
Changes in Representation of Thalamic Projection Neurons within Prefrontal-Thalamic-Hippocampal Circuitry in a Rat Model of Third Trimester Binge Drinking


Abstract
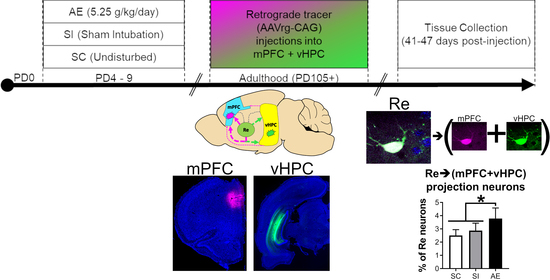
1. Introduction
1.1. Gestational Alcohol Exposure Is a Global Public Health Concern, Often Resulting in Impaired Executive Functioning
1.2. Thalamic Nucleus Reuniens Supports EF by Coordinating Prefrontal Cortex and Hippocampus
1.3. Neuroanatomical Alterations to mPFC-Re-HPC Circuitry in FASD
2. Materials and Methods
2.1. Experimental Subjects
2.1.1. Experimental Postnatal Treatments
2.1.2. Blood Alcohol Concentration Analysis
2.1.3. Weaning
2.2. Stereotaxic Surgery
AAVrg-CAG Characteristics
2.3. Tissue Fixation
2.4. Immunofluorescent Labeling
2.5. Fluorescence Microscopy
2.6. Unbiased Stereological Estimation
2.7. Statistical Analyses
3. Results
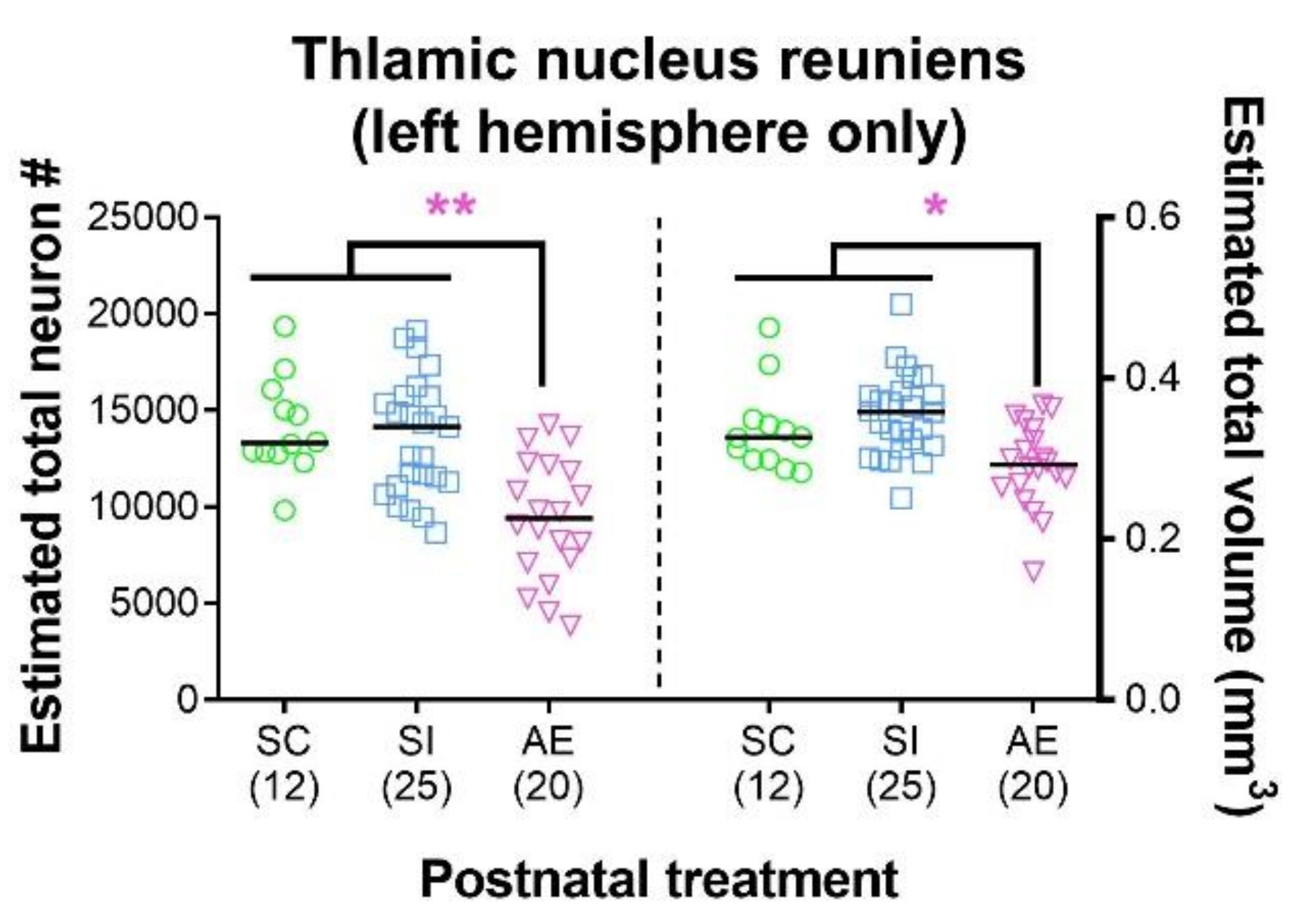
3.1. High-Dose AE Reduces Re Neuron Number and Volume
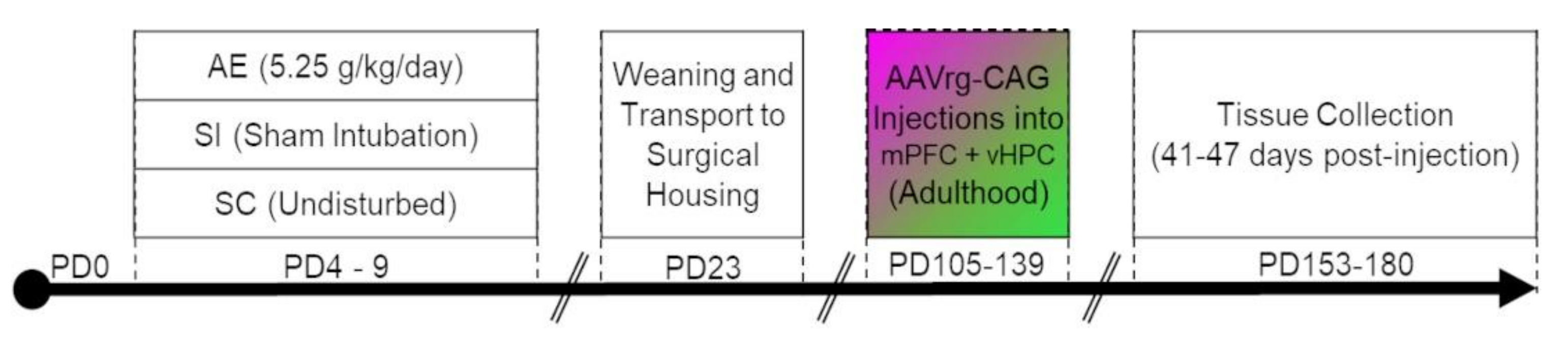
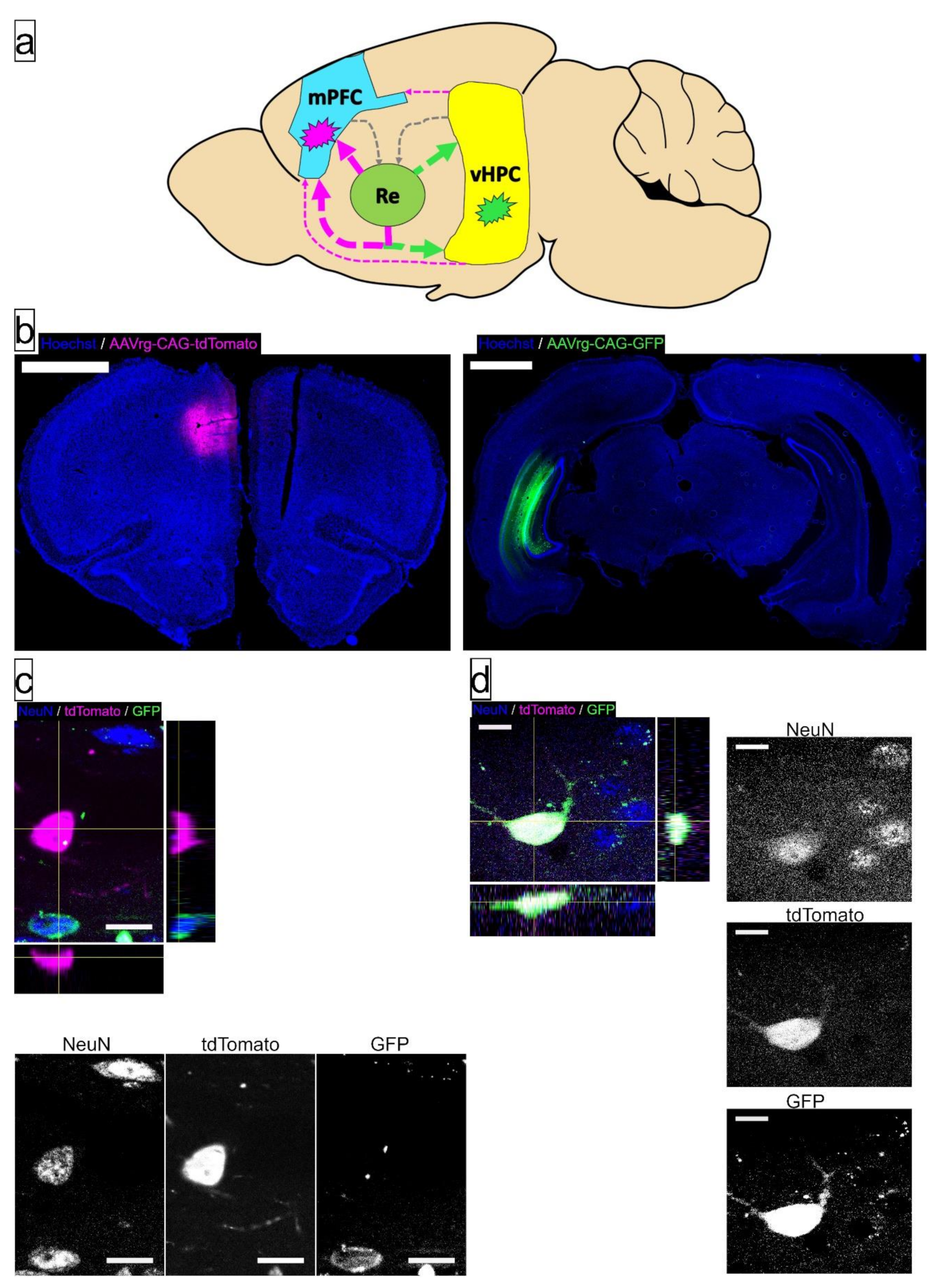
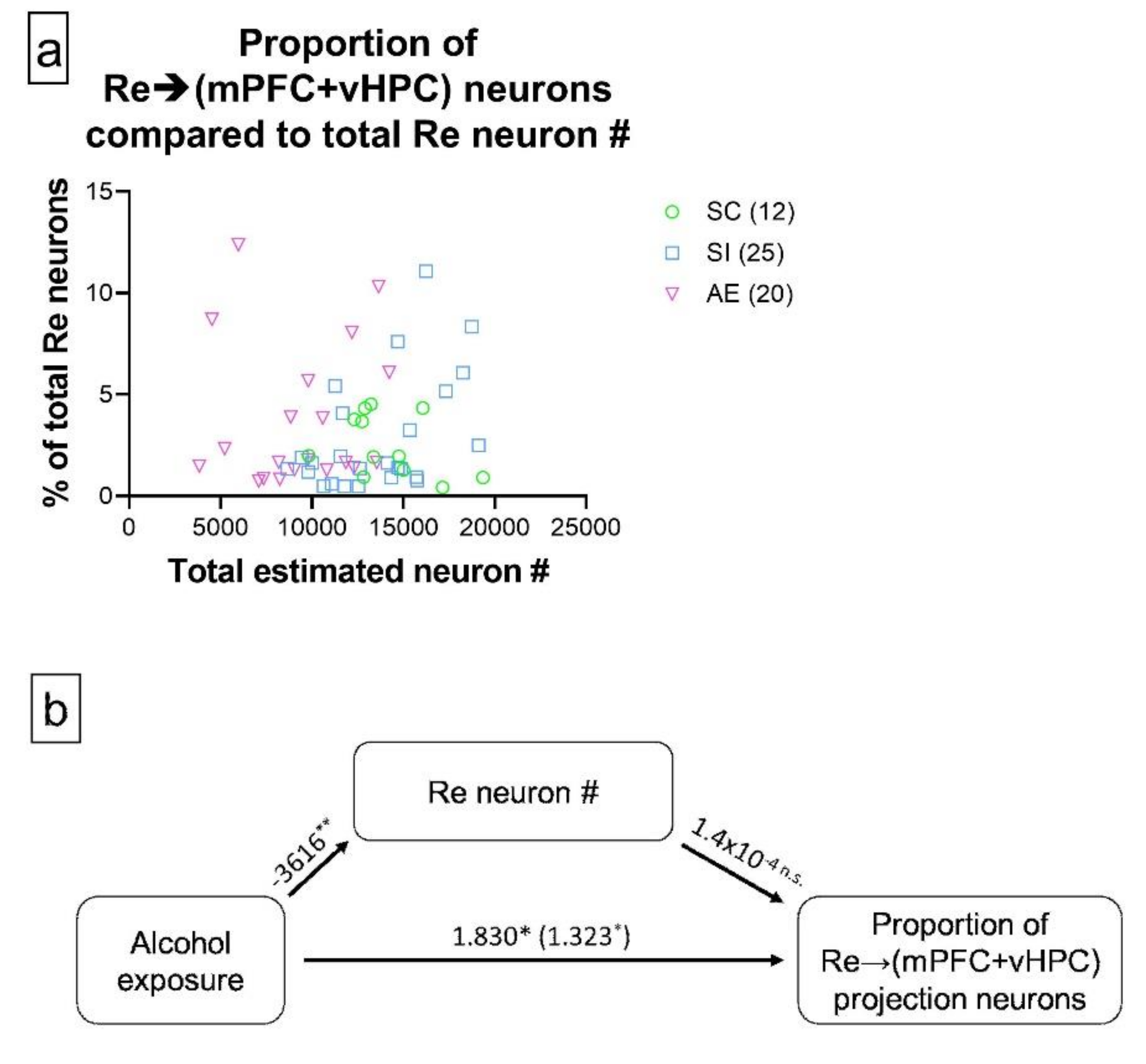
3.2. AE Selectively Increased the Proportion of Re Projection Neurons That Concurrently Innervate mPFC and vHPC, but Not Those That Innervate either Region Individually
3.3. AE-Induced Changes in Re→(mPFC + vHPC) Projection Neuron Representation Are Not Mediated by AE-Induced Re Neuron loss
4. Discussion
4.1. Summary of Key Findings
4.2. The Pressing Need to Identify Primary versus Secondary Consequences of Developmental AE
4.3. A Proposed Cellular Mechanism by Which Re Damage May Impair PFC Function through Altered Timing of HPC and Re Input
4.4. Limitations of the Current Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burton, R.; Sheron, N. No level of alcohol consumption improves health. Lancet 2018, 392, 987–988. [Google Scholar] [CrossRef]
- Bilbo, S.D.; Schwarz, J.M. The immune system and developmental programming of brain and behavior. Front. Neuroendocr. 2012, 33, 267–286. [Google Scholar] [CrossRef] [PubMed]
- Baram, T.Z.; Donato, F.; Holmes, G.L. Construction and disruption of spatial memory networks during development. Learn. Mem. 2019, 26, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Klintsova, A.Y.; Hamilton, G.F.; Boschen, K.E. Long-Term Consequences of Developmental Alcohol Exposure on Brain Structure and Function: Therapeutic Benefits of Physical Activity. Brain Sci. 2012, 3, 1–38. [Google Scholar] [CrossRef]
- Astley, S.J.; Clarren, S.K. Diagnosing the full spectrum of fetal alcohol-exposed individuals: introducing the 4-digit diagnostic code. Alcohol Alcohol. 2000, 35, 400–410. [Google Scholar] [CrossRef]
- Hoyme, H.E.; Kalberg, W.O.; Elliott, A.J.; Blankenship, J.; Buckley, D.; Marais, A.-S.; Manning, M.A.; Robinson, L.K.; Adam, M.P.; Abdul-Rahman, O.; et al. Updated Clinical Guidelines for Diagnosing Fetal Alcohol Spectrum Disorders. Pediatrics 2016, 138, e20154256. [Google Scholar] [CrossRef] [PubMed]
- Dobbing, J.; Sands, J. Comparative aspects of the brain growth spurt. Early Hum. Dev. 1979, 3, 79–83. [Google Scholar] [CrossRef]
- Umer, A.; Lilly, C.; Hamilton, C.; Baldwin, A.; Breyel, J.; Tolliver, A.; Mullins, C.; John, C.; Maxwell, S. Prevalence of alcohol use in late pregnancy. Pediatr. Res. 2020, 88, 312–319. [Google Scholar] [CrossRef]
- Kelly, S.J.; Bonthius, D.J.; West, J.R. Developmental Changes in Alcohol Pharmacokinetics in Rats. Alcohol. Clin. Exp. Res. 1987, 11, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.J.; Lawrence, C.R. Intragastric Intubation of Alcohol During the Perinatal Period. In Methods in Molecular Biology; Springer: Berlin, Germany, 2008; Volume 447, pp. 101–110. [Google Scholar]
- Khoury, J.E.; Milligan, K.; Girard, T.A. Executive Functioning in Children and Adolescents Prenatally Exposed to Alcohol: A Meta-Analytic Review. Neuropsychol. Rev. 2015, 25, 149–170. [Google Scholar] [CrossRef] [PubMed]
- Suchy, Y. Executive Functioning: Overview, Assessment, and Research Issues for Non-Neuropsychologists. Ann. Behav. Med. 2009, 37, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Gursky, Z.; Savage, L.; Klintsova, A. Executive functioning-specific behavioral impairments in a rat model of human third trimester binge drinking implicate prefrontal-thalamo-hippocampal circuitry in Fetal Alcohol Spectrum Disorders. Behav. Brain Res. 2021, 113208. [Google Scholar] [CrossRef]
- Spellman, T.; Rigotti, M.; Ahmari, S.E.; Fusi, S.; Gogos, J.A.; Gordon, J.A. Hippocampal–prefrontal input supports spatial encoding in working memory. Nat. Cell Biol. 2015, 522, 309–314. [Google Scholar] [CrossRef]
- Hallock, H.L.; Wang, A.; Griffin, A.L. Ventral Midline Thalamus Is Critical for Hippocampal-Prefrontal Synchrony and Spatial Working Memory. J. Neurosci. 2016, 36, 8372–8389. [Google Scholar] [CrossRef]
- Dolleman-van der Weel, M.J.; Griffin, A.L.; Ito, H.T.; Shapiro, M.L.; Witter, M.P.; Vertes, R.P.; Allen, T.A. The nucleus reuniens of the thalamus sits at the nexus of a hippocampus and medial prefrontal cortex circuit enabling memory and behavior. Learn Mem. 2019, 26, 191–205. [Google Scholar] [CrossRef]
- Varela, C.; Kumar, S.; Yang, J.Y.; Wilson, M.A. Anatomical substrates for direct interactions between hippocampus, medial prefrontal cortex, and the thalamic nucleus reuniens. Brain Struct. Funct. 2014, 219, 911–929. [Google Scholar] [CrossRef]
- Hoover, W.B.; Vertes, R.P. Collateral projections from nucleus reuniens of thalamus to hippocampus and medial prefrontal cortex in the rat: A single and double retrograde fluorescent labeling study. Brain Struct. Funct. 2011, 217, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Banks, P.J.; Warburton, E.C.; Bashir, Z.I. Plasticity in prefrontal cortex induced by coordinated nucleus reuniens and hippocampal synaptic transmission. BioarXiv 2020. [Google Scholar] [CrossRef]
- Klein, M.M.; Cholvin, T.; Cosquer, B.; Salvadori, A.; Le Mero, J.; Kourouma, L.; Boutillier, A.-L.; De Vasconcelos, A.P.; Cassel, J.-C. Ventral midline thalamus lesion prevents persistence of new (learning-triggered) hippocampal spines, delayed neocortical spinogenesis, and spatial memory durability. Brain Struct. Funct. 2019, 224, 1659–1676. [Google Scholar] [CrossRef]
- Gursky, Z.H.; Savage, L.M.; Klintsova, A.Y. Nucleus reuniens of the midline thalamus of a rat is specifically damaged after early postnatal alcohol exposure. NeuroReport 2019, 30, 748–752. [Google Scholar] [CrossRef]
- Gursky, Z.H.; Spillman, E.C.; Klintsova, A.Y. Single-day Postnatal Alcohol Exposure Induces Apoptotic Cell Death and Causes long-term Neuron Loss in Rodent Thalamic Nucleus Reuniens. Neuroscience 2020, 435, 124–134. [Google Scholar] [CrossRef]
- Helfer, J.L.; Calizo, L.H.; Dong, W.K.; Goodlett, C.R.; Greenough, W.T.; Klintsova, A.Y. Binge-like postnatal alcohol exposure triggers cortical gliogenesis in adolescent rats. J. Comp. Neurol. 2009, 514, 259–271. [Google Scholar] [CrossRef]
- Goodlett, C.R.; Johnson, T.B. Neonatal Binge Ethanol Exposure Using Intubation: Timing and Dose Effects on Place Learning. Neurotoxicology Teratol. 1997, 19, 435–446. [Google Scholar] [CrossRef]
- Stanton, M.E.; Goodlett, C.R. Neonatal Ethanol Exposure Impairs Eyeblink Conditioning in Weanling Rats. Alcohol. Clin. Exp. Res. 1998, 22, 270. [Google Scholar] [CrossRef]
- Ryan, S.H.; Williams, J.K.; Thomas, J.D. Choline supplementation attenuates learning deficits associated with neonatal alcohol exposure in the rat: Effects of varying the timing of choline administration. Brain Res. 2008, 1237, 91–100. [Google Scholar] [CrossRef]
- Gursky, Z.H.; Klintsova, A.Y. Wheel Running and Environmental Complexity as a Therapeutic Intervention in an Animal Model of FASD. J. Vis. Exp. 2017, 2017. [Google Scholar] [CrossRef]
- Galbraith, S.; Daniel, J.A.; Vissel, B. A Study of Clustered Data and Approaches to Its Analysis. J. Neurosci. 2010, 30, 10601–10608. [Google Scholar] [CrossRef]
- Giesbrecht, F.G.; Burns, J.C. Two-Stage Analysis Based on a Mixed Model: Large-Sample Asymptotic Theory and Small-Sample Simulation Results. Biometrics 1985, 41, 477. [Google Scholar] [CrossRef]
- West, J.R.; Hamre, K.M.; Pierce, D.R. Delay in brain growth induced by alcohol in artificially reared rat pups. Alcohol 1984, 1, 213–222. [Google Scholar] [CrossRef]
- Bonthius, D.J.; West, J.R. Blood alcohol concentration and microencephaly: A dose-response study in the neonatal rat. Teratology 1988, 37, 223–231. [Google Scholar] [CrossRef]
- Boschen, K.; Criss, K.; Palamarchouk, V.; Roth, T.; Klintsova, A. Effects of developmental alcohol exposure vs. intubation stress on BDNF and TrkB expression in the hippocampus and frontal cortex of neonatal rats. Int. J. Dev. Neurosci. 2015, 43, 16–24. [Google Scholar] [CrossRef]
- Heroux, N.A.; Robinson-Drummer, P.A.; Kawan, M.; Rosen, J.B.; Stanton, M.E. Neonatal ethanol exposure impairs long-term context memory formation and prefrontal immediate early gene expression in adolescent rats. Behav. Brain Res. 2019, 359, 386–395. [Google Scholar] [CrossRef]
- Otero, N.K.H.; Thomas, J.D.; Saski, C.A.; Xia, X.; Kelly, S.J. Choline Supplementation and DNA Methylation in the Hippocampus and Prefrontal Cortex of Rats Exposed to Alcohol During Development. Alcohol. Clin. Exp. Res. 2012, 36, 1701–1709. [Google Scholar] [CrossRef]
- Thomas, J.D.; Sather, T.M.; Whinery, L.A. Voluntary exercise influences behavioral development in rats exposed to alcohol during the neonatal brain growth spurt. Behav. Neurosci. 2008, 122, 1264–1273. [Google Scholar] [CrossRef]
- Wagner, A.F.; Hunt, P.S. Impaired trace fear conditioning following neonatal ethanol: Reversal by choline. Behav. Neurosci. 2006, 120, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Bonthius, D.J.; West, J.R. Alcohol-Induced Neuronal Loss in Developing Rats: Increased Brain Damage with Binge Exposure. Alcohol. Clin. Exp. Res. 1990, 14, 107–118. [Google Scholar] [CrossRef]
- Milbocker, K.A.; Klintsova, A.Y. Examination of cortically projecting cholinergic neurons following exercise and environmental intervention in a rodent model of fetal alcohol spectrum disorders. Birth Defects Res. 2021, 113, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Angulo-Garcia, D.; Ferraris, M.; Ghestem, A.; Bernard, C.; Quilichini, P.P. Spatio-temporal organization of cell assemblies in Nucleus Reuniens during slow oscillations. BiorXiv 2018. [Google Scholar] [CrossRef]
- O’Neill, P.-K.; Gordon, J.A.; Sigurdsson, T. Theta Oscillations in the Medial Prefrontal Cortex Are Modulated by Spatial Working Memory and Synchronize with the Hippocampus through Its Ventral Subregion. J. Neurosci. 2013, 33, 14211–14224. [Google Scholar] [CrossRef]
- Swanson, L.W. A direct projection from Ammon’s horn to prefrontal cortex in the rat. Brain Res. 1981, 217, 150–154. [Google Scholar] [CrossRef]
- Hauer, B.E.; Pagliardini, S.; Dickson, C.T. The Reuniens Nucleus of the Thalamus Has an Essential Role in Coordinating Slow-Wave Activity between Neocortex and Hippocampus. eneuro 2019, 6. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C.R.; Emson, P.C. AChE-stained horizontal sections of the rat brain in stereotaxic coordinates. J. Neurosci. Methods 1980, 3, 129–149. [Google Scholar] [CrossRef]
- Clarren, S.K.; Alvord, E.C.; Sumi, S.M.; Streissguth, A.P.; Smith, D.W. Brain malformations related to prenatal exposure to ethanol. J. Pediatr. 1978, 92, 64–67. [Google Scholar] [CrossRef]
- Vertes, R.P.; Hoover, W.B.; Valle, A.C.D.; Sherman, A.; Rodriguez, J.J. Efferent projections of reuniens and rhomboid nuclei of the thalamus in the rat. J. Comp. Neurol. 2006, 499, 768–796. [Google Scholar] [CrossRef]
- Scheimann, J.R.; Moloney, R.D.; Mahbod, P.; Morano, R.L.; Fitzgerald, M.; Hoskins, O.; Packard, B.A.; Cotella, E.M.; Hu, Y.-C.; Herman, J.P. Conditional deletion of glucocorticoid receptors in rat brain results in sex-specific deficits in fear and coping behaviors. eLife 2019, 8, 8. [Google Scholar] [CrossRef]
- Lind, D.; Franken, S.; Kappler, J.; Jankowski, J.; Schilling, K. Characterization of the neuronal marker NeuN as a multiply phosphorylated antigen with discrete subcellular localization. J. Neurosci. Res. 2005, 79, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Kapeller-Libermann, D.; Travaglia, A.; Inda, M.C.; Alberini, C.M. Direct dorsal hippocampal–prelimbic cortex connections strengthen fear memories. Nat. Neurosci. 2016, 20, 52–61. [Google Scholar] [CrossRef]
- Cassel, J.C.; Pereira de Vasconcelos, A. Importance of the ventral midline thalamus in driving hippocampal functions. Prog. Brain Res. 2015, 219, 145–161. [Google Scholar]
- Mitala, C.M.; Wang, Y.; Borland, L.M.; Jung, M.; Shand, S.; Watkins, S.; Weber, S.G.; Michael, A.C. Impact of microdialysis probes on vasculature and dopamine in the rat striatum: A combined fluorescence and voltammetric study. J. Neurosci. Methods 2008, 174, 177–185. [Google Scholar] [CrossRef]
- Napper, R.M.A. Total Number Is Important: Using the Disector Method in Design-Based Stereology to Understand the Structure of the Rodent Brain. Front. Neuroanat. 2018, 12, 16. [Google Scholar] [CrossRef]
- Sahin, B.; Acer, N.; Sonmez, O.; Emirzeoglu, M.; Başaloğlu, H.; Uzun, A.; Bilgiç, S. Comparison of four methods for the estimation of intracranial volume: A gold standard study. Clin. Anat. 2007, 20, 766–773. [Google Scholar] [CrossRef]
- Slomianka, L.; West, M. Estimators of the precision of stereological estimates: An example based on the CA1 pyramidal cell layer of rats. Neuroscience 2005, 136, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Gundersen, H.J.G.; Jensen, E.B. The efficiency of systematic sampling in stereology and its prediction. J. Microsc. 1987, 147, 229–263. [Google Scholar] [CrossRef]
- Barker, G.R.I.; Warburton, E.C. Object-in-Place Associative Recognition Memory Depends on Glutamate Receptor Neurotransmission Within Two Defined Hippocampal-Cortical Circuits: A Critical Role for AMPA and NMDA Receptors in the Hippocampus, Perirhinal, and Prefrontal Cortices. Cereb. Cortex 2013, 25, 472–481. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- RStudio Team. RStudio: Integrated Development Environment for R; RStudio: Boston, MA, USA, 2016. [Google Scholar]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Software 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Kuznetsova, A.; Brockhoff, P.B.; Christensen, R.H.B. lmerTest Package: Tests in Linear Mixed Effects Models. J. Stat. Softw. 2017, 82, 1–26. [Google Scholar] [CrossRef]
- Tingley, D.; Yamamoto, T.; Hirose, K.; Keele, L.; Imai, K. mediation: R Package for Causal Mediation Analysis. J. Stat. Software 2014, 59, 1–38. [Google Scholar] [CrossRef]
- Wickham, H.; Bryan, J. readxl: Read Excel Files. Available online: https://cran.r-project.org/web/packages/readxl/index.html (accessed on 16 January 2021).
- Lüdecke, D. sjPlot: Data Visualization for Statistics in Social Science. Available online: https://cran.r-project.org/web/packages/sjPlot/index.html (accessed on 16 January 2021).
- Wickham, H. tidyverse: Easily Install and Load the ‘Tidyverse’. Available online: https://cran.r-project.org/web/packages/tidyverse/index.html (accessed on 16 January 2021).
- Gursky, Z.H.; Johansson, J.R.; Klintsova, A.Y. Postnatal alcohol exposure and adolescent exercise have opposite effects on cerebellar microglia in rat. Int. J. Dev. Neurosci. 2020, 80, 558–571. [Google Scholar] [CrossRef]
- Granato, A.; Dering, B. Alcohol and the Developing Brain: Why Neurons Die and How Survivors Change. Int. J. Mol. Sci. 2018, 19, 2992. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidou, C. Ethanol-Induced Apoptotic Neurodegeneration and Fetal Alcohol Syndrome. Science 2000, 287, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Südhof, T.C. A Neural Circuit for Memory Specificity and Generalization. Science 2013, 339, 1290–1295. [Google Scholar] [CrossRef]
- McDonald, R.J.; Devan, B.D.; Hong, N.S. Multiple memory systems: The power of interactions. Neurobiol. Learn. Mem. 2004, 82, 333–346. [Google Scholar] [CrossRef]
- Baum, G.L.; Cui, Z.; Roalf, D.R.; Ciric, R.; Betzel, R.F.; Larsen, B.; Cieslak, M.; Cook, P.A.; Xia, C.H.; Moore, T.M.; et al. Development of structure–function coupling in human brain networks during youth. Proc. Natl. Acad. Sci. USA 2020, 117, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Dan, Y.; Poo, M.-M. Spike Timing-Dependent Plasticity of Neural Circuits. Neuron 2004, 44, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Basu, J.; Zaremba, J.D.; Cheung, S.K.; Hitti, F.L.; Zemelman, B.V.; Losonczy, A.; Siegelbaum, S.A. Gating of hippocampal activity, plasticity, and memory by entorhinal cortex long-range inhibition. Science 2016, 351, aaa5694. [Google Scholar] [CrossRef]
- Briner, A.; Nikonenko, I.; De Roo, M.; Dayer, A.; Muller, D.; Vutskits, L. Developmental Stage-dependent Persistent Impact of Propofol Anesthesia on Dendritic Spines in the Rat Medial Prefrontal Cortex. Anesthesiology 2011, 115, 282–293. [Google Scholar] [CrossRef]
- Huttenlocher, P.R.; Dabholkar, A.S. Regional differences in synaptogenesis in human cerebral cortex. J. Comparat. Neurology 1997, 387, 167–178. [Google Scholar] [CrossRef]
- Heroux, N.A.; Horgan, C.J.; Pinizzotto, C.C.; Rosen, J.B.; Stanton, M.E. Medial prefrontal and ventral hippocampal contributions to incidental context learning and memory in adolescent rats. Neurobiol. Learn. Mem. 2019, 166, 107091. [Google Scholar] [CrossRef]
- Viena, T.D.; Linley, S.B.; Vertes, R.P. Inactivation of nucleus reuniens impairs spatial working memory and behavioral flexibility in the rat. Hippocampus 2018, 28, 297–311. [Google Scholar] [CrossRef]
- Hellemans, K.G.; Sliwowska, J.H.; Verma, P.; Weinberg, J. Prenatal alcohol exposure: Fetal programming and later life vulnerability to stress, depression and anxiety disorders. Neurosci. Biobehav. Rev. 2010, 34, 791–807. [Google Scholar] [CrossRef] [PubMed]
- Raineki, C.; Bodnar, T.S.; Holman, P.J.; Baglot, S.L.; Lan, N.; Weinberg, J. Effects of early-life adversity on immune function are mediated by prenatal environment: Role of prenatal alcohol exposure. Brain Behav. Immun. 2017, 66, 210–220. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Postnatal Treatment | Sex | Number of Litters Represented | Total Sample Size |
|---|---|---|---|
| SC | Female | 2 | 8 |
| SC | Male | 2 | 4 |
| SI | Female | 7 | 14 |
| SI | Male | 7 | 11 |
| AE | Female | 5 | 12 |
| AE | Male | 6 | 8 |
| Postnatal Treatment | Sex | Sample Size | mPFC Injection Diffusion Volume (Mean CE ± SEM) | vHPC Injection Diffusion Volume (Mean CE ± SEM) | Re Neuron # (Mean CE ± SEM) |
|---|---|---|---|---|---|
| SC | Female | 08 | 0.075 ± 0.010 | 0.048 ± 0.004 | 0.074 ± 0.002 |
| SC | Male | 04 | 0.068 ± 0.021 | 0.044 ± 0.006 | 0.070 ± 0.000 |
| SI | Female | 14 | 0.074 ± 0.010 | 0.057 ± 0.010 | 0.074 ± 0.002 |
| SI | Male | 11 | 0.063 ± 0.007 | 0.052 ± 0.008 | 0.071 ± 0.003 |
| AE | Female | 12 | 0.071 ± 0.013 | 0.052 ± 0.007 | 0.084 ± 0.005 |
| AE | Male | 08 | 0.056 ± 0.007 | 0.060 ± 0.019 | 0.091 ± 0.005 |
| Re Neuron Number | Re Volume (mm3) | |||||||
|---|---|---|---|---|---|---|---|---|
| Predictors | Estimates | 95% CI | p | df | Estimates | 95% CI | p | df |
| (Intercept) | 13884.911 | 12109.685–15660.138 | <0.001 | 17.265 | 0.352 | 0.324–0.379 | <0.001 | 51.000 |
| SC | −461.776 | −3780.639–2857.086 | 0.790 | 10.724 | −0.029 | −0.074–0.016 | 0.217 | 51.000 |
| AE | −3615.875 | −5673.838–−1557.913 | 0.001 | 45.344 | −0.050 | −0.090–−0.010 | 0.019 | 51.000 |
| Male | −125.126 | −2216.416–1966.163 | 0.907 | 44.351 | 0.005 | −0.036–0.046 | 0.815 | 51.000 |
| SC × Male | 2800.211 | −1040.931–6641.354 | 0.160 | 45.444 | 0.037 | −0.038–0.112 | 0.340 | 51.000 |
| AE × Male | −1074.289 | −4294.209–2145.631 | 0.516 | 46.255 | −0.031 | −0.093–0.032 | 0.339 | 51.000 |
| Random Effects | ||||||||
| σ2 | 6925310.96 | 0.00 | ||||||
| τ00 | 2151974.33 Litter | 0.00 Litter | ||||||
| ICC | 0.24 | |||||||
| N | 9 Litter | 9 Litter | ||||||
| Observations | 57 | 57 | ||||||
| Marginal R2/Conditional R2 | 0.336/0.493 | 0.255/NA | ||||||
| Number of Projection Neurons: Re→(mPFC + vHPC) | % of total Re Neurons: Re→(mPFC + vHPC) | |||||||
|---|---|---|---|---|---|---|---|---|
| Predictors | Estimates | 95% CI | p | df | Estimates | 95% CI | p | df |
| (Intercept) | 397.355 | 117.713–676.997 | 0.005 | 49.000 | 2.864 | 0.725–5.002 | 0.009 | 49.000 |
| SC | 9.298 | −544.136–562.733 | 0.974 | 49.000 | 0.102 | −4.196–4.400 | 0.963 | 49.000 |
| AE | 116.009 | −133.738–365.756 | 0.363 | 49.000 | 2.006 | 0.308–3.705 | 0.021 | 49.000 |
| Male | 123.429 | −129.498–376.356 | 0.339 | 49.000 | 0.453 | −1.266–2.171 | 0.606 | 49.000 |
| SC × Male | −280.910 | −747.112–185.292 | 0.238 | 49.000 | −1.686 | −4.856–1.485 | 0.297 | 49.000 |
| AE × Male | −322.897 | −714.791–68.996 | 0.106 | 49.000 | −1.657 | −4.323–1.010 | 0.223 | 49.000 |
| Random Effects | ||||||||
| σ2 | 100976.98 | 4.66 | ||||||
| τ00 | 89389.05 Litter | 5.87 Litter | ||||||
| ICC | 0.47 | 0.56 | ||||||
| N | 9 Litter | 9 Litter | ||||||
| Observations | 57 | 57 | ||||||
| Marginal R2/Conditional R2 | 0.039/0.490 | 0.068/0.588 | ||||||
| Number of Projection Neurons: Re→mPFC | % of toTal Re Neurons: Re→mPFC | |||||||
|---|---|---|---|---|---|---|---|---|
| Predictors | Estimates | 95% CI | p | df | Estimates | 95% CI | p | df |
| (Intercept) | 323.757 | 131.349–516.165 | 0.001 | 49.000 | 2.441 | 0.764–4.119 | 0.004 | 49.000 |
| SC | 10.852 | −308.221–329.925 | 0.947 | 49.000 | 0.071 | −2.779–2.922 | 0.961 | 49.000 |
| AE | 37.432 | −245.785–320.649 | 0.796 | 49.000 | 1.036 | −1.356–3.429 | 0.396 | 49.000 |
| Male | −171.805 | −461.872–118.261 | 0.246 | 49.000 | −1.250 | −3.697–1.197 | 0.317 | 49.000 |
| SC × Male | 110.667 | −417.063–638.396 | 0.681 | 49.000 | 0.550 | −3.910–5.010 | 0.809 | 49.000 |
| AE × Male | 129.919 | −308.392–568.229 | 0.561 | 49.000 | 1.476 | −2.238–5.190 | 0.436 | 49.000 |
| Random Effects | ||||||||
| σ2 | 134920.91 | 9.57 | ||||||
| τ00 | 0.00 Litter | 0.31 Litter | ||||||
| ICC | 0.03 | |||||||
| N | 9 Litter | 9 Litter | ||||||
| Observations | 57 | 57 | ||||||
| Marginal R2/Conditional R2 | 0.039/NA | 0.073/0.102 | ||||||
| Number of Projection Neurons: Re→vHPC | % of Total Re Neurons: Re→vHPC | |||||||
|---|---|---|---|---|---|---|---|---|
| Predictors | Estimates | 95% CI | p | df | Estimates | 95% CI | p | df |
| (Intercept) | 1885.348 | 584.630–3186.066 | 0.004 | 49.000 | 12.205 | 3.688–20.721 | 0.005 | 49.000 |
| SC | 1081.260 | −1611.037–3773.558 | 0.431 | 49.000 | 8.259 | −9.383–25.900 | 0.359 | 49.000 |
| AE | −557.028 | −1267.687–153.631 | 0.124 | 49.000 | 0.057 | −4.523–4.636 | 0.981 | 49.000 |
| Male | −160.655 | −878.649–557.339 | 0.661 | 49.000 | −0.626 | −5.252–4.001 | 0.791 | 49.000 |
| SC × Male | −503.139 | −1829.473–823.195 | 0.457 | 49.000 | −6.417 | −14.964–2.130 | 0.141 | 49.000 |
| AE × Male | 281.193 | −835.784–1398.170 | 0.622 | 49.000 | 3.208 | −3.990–10.406 | 0.382 | 49.000 |
| Random Effects | ||||||||
| σ2 | 812289.37 | 33.73 | ||||||
| τ00 | 2651619.58 Litter | 114.25 Litter | ||||||
| ICC | 0.77 | 0.77 | ||||||
| N | 9 Litter | 9 Litter | ||||||
| Observations | 57 | 57 | ||||||
| Marginal R2/Conditional R2 | 0.074/0.783 | 0.055/0.785 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gursky, Z.H.; Klintsova, A.Y. Changes in Representation of Thalamic Projection Neurons within Prefrontal-Thalamic-Hippocampal Circuitry in a Rat Model of Third Trimester Binge Drinking. Brain Sci. 2021, 11, 323. https://doi.org/10.3390/brainsci11030323
Gursky ZH, Klintsova AY. Changes in Representation of Thalamic Projection Neurons within Prefrontal-Thalamic-Hippocampal Circuitry in a Rat Model of Third Trimester Binge Drinking. Brain Sciences. 2021; 11(3):323. https://doi.org/10.3390/brainsci11030323
Chicago/Turabian StyleGursky, Zachary H., and Anna Y. Klintsova. 2021. "Changes in Representation of Thalamic Projection Neurons within Prefrontal-Thalamic-Hippocampal Circuitry in a Rat Model of Third Trimester Binge Drinking" Brain Sciences 11, no. 3: 323. https://doi.org/10.3390/brainsci11030323
APA StyleGursky, Z. H., & Klintsova, A. Y. (2021). Changes in Representation of Thalamic Projection Neurons within Prefrontal-Thalamic-Hippocampal Circuitry in a Rat Model of Third Trimester Binge Drinking. Brain Sciences, 11(3), 323. https://doi.org/10.3390/brainsci11030323