
Distinguishing Alzheimer’s Disease Patients and Biochemical Phenotype Analysis Using a Novel Serum Profiling Platform: Potential Involvement of the VWF/ADAMTS13 Axis

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Clinical Samples
- (a)
- Had a receptive or expressive aphasia;
- (b)
- Were in a bedridden state;
- (c)
- Were legally blind;
- (d)
- Had severe dementia (CDR = 3.0, or MMSE < 10);
- (e)
- Had severe depression, anxiety, or psychosis, or Neuropsychiatric Inventory (NPI) score according to Cummings et al. [25], of >5/12;
- (f)
- Had a history of cancer;
- (g)
- Had a diagnosis of MCI (0 < CDR ≤ 0.5);
- (h)
- Were University of Oklahoma (OU).
2.2. Electrospray Mass Spectrometry of Sera from Mild or Moderate AD Patients, and from Control Individuals
2.3. Statistical and Quantitative Analysis
2.4. Test Metrics
3. Results
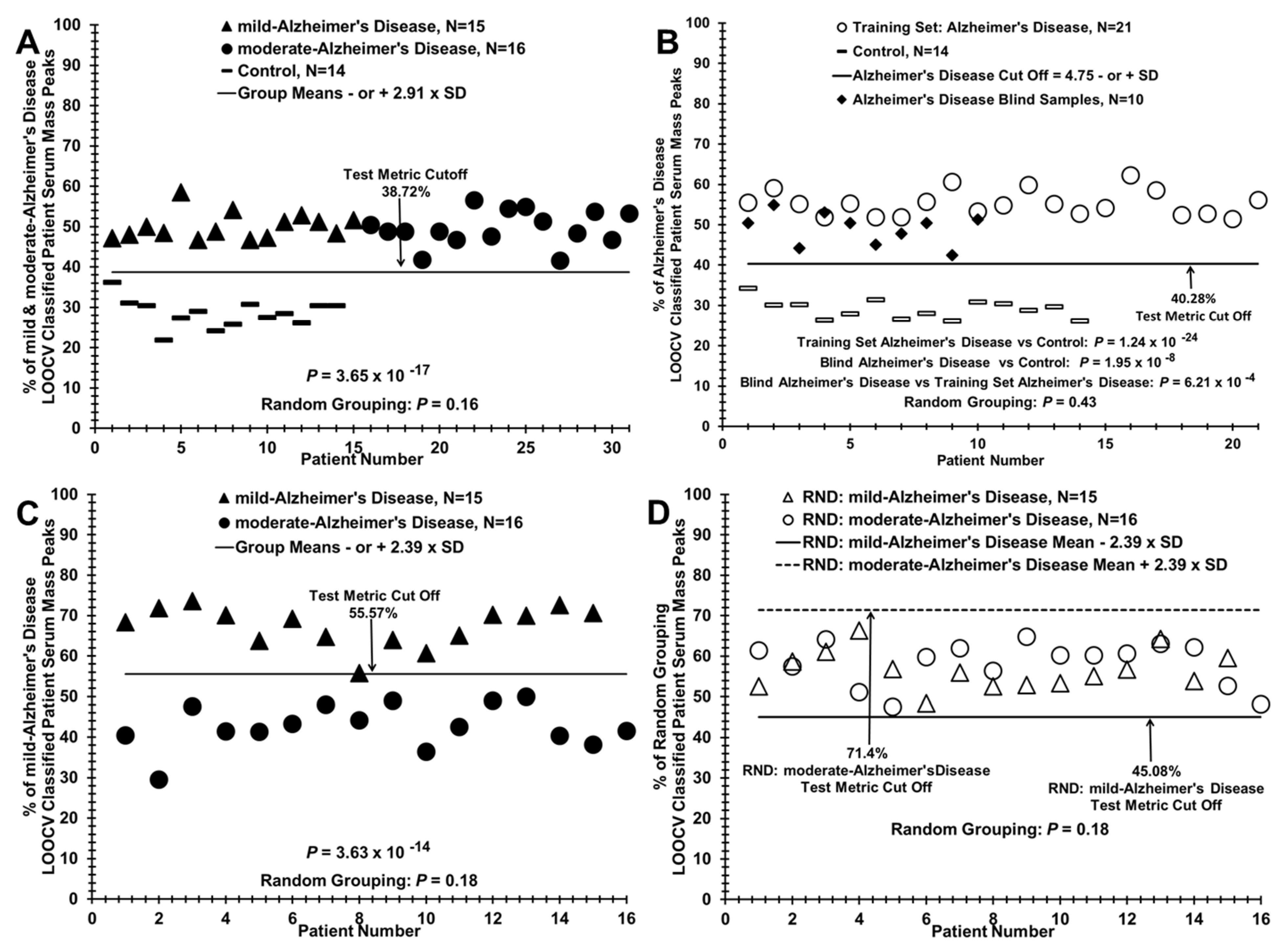
3.1. Discriminating AD Patients from Control Individuals Using ESI-MS Serum Profiling
3.2. Discriminating Patients with Mild or Moderate AD from Control Individuals, from Each Other, and Assigning Group Designation (Control or AD) of a Blinded AD Sub-Group
3.3. Segregation of Traumatic Brain Injury (TBI) Patient Sera with AD Patients when Compared to Control Individuals Using the LOOCV/PCV Process
3.4. Distinguishing AD Patients and Control Individuals Using a Lower-Cost Desktop ESI-MS Instrument
3.5. Test Metric Data for AD, Control, and TBI Serum LOOCV/ESI-MS Profiling Comparisons
3.6. Phenotype Assessments of AD Patients Using Tandem MS/MS of Serum Peptide/Proteins, and Bioinformatic Cell Pathway/Disease Mechanism Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hampel, H.; O’Bryant, S.E.; Molinuevo, J.L.; Zetterberg, H.; Masters, C.L.; Lista, S.; Kiddle, S.J.; Batrla, R.; Blennow, K. Blood-based biomarkers for Alzheimer disease: Mapping the road to the clinic. Nat. Rev. Neurol. 2018, 14, 639–652. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Noh, Y.; Jeon, S.; Lee, J.M.; Seok, Y.B.; Kim, G.H.; Cho, H.; Ye, B.S.; Yoon, C.W.; Kim, H.J.; Chin, J.; et al. Anatomical heterogeneity of Alzheimer disease: Based on cortical thickness on MRIs. Neurol. 2014, 83, 1936–1944. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, S.; Marizzoni, M.; Babiloni, C.; Albani, D.; Antelmi, L.; Bagnoli, C.; Bartres-Faz, D.; Cordone, S.; Didic, M.; Farotti, L.; et al. Clinical and biomarker profiling of prodromal Alzheimer’s disease in workpackage 5 of the Innovative Medicines Initiative PharmaCog project: A ‘European ADNI study’. J. Intern. Med. 2016, 279, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Rabinovici, G.D.; Rosen, H.J.; Alkalay, A.; Kornak, J.; Furst, A.J.; Agarwal, N.; Mormino, E.C.; O’Neil, J.P.; Janabi, M.; Karydas, A.; et al. Amyloid vs FDG-PET in the differential diagnosis of AD and FTLD. Neurol. 2011, 77, 2034–2042. [Google Scholar] [CrossRef]
- Landau, S.M.; Lu, M.; Joshi, A.D.; Pontecorvo, M.; Mintun, M.A.; Trojanowski, J.Q.; Shaw, L.M.; Jagust, W.J. Alzheimer’s Disease Neuroimaging I. Comparing positron emission tomography imaging and cerebrospinal fluid measurements of beta-amyloid. Ann Neurol. 2013, 74, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Rowe, C.C.; Bourgeat, P.; Ellis, K.A.; Brown, B.; Lim, Y.Y.; Mulligan, R.; Jones, G.; Maruff, P.; Woodward, M.; Price, R.; et al. Predicting Alzheimer disease with beta-amyloid imaging: Results from the Australian imaging, biomarkers, and lifestyle study of ageing. Ann. Neurol. 2013, 74, 905–913. [Google Scholar] [CrossRef]
- Palmqvist, S.; Zetterberg, H.; Mattsson, N.; Johansson, P.; For the Alzheimer’s Disease Neuroimaging Initiative; Minthon, L.; Blennow, K.; Olsson, M. For the Swedish BioFINDER study group; Hansson, O. Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurolongy 2015, 85, 1240–1249. [Google Scholar] [CrossRef]
- Okamura, N.; Harada, R.; Furumoto, S.; Arai, H.; Yanai, K.; Kudo, Y. Tau PET imaging in Alzheimer’s disease. Curr. Neurol. Neurosci. Rep. 2014, 14, 500. [Google Scholar] [CrossRef]
- Winblad, B.; Palmer, K.; Kivipelto, M.; Jelic, V.; Fratiglioni, L.; Wahlund, L.-O.; Nordberg, A.; Backman, L.J.; Albert, M.S.; Almkvist, O.; et al. Mild cognitive impairment-beyond controversies, towards a consensus: Report of the International Working Group on Mild Cognitive Impairment. J. Intern. Med. 2004, 256, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Ewers, M.; Buerger, K.; Teipel, S.J.; Scheltens, P.; Schroder, J.; Zinkowski, R.P.; Bouwman, F.H.; Schonknecht, P.; Schoonenboom, N.; Andreasen, N.; et al. Multicenter assessment of CSF-phosphorylated tau for the prediction of conversion of MCI. Neurol. 2007, 69, 2205–2212. [Google Scholar] [CrossRef]
- Moscoso, A.; Grothe, M.J.; Ashton, N.J.; Karikari, T.K.; Rodríguez, J.L.; Snellman, A.; Suárez-Calvet, M.; Blennow, K.; Zetterberg, H.; Schöll, M.; et al. Longitudinal Associations of Blood Phosphorylated Tau181 and Neurofilament Light Chain With Neurodegeneration in Alzheimer Disease. JAMA Neurol. 2021, 78, 396. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA 2020, 324, 772. [Google Scholar] [CrossRef] [PubMed]
- Cedazo-Minguez, A.; Winblad, B. Biomarkers for Alzheimer’s disease and other forms of dementia: Clinical needs, limitations and future aspects. Exp. Gerontol. 2010, 45, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Schiele, F.; De Bacquer, D.; Vincent-Viry, M.; Beisiegel, U.; Ehnholm, C.; Evans, A.; Kafatos, A.; Martins, M.; Sans, S.; Sass, C.; et al. Apolipoprotein E serum concentration and polymorphism in six European countries: The ApoEurope Project. Atheroscler. 2000, 152, 475–488. [Google Scholar] [CrossRef]
- Llano, D.A.; Devanarayan, V.; Simon, A.J. Evaluation of Plasma Proteomic Data for Alzheimer Disease State Classification and for the Prediction of Progression From Mild Cognitive Impairment to Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2013, 27, 233–243. [Google Scholar] [CrossRef]
- Nettiksimmons, J.; Ayonayon, H.; Harris, T.; Phillips, C.; Rosano, C.; Satterfield, S.; Yaffe, K. For the Health ABC Study Development and validation of risk index for cognitive decline using blood-derived markers. Neurology 2015, 84, 696–702. [Google Scholar] [CrossRef]
- Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Li, Y.; Gordon, B.A.; Holtzman, D.M.; Morris, J.C.; Benzinger, T.L.S.; Xiong, C.; et al. High-precision plasma beta-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 2019, 93, e1647–e1659. [Google Scholar] [PubMed]
- Hanas, J.S.; Hocker, J.R.S.; Lerner, M.R.; Couch, J.R. Distinguishing and phenotype monitoring of traumatic brain injury and post-concussion syndrome including chronic migraine in serum of Iraq and Afghanistan war veterans. PLoS ONE 2019, 14, e0215762. [Google Scholar] [CrossRef]
- Hanas, J.; Hocker, J.; Vannarath, C.; Evangeline, B.; Prabhakaran, V.; Oommen, A.; Couch, J.; Anderson, M.; Rajshekhar, V.; Carabin, H.; et al. Distinguishing and Biochemical Phenotype Analysis of Epilepsy Patients Using a Novel Serum Profiling Platform. Brain Sci. 2020, 10, 504. [Google Scholar] [CrossRef]
- Hanas, J.S.; Hocker, J.R.; Ramajayam, G.; Prabhakaran, V.; Rajshekhar, V.; Oommen, A.; Manoj, J.J.; Anderson, M.P.; Drevets, D.A.; Carabin, H. Distinguishing neurocysticercosis epilepsy from epilepsy of unknown etiology using a minimal serum mass profiling platform. Exp. Parasitol. 2018, 192, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Vachani, A.; Pass, H.I.; Rom, W.N.; Midthun, D.E.; Edell, E.S.; LaViolette, M.; Clive, H.; Fong, P.-Y.; Hunsucker, S.W.; Hayward, C.; et al. Validation of a Multiprotein Plasma Classifier to Identify Benign Lung Nodules. J. Thorac. Oncol. 2015, 10, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Edland, S.; Clark, C.; Galasko, D.; Koss, E.; Mohs, R.; van Belle, G.; Fillenbaum, G.; Heyman, A. The consortium to establish a registry for Alzheimer’s disease (CERAD). Part IV. Rates of cognitive change in the longitudinal assessment of probable Alzheimer’s disease. Neurology 1993, 43, 2457–2465. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Cummings, J.L.; Mega, M.; Gray, K.; Rosenberg-Thompson, S.; Carusi, D.A.; Gornbein, J. The Neuropsychiatric Inventory: Comprehensive assessment of psychopathology in dementia. Neurol. 1994, 44, 2308. [Google Scholar] [CrossRef]
- Tuck, M.K.; Chan, D.W.; Chia, D.; Godwin, A.K.; Grizzle, W.E.; Krueger, K.E.; Rom, W.; Sanda, M.; Sorbara, L.; Stass, S.; et al. Standard Operating Procedures for Serum and Plasma Collection: Early Detection Research Network Consensus Statement Standard Operating Procedure Integration Working Group. J. Proteome Res. 2009, 8, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Hocker, J.R.; Deb, S.J.; Li, M.; Lerner, M.R.; Lightfoot, S.A.; Quillet, A.A.; Hanas, R.J.; Reinersman, M.; Thompson, J.L.; Vu, N.T.; et al. Serum Monitoring and Phenotype Identification of Stage I Non-Small Cell Lung Cancer Patients. Cancer Investig. 2017, 35, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Baker, S.G.; Kramer, B.S.; Srivastava, S. Markers for early detection of cancer: Statistical guidelines for nested case-control studies. BMC Med. Res. Methodol. 2002, 2, 4. [Google Scholar] [CrossRef]
- Guan, W.; Zhou, M.; Hampton, C.Y.; Benigno, B.B.; Walker, L.D.; Gray, A.; McDonald, J.F.; Fernández, F.M. Ovarian cancer detection from metabolomic liquid chromatography/mass spectrometry data by support vector machines. BMC Bioinform. 2009, 10, 259. [Google Scholar] [CrossRef]
- Ransohoff, D.F. Evaluating discovery-based research: When biologic reasoning cannot work. Gastroenterology 2004, 127, 1028. [Google Scholar] [CrossRef]
- Cohen, J. Statistical Power Analysis for the Behavioral-Sciences, 2nd ed.; Lawrence Erlbaum Associates, Publishers: Hillsdale, NJ, USA, 1988. [Google Scholar]
- Soper, D.S. Post-hoc Statistical Power Calculator for a Student t-Test [Software]. 2018. Available online: http://www.danielsoper.com/statcalc (accessed on 29 April 2021).
- Altman, D.G.; Bland, J.M. Diagnostic-Tests-2—Predictive Values. Brit Med. J. 1994, 309, 102. [Google Scholar] [CrossRef]
- Altman, D.G.; Bland, J.M. Diagnostic tests. 1: Sensitivity and specificity. BMJ 1994, 308, 1552. [Google Scholar] [CrossRef]
- Ramos-Cejudo, J.; Wisniewski, T.; Marmar, C.; Zetterberg, H.; Blennow, K.; de Leon, M.J.; Fossati, S. Traumatic Brain Injury and Alzheimer’s Disease: The Cerebrovascular Link. EBioMedicine 2018, 28, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zheng, J.; Zheng, F.; Cai, Z.; Yu, Q. Advancing serum peptidomic profiling by data-independent acquisition for clear-cell renal cell carcinoma detection and biomarker discovery. J. Proteom. 2020, 215, 103671. [Google Scholar] [CrossRef]
- Banks, W.A. Peptides and the blood-brain barrier. Peptides. 2015, 72, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Sakka, L.; Delétage, N.; Lalloué, F.; Duval, A.; Chazal, J.; Lemaire, J.-J.; Meiniel, A.; Monnerie, H.; Gobron, S. SCO-Spondin Derived Peptide NX210 Induces Neuroprotection In Vitro and Promotes Fiber Regrowth and Functional Recovery after Spinal Cord Injury. PLoS ONE 2014, 9, e93179. [Google Scholar] [CrossRef]
- Chew, E.G.Y.; Liany, H.; Tan, L.C.S.; Au, W.L.; Prakash, K.M.; Annuar, A.A.; Chan, A.Y.Y.; Lim, S.Y.; Mok, V.; Chung, S.J.; et al. Evaluation of novel Parkinson’s disease candidate genes in the Chinese population. Neurobiol. Aging 2019, 235, e1–e4. [Google Scholar] [CrossRef]
- Randi, A.M.; Smith, K.E.; Castaman, G. von Willebrand factor regulation of blood vessel formation. Blood 2018, 132, 132–140. [Google Scholar] [CrossRef]
- Gragnano, F.; Sperlongano, S.; Golia, E.; Natale, F.; Bianchi, R.; Crisci, M.; Fimiani, F.; Pariggiano, I.; Diana, V.; Carbone, A.; et al. The Role of von Willebrand Factor in Vascular Inflammation: From Pathogenesis to Targeted Therapy. Mediat. Inflamm. 2017, 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Miloudi, K.; Oubaha, M.; Ménard, C.; Dejda, A.; Guber, V.; Cagnone, G.; Wilson, A.M.; Tétreault, N.; Mawambo, G.; Binet, F.; et al. NOTCH1 signaling induces pathological vascular permeability in diabetic retinopathy. PNAS 2019, 116, 4538–4547. [Google Scholar] [CrossRef]
- Dobson-Stone, C.; Hallupp, M.; Shahheydari, H.; Ragagnin, A.M.G.; Chatterton, Z.; Carew-Jones, F.; E Shepherd, C.; Stefen, H.; Paric, E.; Fath, T.; et al. CYLD is a causative gene for frontotemporal dementia—Amyotrophic lateral sclerosis. Brain 2020, 143, 783–799. [Google Scholar] [CrossRef]
- Fontana, G.A.; Reinert, J.K.; Thomä, N.H.; Rass, U. Shepherding DNA ends: Rif1 protects telomeres and chromosome breaks. Microb. Cell 2018, 5, 327–343. [Google Scholar] [CrossRef]
- Wolters, F.J.; Boender, J.; De Vries, P.S.; Sonneveld, M.A.; Koudstaal, P.J.; De Maat, M.P.; Franco, O.H.; Ikram, M.K.; Leebeek, F.W.; Ikram, M.A. Von Willebrand factor and ADAMTS13 activity in relation to risk of dementia: A population-based study. Sci. Rep. 2018, 8, 5474. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-H.; Chen, C.-M.; Chen, Y.-C.; Hsiao, Y.-C.; Huang, C.-C.; Kuo, H.-C.; Hsu, H.-C.; Lee-Chen, G.-J.; Wu, Y.-R. Association between GRN rs5848 Polymorphism and Parkinson′s Disease in Taiwanese Population. PLoS ONE 2013, 8, e54448. [Google Scholar] [CrossRef]
- Pottier, C.; Zhou, X.; Perkerson, R.B., 3rd; Baker, M.; Jenkins, G.D.; Serie, D.J.; Ghidoni, R.; Benussi, L.; Binetti, G.; Lopez de Munain, A.; et al. Potential genetic modifiers of disease risk and age at onset in patients with frontotemporal lobar degeneration and GRN mutations: A genome-wide association study. Lancet Neurol. 2018, 17, 548–558. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Kanekiyo, T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1965. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wang, H.; Schwartz, D.M.; Stoffels, M.; Park, Y.H.; Zhang, Y.; Yang, D.; Demirkaya, E.; Takeuchi, M.; Tsai, W.L.; et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat. Genet. 2016, 48, 67–73. [Google Scholar] [CrossRef]
- Bastaki, F.; Nair, P.; Mohamed, M.; Malik, E.M.; Helmi, M.; Al-Ali, M.T.; Hamzeh, A.R. Identification of a novel CTCF mutation responsible for syndromic intellectual disability—A case report. BMC Med. Genet. 2017, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Huang, Q.; Du, X.; Xu, S.; Li, M.; Ma, S. Early activation of Egr-1 promotes neuroinflammation and dopaminergic neurodegeneration in an experimental model of Parkinson’s disease. Exp. Neurol. 2018, 302, 145–154. [Google Scholar] [CrossRef]
- Manderstedt, E.; Nilsson, R.; Lind-Halldén, C.; Ljung, R.; Astermark, J.; Halldén, C. Targeted re-sequencing of F8, F9 and VWF: Characterization of Ion Torrent data and clinical implications for mutation screening. PLoS ONE 2019, 14, e0216179. [Google Scholar] [CrossRef] [PubMed]
- Badea, A.; Schmalzigaug, R.; Kim, W.; Bonner, P.; Ahmed, U.; Johnson, G.A.; Cofer, G.; Foster, M.; Anderson, R.J.; Badea, C.; et al. Microcephaly with altered cortical layering in GIT1 deficiency revealed by quantitative neuroimaging. Magn. Reson. Imaging 2021, 76, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Katsel, P.; Tan, W.; Haroutunian, V. Gain in Brain Immunity in the Oldest-Old Differentiates Cognitively Normal from Demented Individuals. PLoS ONE 2009, 4, e7642. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Liu, Y.-L.; Tsai, S.-J.; Kuo, P.-H.; Huang, S.-S.; Lee, Y.-S. LRRTM4 and PCSK5 Genetic Polymorphisms as Markers for Cognitive Impairment in A Hypotensive Aging Population: A Genome-Wide Association Study in Taiwan. J. Clin. Med. 2019, 8, 1124. [Google Scholar] [CrossRef]
- Zander, C.B.; Cao, W.; Zheng, X.L. ADAMTS13 and von Willebrand factor interactions. Curr. Opin. Hematol. 2015, 22, 452–459. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Liu, C.C.; Shinohara, M.; Li, J.; Bu, G. LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer’s amyloid-beta. J. Neurosci. 2012, 32, 16458–16465. [Google Scholar] [CrossRef]
- Koldamova, R.; Schug, J.; Lefterova, M.; Cronican, A.A.; Fitz, N.F.; Davenport, F.A.; Carter, A.; Castranio, E.L.; Lefterov, I. Genome-wide approaches reveal EGR1-controlled regulatory networks associated with neurodegeneration. Neurobiol. Dis. 2014, 63, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Lunnon, K.; Keohane, A.; Pidsley, R.; Newhouse, S.; Riddoch-Contreras, J.; Thubron, E.B.; Devall, M.; Soininen, H.; Kloszewska, I.; Mecocci, P.; et al. Mitochondrial genes are altered in blood early in Alzheimer’s disease. Neurobiol. Aging 2017, 53, 36–47. [Google Scholar] [CrossRef]
- Crehan, H.; Holton, P.; Wray, S.; Pocock, J.; Guerreiro, R.; Hardy, J. Complement receptor 1 (CR1) and Alzheimer’s disease. Immunobiology 2012, 217, 244–250. [Google Scholar] [CrossRef]
- Joiner, D.M.; Ke, J.; Zhong, Z.; Xu, H.E.; Williams, B.O. LRP5 and LRP6 in development and disease. Trends Endocrinol. Metab. 2013, 24, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Alliey-Rodriguez, N.; Grey, T.; Shafee, R.; Padmanabhan, J.; Tandon, N.; Klinger, M.; Spring, J.; Coppes, L.; Reis, K.; Keshavan, M.S.; et al. Common variants of NRXN1, LRP1B and RORA are associated with increased ventricular volumes in psychosis-GWAS findings from the B-SNIP deep phenotyping study. bioRxiv 2017. [Google Scholar] [CrossRef]
- Ye, K. PIKE/nuclear PI 3-kinase signaling in preventing programmed cell death. J. Cell. Biochem. 2005, 96, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Linder, B.; Fischer, U.; Gehring, N.H. mRNA metabolism and neuronal disease. FEBS Lett. 2015, 589, 1598–1606. [Google Scholar] [CrossRef]
- Zhu, G.-J.; Gong, S.; Ma, D.-B.; Tao, T.; He, W.-Q.; Zhang, L.; Wang, F.; Qian, X.-Y.; Zhou, H.; Fan, C.; et al. Aldh inhibitor restores auditory function in a mouse model of human deafness. PLoS Genet. 2020, 16, e1009040. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Yan, W.; Cheng, Y. Pou4f3 gene mutation promotes autophagy and apoptosis of cochlear hair cells in cisplatin-induced deafness mice. Arch. Biochem. Biophys. 2020, 680, 108224. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fu, X.; Tang, Z.; Li, C.; Xu, Y.; Zhang, F.; Zhou, D.; Zhu, C. Altered expression of the CSMD1 gene in the peripheral blood of schizophrenia patients. BMC Psychiatry 2019, 19, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.T.; Zhang, Z.X.; Chan, P.; He, X.M.; Tang, M.N.; Wu, C.B.; Hong, Z. Genetic association between low-density lipoprotein receptor-related protein gene polymorphisms and Alzheimer’s disease in Chinese Han population. Neurosci. Lett. 2008, 444, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 1–22. [Google Scholar] [CrossRef]
- Cao, W.; Krishnaswamy, S.; Camire, R.M.; Lenting, P.J.; Zheng, X.L. Factor VIII accelerates proteolytic cleavage of von Willebrand factor by ADAMTS13. Proc. Natl. Acad. Sci. USA 2008, 105, 7416–7421. [Google Scholar] [CrossRef]
- Pop, V.; Sorensen, D.W.; Kamper, J.E.; Ajao, D.O.; Murphy, M.P.; Head, E.; Hartman, R.E.; Badaut, J. Early brain injury alters the blood-brain barrier phenotype in parallel with beta-amyloid and cognitive changes in adulthood. J. Cereb. Blood Flow Metab. 2013, 33, 205–214. [Google Scholar] [CrossRef]
- Zhu, S.; He, M.; Liu, Z.; Qin, Z.; Wang, Z.; Duan, L. Shared genetic susceptibilities for irritable bowel syndrome and depressive disorder in Chinese patients uncovered by pooled whole-exome sequencing. J. Adv. Res. 2020, 23, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Paris, D.; Ganey, N.; Banasiak, M.; Laporte, V.; Patel, N.; Mullan, M.; Murphy, S.F.; Yee, G.T.; Bachmeier, C.; Ganey, C.; et al. Impaired orthotopic glioma growth and vascularization in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2010, 30, 11251–11258. [Google Scholar] [CrossRef]
- Mazza, M.; Marano, G.; Traversi, G.; Bria, P.; Mazza, S. Primary cerebral blood flow deficiency and Alzheimer’s disease: Shadows and lights. J. Alzheimers Dis. 2011, 23, 375–389. [Google Scholar] [CrossRef]
- Frentzou, G.A.; Bradford, C.; Harkness, K.A.; Haddock, G.; Woodroofe, M.N.; Cross, A.K. IL-1beta down-regulates ADAMTS-13 mRNA expression in cells of the central nervous system. J. Mol. Neurosci. 2012, 46, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Xin, S.-H.; Tan, L.; Cao, X.; Yu, J.-T.; Tan, L. Clearance of Amyloid Beta and Tau in Alzheimer’s Disease: From Mechanisms to Therapy. Neurotox. Res. 2018, 34, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Gabin, J.M.; Tambs, K.; Saltvedt, I.; Sund, E.; Holmen, J. Association between blood pressure and Alzheimer disease measured up to 27 years prior to diagnosis: The HUNT Study. Alzheimer’s Res. Ther. 2017, 9, 1–12. [Google Scholar] [CrossRef]
- Stampfer, M.J. Cardiovascular disease and Alzheimer’s disease: Common links. J. Intern. Med. 2006, 260, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, P.L.; Castillo-García, A.; Morales, J.S.; De La Villa, P.; Hampel, H.; Emanuele, E.; Lista, S.; Lucia, A. Exercise benefits on Alzheimer’s disease: State-of-the-science. Ageing Res. Rev. 2020, 62, 101108. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Panel I, Demographic Information about Study Participants | ||||
|---|---|---|---|---|
| Control (n = 14) | Mild AD * (n = 15) | Moderate AD * (n = 16) | All AD * (n = 31) | |
| Age, mean (yrs range) | 74.3 (59−84) | 74.9 (56−90) | 77.3 (61−88) | 76.2 (56−90) |
| Sex, men/women | 7/7 | 5/10 | 7/9 | 12/19 |
| Race **, AA (C) | 0 (14) | 0 (15) | 1 (15) | 1 (44) |
| Smoking ***, men (women) | 0 (2) | 3 (1) | 0 (3) | 3 (4) |
| Clinical Dementia Rating (CDR): | 0 | 0.5 < CDR ≤ 1 | 1< CDR ≤ 2 | >0.5 |
| Mini-Mental State Exam (MMSE) (mean) | 29.14 | 22.6 | 15.5 | 18.9 |
| Instrumental Activities of Daily Living (IADL) | Normal | Below Normal | Below Normal | Below Normal |
| Panel II, Inclusion/Exclusion Criteria for Participants | ||||
| Inclusion Criteria (General) | Exclusion Criteria (General) | Control Inclusion | Alzheimer’s Inclusion | |
|
|
|
| |
| ESI-Ion Trap MS Group 1 vs. Group 2 | Mean (SD) Group 1 | Mean (SD) Group 2 | “Effect Size” | p Value | N Group 1:Group 2 | Sensitivity | Specificity | Figure# |
|---|---|---|---|---|---|---|---|---|
| moderate-Alzheimer’s disease vs. Control, N = 14 | 62.73% (7.34%) | 28.07% (6.85%) | 4.88 | 6.06 × 10−14 | 16:14 | 1 | 1 | Figure 2A |
| mild-Alzheimer’s disease vs. Control, N = 14 | 63.67% (5.01%) | 37.29% (5.86%) | 4.83 | 4.24 × 10−13 | 15:14 | 1 | 1 | Figure 2C |
| Alzheimer’s disease, N = 31 vs. Control, N = 14 | 49.81% (3.81%) | 28.53% (3.50%) | 5.81 | 3.63 × 10−14 | 31:14 | 1 | 1 | Figure 3A and Figure 4A |
| mild-Alzheimer’s disease vs. moderate-Alzheimer’s disease | 67.33% (4.92%) | 42.66% (5.41%) | 0.77 | 3.25 × 10−14 | 15:16 | 0.93 | 1 | Figure 3C and Figure 4D |
| Training set: Alzheimer’s disease vs. Control | 55.25% (3.15%) | 20.09% (2.36%) | 12.63 | 1.24 × 10−24 | 21:14 | 1 | 1 | Figure 3B |
| mild and moderate-Alzheimer’s disease vs. TBI | 64.12% (4.17%) | 26.78% (6.13%) | 7.12 | 1.53 × 10−14 | 18:13 | 1 | 1 | Figure 4B |
| ESI-Single-Quad MS Group 1 vs. Group 2 | Mean (SD) Group 1 | Mean (SD) Group 2 | “effect size” | pValue | N Group 1:Group 2 | Sensitivity | Specificity | Figure# |
| mild-Alzheimer’s diseasevs. Control, N = 14 | 66.67% (8.80%) | 26.07% (12.74%) | 3.70 | 4.58 × 10−10 | 15:14 | 1 | 1 | Figure 5A |
| moderate-Alzheimer’s diseasevs. Control | 73.14% (7.11%) | 49.35% (11.36%) | 2.51 | 5.12 × 10−7 | 16:14 | 0.93 | 0.86 | Figure 5B |
| Alzheimer’s diseasevs. Control, N = 14 | 44.82% (8.92%) | 27.91% (6.45%) | 2.17 | 1.28 × 10−8 | 31:14 | 0.93 | 0.71 | Figure 5C |
| moderate-Alzheimer’s disease vs. mild-Alzheimer’s disease | 75.56% (8.35%) | 39.42% (11.14%) | 3.67 | 7.75 × 10−11 | 15:16 | 0.93 | 0.94 | Figure 5D |
| Mild AD > Control, 2x Sera, 1.5x Hits | ||||||||
|---|---|---|---|---|---|---|---|---|
| Symbol | Mild: Control | Symbol | Mild: Control | Symbol | Mild: Control | |||
| [#Sera(#Hits)] | [#Sera(#Hits)] | [#Sera(#Hits)] | ||||||
| 1 | SSPO 2,3,7 | 8(162): 4(57) | 31 | STXBP2 2,3 | 3(40): 0(0) | 61 | ZC3H4 | 3(12): 0(0) |
| 2 | VWF 1,2,3,4,5,6 | 6(158): 1(15) | 32 | PKD1P6 | 3(39): 0(0) | 62 | NOXP20 | 3(11): 0(0) |
| 3 | IGKC 1,2 | 5(130): 1(3) | 33 | KMT2E 2,3 | 3(37): 0(0) | 63 | ARIH1 | 3(9): 0(0) |
| 4 | F8 1,2,3 | 4(84): 0(0) | 34 | SRRM2 1 | 3(37): 0(0) | 64 | OIT3 4 | 3(8): 0(0) |
| 5 | CR1 1,2,3,5,7 | 4(30): 0(0) | 35 | CNOT1 | 3(35): 0(0) | 65 | IGLC3 | 3(259): 1(2) |
| 6 | UPF2 2,3 | 4(30): 0(0) | 36 | POU4F3 2,3 | 3(35): 0(0) | 66 | CHFR 2,3,6 | 3(169): 1(22) |
| 7 | LTBP2 | 4(66): 1(5) | 37 | CYLD 2,3,4,6,7 | 3(33): 0(0) | 67 | POGZ 3 | 3(126): 1(26) |
| 8 | ADGRV1 3 | 4(66): 1(15) | 38 | CAPN3 4 | 3(32): 0(0) | 68 | AGAP6 3 | 3(119): 1(3) |
| 9 | SVEP1 | 4(56): 1(7) | 39 | ITGAX 1,2,6 | 3(32): 0(0) | 69 | LTBP1 1 | 3(118): 1(25) |
| 10 | LRP4 2,3 | 4(42): 1(5) | 40 | SMCHD1 | 3(30): 0(0) | 70 | HSPG2 1,3,5,6 | 3(73): 1(3) |
| 11 | ZNF469 | 4(37): 1(5) | 41 | GIT1 1,2,4,5,6,7 | 3(29): 0(0) | 71 | PRUNE2 | 3(59): 1(7) |
| 12 | FBN1 1,2,3,4,5 | 4(28): 1(14) | 42 | ACACB 1,2,3 | 3(27): 0(0) | 72 | FRMPD1 | 3(32): 1(9) |
| 13 | MT1B | 4(17): 1(3) | 43 | C5orf42 4 | 3(26): 0(0) | 73 | RYR3 1,2,3,4,6 | 3(32): 1(15) |
| 14 | MT-ND1 1 | 4(127): 2(25) | 44 | KHSRP 2 | 3(26): 0(0) | 74 | MIDN 2,3,5,7 | 3(27): 1(6) |
| 15 | OTOGL | 4(74): 2(25) | 45 | OR7G1 | 3(26): 0(0) | 75 | CELSR2 3 | 3(25): 1(4) |
| 16 | NOTCH1 1,2,3,4,5,6,7 | 4(39): 2(22) | 46 | ASCC1 2 | 3(25): 0(0) | 76 | SEC24C 1,7 | 3(25): 1(5) |
| 17 | LRP1 1,2,3,4,5,6,7 | 4(34): 2(13) | 47 | MFRP | 3(25): 0(0) | 77 | NBPF10 1,7 | 3(24): 1(3) |
| 18 | DLGAP5 7 | 3(115): 0(0) | 48 | SSC5D2 | 3(25): 0(0) | 78 | FREM2 | 3(24): 1(7) |
| 19 | BSG 1,2,7 | 3(72): 0(0) | 49 | HNRNPA3 1,3 | 3(23): 0(0) | 79 | KMT2C 3 | 3(24): 1(11) |
| 20 | NFXL1 | 3(59): 0(0) | 50 | RXFP3 1,6 | 3(23): 0(0) | 80 | MT-ND6 1 | 3(22): 1(8) |
| 21 | KIAA119 | 3(55): 0(0) | 51 | AGAP2 1,7 | 3(22): 0(0) | 81 | PDZD2 | 3(22): 1(9) |
| 22 | TNRC6A 6 | 3(53): 0(0) | 52 | NAAA 1,2 | 3(22): 0(0) | 82 | ADAMTS6 | 3(19): 1(3) |
| 23 | ATP1A1 1,2,4,6 | 3(52): 0(0) | 53 | MEGF11 | 3(21): 0(0) | 83 | MALRD1 3 | 3(17): 1(8) |
| 24 | SPINK5 2 | 3(51): 0(0) | 54 | ATMIN 3,6 | 3(19): 0(0) | 84 | PCDHB7 | 3(16): 1(7) |
| 25 | PCDH8 4 | 3(46): 0(0) | 55 | EGR1 1,2,3,4,5,6,7 | 3(18): 0(0) | 85 | SPEN 3,7 | 3(15): 1(5) |
| 26 | ARHGEF2 2,3 | 3(45): 0(0) | 56 | GOLGA2P11 | 3(16): 0(0) | 86 | MYCBP2 3 | 3(15): 1(6) |
| 27 | CFAP221 | 3(45): 0(0) | 57 | LOC4682 | 3(16): 0(0) | 87 | HLA-DQA1 | 3(14): 1(5) |
| 28 | EYA1 3,6 | 3(45): 0(0) | 58 | TENM1 | 3(15): 0(0) | 88 | TRIP11 | 3(13): 1(5) |
| 29 | SARDH | 3(44): 0(0) | 59 | PCDHA4 1 | 3(13): 0(0) | 89 | POLA1 | 3(9): 1(3) |
| 30 | RALGAPA2 2 | 3(41): 0(0) | 60 | TNFAIP3 2,3 | 3(13): 0(0) | 90 | CDCA2 3 | 3(8): 1(3) |
| Control > Mild AD, 2x Sera, 1.5x Hits | ||||||||
|---|---|---|---|---|---|---|---|---|
| Symbol | Control: Mild | Symbol | Control: Mild | Symbol | Control: Mild | |||
| [#Sera(#Hits)] | [#Sera(#Hits)] | [#Sera(#Hits)] | ||||||
| 1 | RIF1 6 | 5(105): 2(43) | 23 | ADAMTS13 1,2,6 | 3(29): 0(0) | 45 | BOC 3 | 3(14): 0(0) |
| 2 | ZNF142 | 4(67): 0(0) | 24 | AMBP 7 | 3(28): 0(0) | 46 | LRRC4 | 3(14): 0(0) |
| 3 | BAZ1A | 4(39): 0(0) | 25 | PDE9A 1,5 | 3(27): 0(0) | 47 | SNTB2 | 3(11): 0(0) |
| 4 | LAMA3 1,2 | 4(46): 1(2) | 26 | SH3PXD2A 1,5 | 3(27): 0(0) | 48 | MC1R 1,2,4,5,6 | 3(10): 0(0) |
| 5 | NAV2 1,3,4 | 4(70): 2(34) | 27 | STK36 3,4 | 3(27): 0(0) | 49 | RAB3GAP2 1 | 3(9): 0(0) |
| 6 | OBSCN | 4(32): 2(17) | 28 | STXBP5L 1 | 3(26): 0(0) | 50 | POTEB 2 | 3(3): 0(0) |
| 7 | MT-CO1 1,6 | 4(22): 2(9) | 29 | PLXNB2 2,3,6 | 3(25): 0(0) | 51 | CYP4F11 | 3(143): 1(8) |
| 8 | LRP1B 1,6 | 4(17): 2(6) | 30 | ZNF592 1 | 3(23): 0(0) | 52 | KMT2A 3 | 3(124): 1(9) |
| 9 | PITRM1 1,4,7 | 3(94): 0(0) | 31 | CLDN7 2,6 | 3(22): 0(0) | 53 | ADAMTS7 2,6 | 3(50): 1(18) |
| 10 | PCSK5 1,2 | 3(56): 0(0) | 32 | CRB2 2,3,6,7 | 3(22): 0(0) | 54 | KAT5 3 | 3(48): 1(5) |
| 11 | ITPR1 3,4,7 | 3(52): 0(0) | 33 | YEATS2 | 3(22): 0(0) | 55 | PTPRQ 1 | 3(40): 1(5) |
| 12 | OAS1 1 | 3(51): 0(0) | 34 | CRYBG2 1 | 3(21): 0(0) | 56 | TSHZ2 1 | 3(36): 1(9) |
| 13 | UNC8 4 | 3(51): 0(0) | 35 | CSMD1 1,2,3 | 3(21): 0(0) | 57 | KIF13B 3,4 | 3(35): 1(9) |
| 14 | KLHL29 1,2,7 | 3(50): 0(0) | 36 | DNMBP 1 | 3(21): 0(0) | 58 | RGS12 4 | 3(33): 1(11) |
| 15 | DIAPH3 3 | 3(49): 0(0) | 37 | WNT8A 2,3,6 | 3(21): 0(0) | 59 | REXO1 | 3(32): 1(3) |
| 16 | POTEB3 1 | 3(43): 0(0) | 38 | LILRA2 2 | 3(20): 0(0) | 60 | CASZ1 2,3 | 3(26): 1(8) |
| 17 | STAB2 2 | 3(39): 0(0) | 39 | ADAMTS16 | 3(19): 0(0) | 61 | BCR6 | 3(25): 1(5) |
| 18 | CASP8AP2 | 3(32): 0(0) | 40 | IGLL1 | 3(19): 0(0) | 62 | LRP6 1,2,3,4,5,6 | 3(24): 1(11) |
| 19 | GATAD2A 3 | 3(32): 0(0) | 41 | ITGB2 1 | 3(19): 0(0) | 63 | DNAH12 | 3(22): 1(6) |
| 20 | GRN 1,2,3,4,5,6,7 | 3(32): 0(0) | 42 | PARP4 1 | 3(19): 0(0) | 64 | ITGB1BP2 1 | 3(11): 1(3) |
| 21 | TUBGCP6 | 3(31): 0(0) | 43 | CTCF 1,2,3,7 | 3(16): 0(0) | |||
| 22 | THSD7B | 3(30): 0(0) | 44 | IGSF1 | 3(16): 0(0) | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanas, J.S.; Hocker, J.R.S.; Vannarath, C.A.; Lerner, M.R.; Blair, S.G.; Lightfoot, S.A.; Hanas, R.J.; Couch, J.R.; Hershey, L.A. Distinguishing Alzheimer’s Disease Patients and Biochemical Phenotype Analysis Using a Novel Serum Profiling Platform: Potential Involvement of the VWF/ADAMTS13 Axis. Brain Sci. 2021, 11, 583. https://doi.org/10.3390/brainsci11050583
Hanas JS, Hocker JRS, Vannarath CA, Lerner MR, Blair SG, Lightfoot SA, Hanas RJ, Couch JR, Hershey LA. Distinguishing Alzheimer’s Disease Patients and Biochemical Phenotype Analysis Using a Novel Serum Profiling Platform: Potential Involvement of the VWF/ADAMTS13 Axis. Brain Sciences. 2021; 11(5):583. https://doi.org/10.3390/brainsci11050583
Chicago/Turabian StyleHanas, Jay S., James R. S. Hocker, Christian A. Vannarath, Megan R. Lerner, Scott G. Blair, Stan A. Lightfoot, Rushie J. Hanas, James R. Couch, and Linda A. Hershey. 2021. "Distinguishing Alzheimer’s Disease Patients and Biochemical Phenotype Analysis Using a Novel Serum Profiling Platform: Potential Involvement of the VWF/ADAMTS13 Axis" Brain Sciences 11, no. 5: 583. https://doi.org/10.3390/brainsci11050583
APA StyleHanas, J. S., Hocker, J. R. S., Vannarath, C. A., Lerner, M. R., Blair, S. G., Lightfoot, S. A., Hanas, R. J., Couch, J. R., & Hershey, L. A. (2021). Distinguishing Alzheimer’s Disease Patients and Biochemical Phenotype Analysis Using a Novel Serum Profiling Platform: Potential Involvement of the VWF/ADAMTS13 Axis. Brain Sciences, 11(5), 583. https://doi.org/10.3390/brainsci11050583