
The Dying Forward Hypothesis of ALS: Tracing Its History

{kind=link}
Abstract
1. Introduction
2. Nineteenth Century
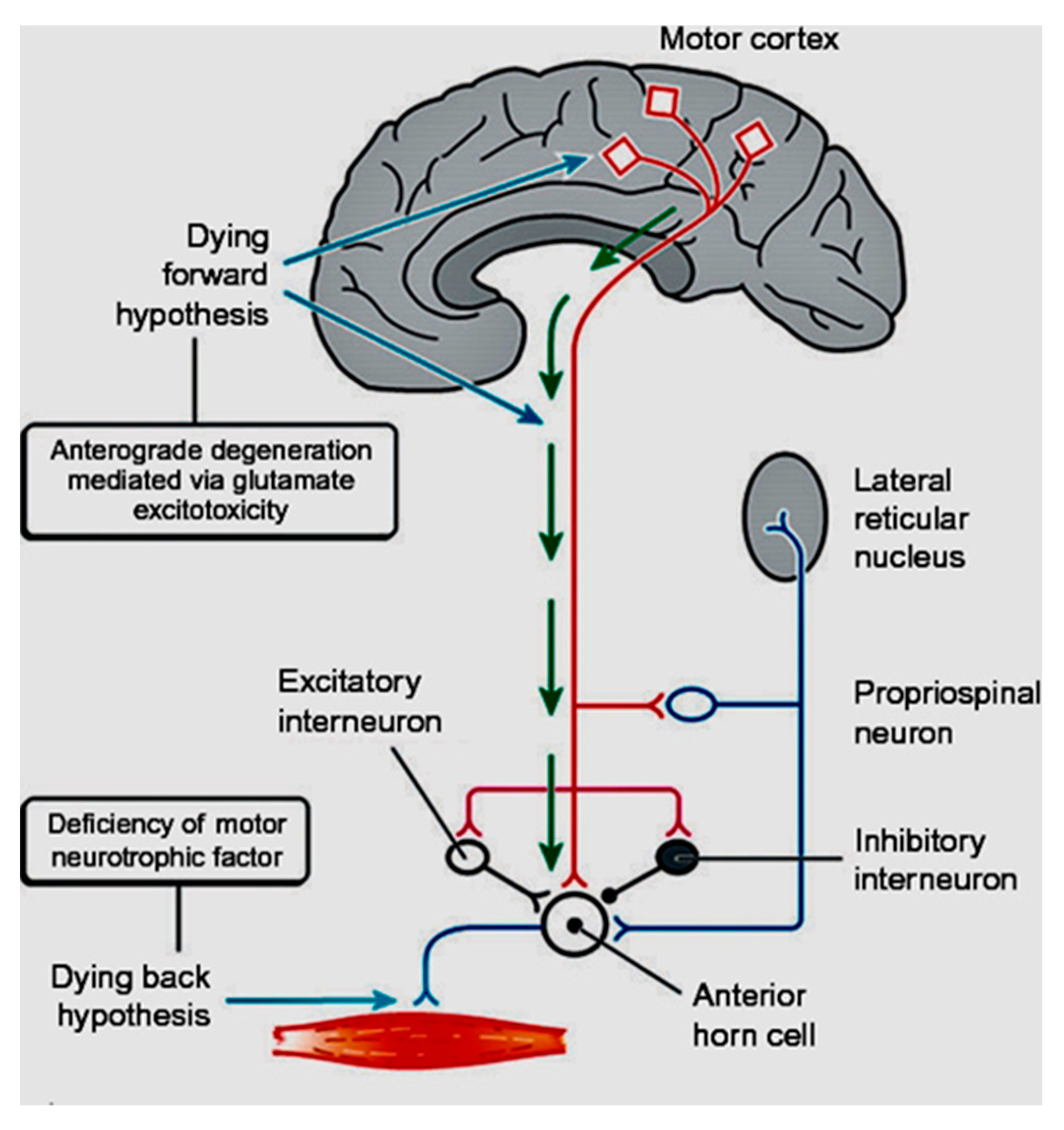
3. Twentieth Century
4. The Growth of Electrophysiological Support
5. Beyond the Motor System
6. Conclusions
Funding
Conflicts of Interest
References
- Lemon, R.N.; Griffiths, J. Comparing the function of the corticospinal system in different species: Organizational differences for motor specialization? Muscle Nerve 2005, 32, 261–279. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Eisen, A. Amyotrophic lateral sclerosis: A 40-year personal perspective. J. Clin. Neurosci. 2009, 16, 505–512. [Google Scholar] [CrossRef]
- Lemon, R.N. Descending pathways in motor control. Annu. Rev. Neurosci. 2008, 31, 195–218. [Google Scholar] [CrossRef]
- Snowden, J.S.; Harris, J.; Richardson, A.; Rollinson, S.; Thompson, J.C.; Neary, D.; Mann, D.M.; Pickering-Brown, S. Frontotemporal dementia with amyotrophic lateral sclerosis: A clinical comparison of patients with and without repeat expansions in C9orf72. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 172–176. [Google Scholar] [CrossRef]
- Woolley, S.C.; Strong, M.J. Frontotemporal Dysfunction and Dementia in Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 787–805. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A.; Kim, S.; Pant, B. Amyotrophic lateral sclerosis (ALS): A phylogenetic disease of the corticomotoneuron? Muscle Nerve 1992, 15, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A.; Braak, H.; Del Tredici, K.; Lemon, R.; Ludolph, A.C.; Kiernan, M.C. Cortical influences drive amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 917–924. [Google Scholar] [CrossRef]
- Baker, M.R. ALS—Dying forward, backward or outward? Nat. Rev. Neurol. 2014, 10, 660. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Brettschneider, J.; Ludolph, A.C.; Lee, V.M.; Trojanowski, J.Q.; Del Tredici, K. Amyotrophic lateral sclerosis—A model of corticofugal axonal spread. Nat. Rev. Neurol. 2013, 9, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Ludolph, A.C.; Emilian, S.; Dreyhaupt, J.; Rosenbohm, A.; Kraskov, A.; Lemon, R.N.; Del Tredici, K.; Braak, H. Pattern of paresis in ALS is consistent with the physiology of the corticomotoneuronal projections to different muscle groups. J. Neurol. Neurosurg. Psychiatry 2020, 91, 991–998. [Google Scholar] [CrossRef]
- Eisen, A.; Weber, M. The motor cortex and amyotrophic lateral sclerosis. Muscle Nerve 2001, 24, 564–573. [Google Scholar] [CrossRef]
- Braak, H.; Ludolph, A.C.; Neumann, M.; Ravits, J.; Del Tredici, K. Pathological TDP-43 changes in Betz cells differ from those in bulbar and spinal alpha-motoneurons in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Kiernan, M.C. Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain J. Neurol. 2006, 129, 2436–2446. [Google Scholar] [CrossRef]
- Vucic, S.; Kiernan, M.C. Utility of transcranial magnetic stimulation in delineating amyotrophic lateral sclerosis pathophysiology. Handb. Clin. Neurol. 2013, 116, 561–575. [Google Scholar] [PubMed]
- Vucic, S.; Ziemann, U.; Eisen, A.; Hallett, M.; Kiernan, M.C. Transcranial magnetic stimulation and amyotrophic lateral sclerosis: Pathophysiological insights. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1161–1170. [Google Scholar] [CrossRef]
- Turner, M.R.; Agosta, F.; Bede, P.; Govind, V.; Lule, D.; Verstraete, E. Neuroimaging in amyotrophic lateral sclerosis. Biomark. Med. 2012, 6, 319–337. [Google Scholar] [CrossRef] [PubMed]
- Brownell, B.; Oppenheimer, D.R.; Hughes, J.T. The central nervous system in motor neurone disease. J. Neurol. Neurosurg. Psychiatry 1970, 33, 338–357. [Google Scholar] [CrossRef]
- Tu, S.; Wang, C.; Menke, R.A.L.; Talbot, K.; Barnett, M.; Kiernan, M.C.; Turner, M.R. Regional callosal integrity and bilaterality of limb weakness in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A.; Turner, M.R.; Lemon, R. Tools and talk: An evolutionary perspective on the functional deficits associated with amyotrophic lateral sclerosis. Muscle Nerve 2014, 49, 469–477. [Google Scholar] [CrossRef]
- Charcot, J. Sclerose laterale amytrophique. Oeuvres Compltes. Bur. Proges Med. 1874, 2, 249–266. [Google Scholar]
- Charcot, J.; Hadden, W.B. Lectures on the Localization of Cerebral and Spinal Diseases; The New Sydenham Society: London, UK, 1883. [Google Scholar]
- Kushchayev, S.V.; Moskalenko, V.F.; Wiener, P.C.; Tsymbaliuk, V.I.; Cherkasov, V.G.; Dzyavulska, I.V.; Kovalchuk, O.I.; Sonntag, V.K.; Spetzler, R.F.; Preul, M.C. The discovery of the pyramidal neurons: Vladimir Betz and a new era of neuroscience. Brain 2012, 135, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Betz, W. Anatomischer Nachweis zweier Gehirncentra. Cent. Die Med. Wiss. 1874, 12, 578–580. [Google Scholar]
- Lewis, B. On the comparitive structure of the cortex cerebri. Brain 1878, 1, 79–96. [Google Scholar] [CrossRef]
- Walshe, F.M.R. The giant cells of Betz, the motor cortex and the prymaidal tract: A critical review. Brain 1942, 65, 409–461. [Google Scholar] [CrossRef]
- Walshe, F.M.R. The pyamidal tract. Brain 1955, 78, 149–150. [Google Scholar] [CrossRef]
- Gowers, W.R. A Manual of Diseases of the Nervous System; Churchill: London, UK, 1893. [Google Scholar]
- Mott, F.W. A case of amyotrophc lateral sclerosis with degeneration of the motor path from cortex to the periphery. Brain J. Neurol. 1895, 18, 21–36. [Google Scholar] [CrossRef]
- Wilson, S.A. Kinnier: Neurology; Arnold: London, UK, 1940. [Google Scholar]
- Hudson, A.J.; Kiernan, J.N. Preservation of certain voluntary muscles in motoneurone disease. Lancet 1988, 1, 652–653. [Google Scholar] [CrossRef]
- Kiernan, J.A.; Hudson, A.J. Changes in sizes of cortical and lower motor neurons in amyotrophic lateral sclerosis. Brain J. Neurol. 1991, 114 Pt 2, 843–853. [Google Scholar] [CrossRef]
- Rathelot, J.A.; Strick, P.L. Subdivisions of primary motor cortex based on cortico-motoneuronal cells. Proc. Natl. Acad. Sci. USA 2009, 106, 918–923. [Google Scholar] [CrossRef]
- Witham, C.L.; Fisher, K.M.; Edgley, S.A.; Baker, S.N. Corticospinal Inputs to Primate Motoneurons Innervating the Forelimb from Two Divisions of Primary Motor Cortex and Area 3a. J. Neurosci. 2016, 36, 2605–2616. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. The pyramidal cells of Betz within the cingulate and precentral gigantopyramidal field in the human brain. A Golgi and pigmentarchitectonic study. Cell Tissue Res. 1976, 172, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Kremer, S.; Chassagnon, S.; Hoffmann, D.; Benabid, A.L.; Kahane, P. The cingulate hidden hand. J. Neurol. Neurosurg. Psychiatry 2001, 70, 264–265. [Google Scholar] [CrossRef] [PubMed]
- Sanides, F. Evolutionary aspect of the primate neocortex. In Proceedings of the International Primatological Society Third International Congress of Primatology, Zurich, Switzerland, 2–5 August 1970; Karger: Basel, Switzerland, 1970; pp. 92–98. [Google Scholar]
- Sanides, F.; Sanides, D. The “extraverted neurons” of the mammalian cerebral cortex. Z. Anat. Entwickl. 1972, 136, 272–293. [Google Scholar] [CrossRef]
- Morecraft, R.J.; Ge, J.; Stilwell-Morecraft, K.S.; McNeal, D.W.; Pizzimenti, M.A.; Darling, W.G. Terminal distribution of the corticospinal projection from the hand/arm region of the primary motor cortex to the cervical enlargement in rhesus monkey. J. Comp. Neurol. 2013, 521, 4205–4235. [Google Scholar] [CrossRef]
- Ralston, D.D.; Ralston, H.J., 3rd. The terminations of corticospinal tract axons in the macaque monkey. J. Comp. Neurol. 1985, 242, 325–337. [Google Scholar] [CrossRef]
- Graf von Keyserlingk, D.; Schramm, U. Diameter of axons and thickness of myelin sheaths of the pyramidal tract fibres in the adult human medullary pyramid. Anat. Anz. 1984, 157, 97–111. [Google Scholar] [PubMed]
- Porter, R.; Lemon, R.N. Corticospinal Neurones and Voluntary Movement; Clarendon: Oxford UK, 1993. [Google Scholar]
- Iwatsubo, T.; Kuzuhara, S.; Kanemitsu, A.; Shimada, H.; Toyokura, Y. Corticofugal projections to the motor nuclei of the brainstem and spinal cord in humans. Neurology 1990, 40, 309–312. [Google Scholar] [CrossRef]
- Fornia, L.; Ferpozzi, V.; Montagna, M.; Rossi, M.; Riva, M.; Pessina, F.; Martinelli Boneschi, F.; Borroni, P.; Lemon, R.N.; Bello, L.; et al. Functional Characterization of the Left Ventrolateral Premotor Cortex in Humans: A Direct Electrophysiological Approach. Cereb. Cortex 2016, 28, 167–183. [Google Scholar] [CrossRef]
- Eisen, A.; Kuwabara, S. The split hand syndrome in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2012, 83, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Menon, P.; Kiernan, M.C.; Vucic, S. Cortical excitability differences in hand muscles follow a split-hand pattern in healthy controls. Muscle Nerve 2014, 49, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.G.; Lee, M.; Bae, J.S.; Mioshi, E.; Lin, C.S.; Pfluger, C.M.; Henderson, R.D.; Vucic, S.; Swash, M.; Burke, D.; et al. Dissociated lower limb muscle involvement in amyotrophic lateral sclerosis. J. Neurol. 2015, 262, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, R.; Martin, S.; Ellis, C.; Burman, R.; Sreedharan, J.; Shaw, C.; Leigh, P.N.; Turner, M.R.; Al-Chalabi, A. Relative preservation of triceps over biceps strength in upper limb-onset ALS: The ‘split elbow’. J. Neurol. Neurosurg. Psychiatry 2019, 90, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S. Split elbow sign: More evidence for the importance of cortical dysfunction in ALS. J. Neurol. Neurosurg. Psychiatry 2019, 90, 729. [Google Scholar] [CrossRef]
- Lemon, R.N.; Landau, W.; Tutssel, D.; Lawrence, D.G. Lawrence and Kuypers (1968a, b) revisited: Copies of the original filmed material from their classic papers in Brain. Brain J. Neurol. 2012, 135, 2290–2295. [Google Scholar] [CrossRef]
- Kuypers, H. Anatomy of the descending pathways. In Handbook of Physiology—The Nervous System II; Brookhart, J., Mountcastle, V.B., Eds.; American Physiological Society; Williams and Wilkins: Bethesda, MD, USA, 1981; pp. 597–666. [Google Scholar]
- Marinacci, A.A.; VonHagen, K.O. Electromyography in amyotrophic lateral sclerosis. A review. Bull. Los Angel. Neurol. Soc. 1974, 39, 17–29. [Google Scholar]
- Daube, J.R. Electrophysiologic studies in the diagnosis and prognosis of motor neuron diseases. Neurol. Clin. 1985, 3, 473–493. [Google Scholar] [CrossRef]
- Swash, M. Why are upper motor neuron signs difficult to elicit in amyotrophic lateral sclerosis? J. Neurol. Neurosurg. Psychiatry 2012, 83, 659–662. [Google Scholar] [CrossRef]
- Swash, M.; Burke, D.; Turner, M.R.; Grosskreutz, J.; Leigh, P.N.; deCarvalho, M.; Kiernan, M.C. Occasional essay: Upper motor neuron syndrome in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2020, 91, 227–234. [Google Scholar] [CrossRef]
- van Es, M.A.; Goedee, H.S.; Westeneng, H.J.; Nijboer, T.C.W.; van den Berg, L.H. Is it accurate to classify ALS as a neuromuscular disorder? Expert Rev. Neurother. 2020, 20, 895–906. [Google Scholar] [CrossRef]
- Eisen, A.; Calne, D. Amyotrophic lateral sclerosis, Parkinson’s disease and Alzheimer’s disease: Phylogenetic disorders of the human neocortex sharing many characteristics. Can. J. Neurol. Sci. 1992, 19, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.H.; Kim, J.I.; Lim, Y.M.; Kim, K.K. Abnormal Oculomotor Functions in Amyotrophic Lateral Sclerosis. J. Clin. Neurol. 2018, 14, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.; Schwartz, M.S.; Swash, M. Involvement of the external anal sphincter in amyotrophic lateral sclerosis. Muscle Nerve 1995, 18, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Goodin, D.S.; Rowley, H.A.; Olney, R.K. Magnetic resonance imaging in amyotrophic lateral sclerosis. Ann. Neurol. 1988, 23, 418–420. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Kinoshita, M.; Ikeda, K.; Takamiya, K. Central nervous system magnetic resonance imaging findings in amyotrophic lateral sclerosis. Eur. Arch. Psychiatry Neurol. Sci. 1989, 239, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Verstraete, E. What does imaging reveal about the pathology of amyotrophic lateral sclerosis? Curr. Neurol. Neurosci. Rep. 2015, 15, 45. [Google Scholar] [CrossRef]
- Mills, K.R.; Murray, N.M.; Hess, C.W. Magnetic and electrical transcranial brain stimulation: Physiological mechanisms and clinical applications. Neurosurgery 1987, 20, 164–168. [Google Scholar] [CrossRef]
- Eisen, A.A.; Shtybel, W. AAEM minimonograph #35: Clinical experience with transcranial magnetic stimulation. Muscle Nerve 1990, 13, 995–1011. [Google Scholar]
- Eisen, A.; Shytbel, W.; Murphy, K.; Hoirch, M. Cortical magnetic stimulation in amyotrophic lateral sclerosis. Muscle Nerve 1990, 13, 146–151. [Google Scholar] [CrossRef]
- Brown, W.F.; Ebers, G.C.; Hudson, A.J.; Pringle, C.E.; Veitch, J. Motor-evoked responses in primary lateral sclerosis. Muscle Nerve 1992, 15, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Howells, J.; Trevillion, L.; Kiernan, M.C. Assessment of cortical excitability using threshold tracking techniques. Muscle Nerve 2006, 33, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Nicholson, G.A.; Kiernan, M.C. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain J. Neurol. 2008, 131, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- van der Graaff, M.M.; de Jong, J.M.; Baas, F.; de Visser, M. Upper motor neuron and extra-motor neuron involvement in amyotrophic lateral sclerosis: A clinical and brain imaging review. Neuromuscul. Disord. 2009, 19, 53–58. [Google Scholar] [CrossRef]
- Agosta, F.; Chio, A.; Cosottini, M.; De Stefano, N.; Falini, A.; Mascalchi, M.; Rocca, M.A.; Silani, V.; Tedeschi, G.; Filippi, M. The present and the future of neuroimaging in amyotrophic lateral sclerosis. Ajnr. Am. J. Neuroradiol. 2010, 31, 1769–1777. [Google Scholar] [CrossRef]
- Mezzapesa, D.M.; D’Errico, E.; Tortelli, R.; Distaso, E.; Cortese, R.; Tursi, M.; Federico, F.; Zoccolella, S.; Logroscino, G.; Dicuonzo, F.; et al. Cortical thinning and clinical heterogeneity in amyotrophic lateral sclerosis. PLoS ONE 2013, 8, e80748. [Google Scholar] [CrossRef]
- Turner, M.R.; Modo, M. Advances in the application of MRI to amyotrophic lateral sclerosis. Expert Opin. Med. Diagn. 2010, 4, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Lillo, P.; Hodges, J.R. Frontotemporal dementia and motor neurone disease: Overlapping clinic-pathological disorders. J. Clin. Neurosci. 2009, 16, 1131–1135. [Google Scholar] [CrossRef]
- Neary, D.; Snowden, J. Frontal lobe dementia, motor neuron disease, and clinical and neuropathological criteria. J. Neurol. Neurosurg. Psychiatry 2013, 84, 713–714. [Google Scholar] [CrossRef]
- Hudson, A.J. Amyotrophic lateral sclerosis and its association with dementia, parkinsonism and other neurological disorders: A review. Brain J. Neurol. 1981, 104, 217–247. [Google Scholar] [CrossRef] [PubMed]
- Hudson, A.J.; Martzke, J. Aphasic dementia and motor neuron disease. Ann. Neurol. 1993, 34, 417–418. [Google Scholar] [CrossRef]
- Strong, M.J. The syndromes of frontotemporal dysfunction in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2008, 9, 323–338. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Uddin, L.Q.; Yeo, B.T.T.; Spreng, R.N. Towards a Universal Taxonomy of Macro-scale Functional Human Brain Networks. Brain Topogr. 2019, 32, 926–942. [Google Scholar] [CrossRef]
- Verstraete, E.; van den Heuvel, M.P.; Veldink, J.H.; Blanken, N.; Mandl, R.C.; Hulshoff Pol, H.E.; van den Berg, L.H. Motor network degeneration in amyotrophic lateral sclerosis: A structural and functional connectivity study. PLoS ONE 2010, 5, e13664. [Google Scholar] [CrossRef]
- Buchanan, C.R.; Pettit, L.D.; Storkey, A.J.; Abrahams, S.; Bastin, M.E. Reduced structural connectivity within a prefrontal-motor-subcortical network in amyotrophic lateral sclerosis. J. Magn. Reson. Imaging 2015, 41, 1342–1352. [Google Scholar] [CrossRef][Green Version]
- Ravits, J.M.; La Spada, A.R. ALS motor phenotype heterogeneity, focality, and spread: Deconstructing motor neuron degeneration. Neurology 2009, 73, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A.; Kiernan, M.; Mitsumoto, H.; Swash, M. Amyotrophic lateral sclerosis: A long preclinical period? Neurol. Neurosurg. Psychiatry 2014, 85, 1232–1238. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eisen, A. The Dying Forward Hypothesis of ALS: Tracing Its History. Brain Sci. 2021, 11, 300. https://doi.org/10.3390/brainsci11030300
Eisen A. The Dying Forward Hypothesis of ALS: Tracing Its History. Brain Sciences. 2021; 11(3):300. https://doi.org/10.3390/brainsci11030300
Chicago/Turabian StyleEisen, Andrew. 2021. "The Dying Forward Hypothesis of ALS: Tracing Its History" Brain Sciences 11, no. 3: 300. https://doi.org/10.3390/brainsci11030300
APA StyleEisen, A. (2021). The Dying Forward Hypothesis of ALS: Tracing Its History. Brain Sciences, 11(3), 300. https://doi.org/10.3390/brainsci11030300