) | A more specific form of ELS restrictively referring to only physically or emotionally painful or distressful interpersonal traumatic events during childhood (e.g., physical/sexual/emotional abuse, physical/emotional neglect) [13]. |
| Childhood Adverse Experiences (ACEs) | This broader term includes both ELS and CT. All ACEs exhibit a dose-response effect between number and duration of ACEs and related negative health effects [14]. |
| Severe Life Stress (SLS) | A serious psychosocial event of random duration, with the potential of causing an impacting psychological traumatism and producing severe strain (e.g., loss of loved ones, job loss, prolonged social isolation, etc.) [15]. |
| Posttraumatic Stress Disorder (PTSD) | A trauma- and stress-related disorder with distinctive symptoms following a psychologically distressing event outside the range of usual human experience [10]. Diagnostic criteria include current symptoms from each of four symptom clusters: intrusion, avoidance, negative alterations in cognitions and mood, and alterations in arousal and reactivity including sleep disturbances. The estimated lifetime prevalence of PTSD in the general U.S. population lies between 5–6% in men and 10–14% in women [16]. The previously defined as Secondary Traumatic Stress (STS) condition is now considered a valid DSM-5 Criterion A for PTSD. |
| Redox Index | Outcome | References | Stress Model |
|---|---|---|---|
| ROS/RNS | ROS ↑ | [105,107] [117] [125] | MS PPE Combined model |
| O2●− ↑ | [105] [117] | MS PPE | |
| H2O2 ↑ | [112] | SI | |
| NO ↑ | [116] | PPE | |
| ONOO− ↑ | [117] | PPE | |
| NOX ↑ | [106] [116] [119] [122] [123] | MS PPE SPS IFS RSE | |
| NOS ↑ | [115,116] | PPE | |
| iNOS ↑ | [121] [123] | SPS RSE | |
| Mitochondrial activity ↑ | [103] | MS | |
| Antioxidants levels | GSH ↑ | [105] | MS |
| GSH ↓ | [119] | SPS | |
| NO ↑ | [118] | SPS | |
| Antioxidant activity | CAT ↓ | [104,108] [112] [113] | MS SI MCD |
| CAT ↑ | [129] [130] | MS SI | |
| TAC ↑ | [108] [112] | MS SI | |
| SOD ↑ | [129] [130] | MS SI | |
| SOD ↓ | [112] [113] [119] | SI MCD SPS | |
| GSH-Px ↓ | [107,108] [112,131] [119] | MS SI SPS | |
| GSH-Px ↑ | [129] [114] | MS Prenatal stress | |
| GSH-Rd ↓ | [131] | SI | |
| Redox end-products | TBARS ↑ | [104,129] | MS |
| Carbonyl ↓ | [113] | MCD | |
| Lipid peroxide ↑ | [115] | PPE | |
| MDA ↑ | [119] | SPS | |
| 8-OH-DG ↑ | [122] | IFS | |
| Other redox-related parameters | PVI activity ↓ | [109] [110] [111] | PMS MS SI |
| pAMPK ↑ | [120] | SPS | |
| GABA progenitors ↓ | [109] | PMS | |
| GLU ↓ | [112] | SI | |
| NAA ↓ | [112] | SI | |
| p-AMPK ↑ | [122] | IFS | |
| COX-2 ↑ | [118] | SPS | |
| PGE-2 ↑ | [118] | SPS | |
| GAD-67 ↓ | [122] | IFS | |
| p38 ↑ | [121] | SPS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karanikas, E.; Daskalakis, N.P.; Agorastos, A. Oxidative Dysregulation in Early Life Stress and Posttraumatic Stress Disorder: A Comprehensive Review. Brain Sci. 2021, 11, 723. https://doi.org/10.3390/brainsci11060723
Karanikas E, Daskalakis NP, Agorastos A. Oxidative Dysregulation in Early Life Stress and Posttraumatic Stress Disorder: A Comprehensive Review. Brain Sciences. 2021; 11(6):723. https://doi.org/10.3390/brainsci11060723
Chicago/Turabian StyleKaranikas, Evangelos, Nikolaos P. Daskalakis, and Agorastos Agorastos. 2021. "Oxidative Dysregulation in Early Life Stress and Posttraumatic Stress Disorder: A Comprehensive Review" Brain Sciences 11, no. 6: 723. https://doi.org/10.3390/brainsci11060723
APA StyleKaranikas, E., Daskalakis, N. P., & Agorastos, A. (2021). Oxidative Dysregulation in Early Life Stress and Posttraumatic Stress Disorder: A Comprehensive Review. Brain Sciences, 11(6), 723. https://doi.org/10.3390/brainsci11060723