
Smoking as a Common Modulator of Sensory Gating and Reward Learning in Individuals with Psychotic Disorders


Abstract
1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Clinical Symptom Scales
2.3. Smoking History and Medication
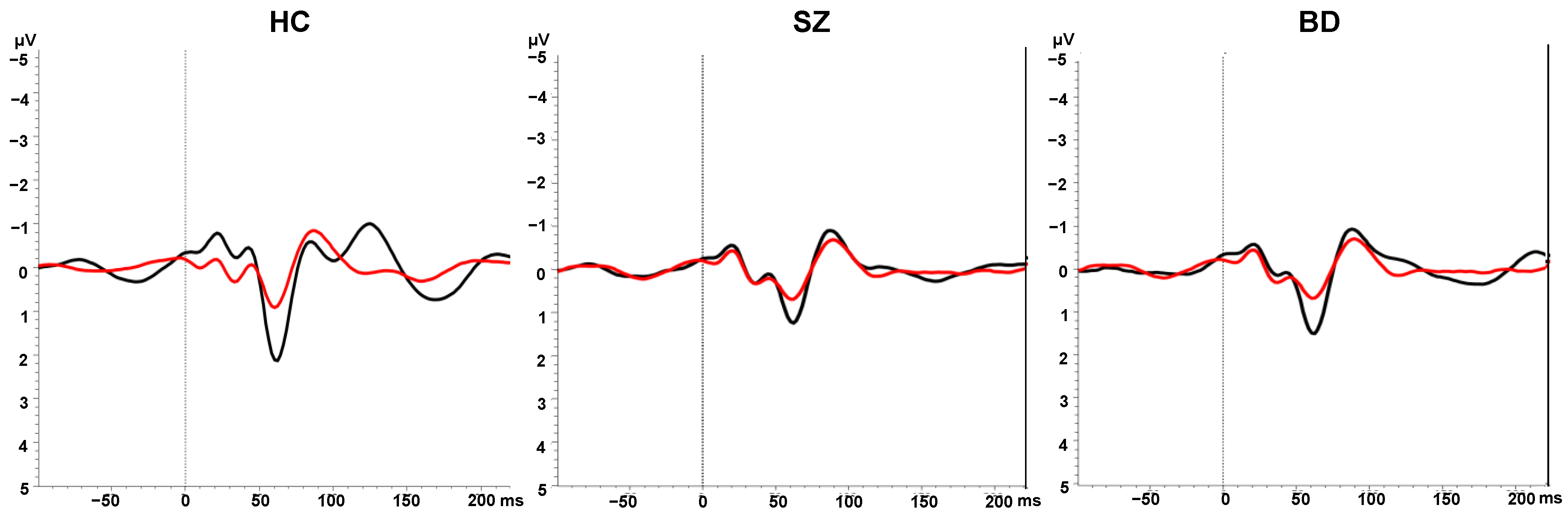
2.4. Sensory Gating
2.4.1. Paired Click Paradigm
2.4.2. EEG Recording and Pre-Processing
2.4.3. Calculation of P50 ERP Components
2.5. Reward Learning
2.6. Statistical Analyses
3. Results
3.1. Sample Characteristics
3.2. Sensory Gating in Patients vs. Controls
3.3. Response Bias and Discriminability in Patients vs. Controls
3.4. Associations between Sensory Gating, Response Bias and Discriminability
3.5. Demographic and Clinical Features Associated with Smoking in the Patient Sample
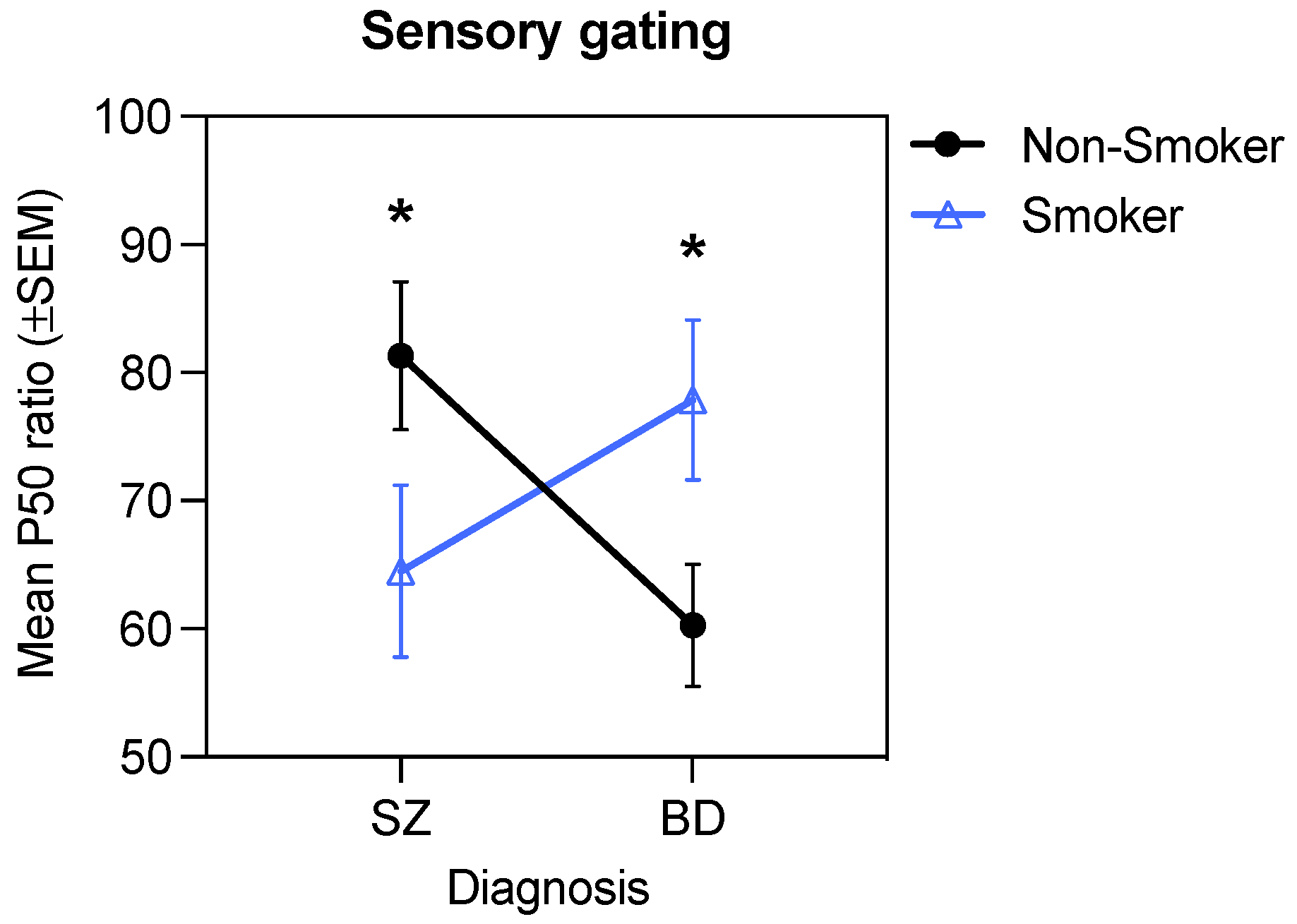
3.6. Interactive Effects of Diagnosis, Smoking, and D2 Antagonism on Sensory Gating
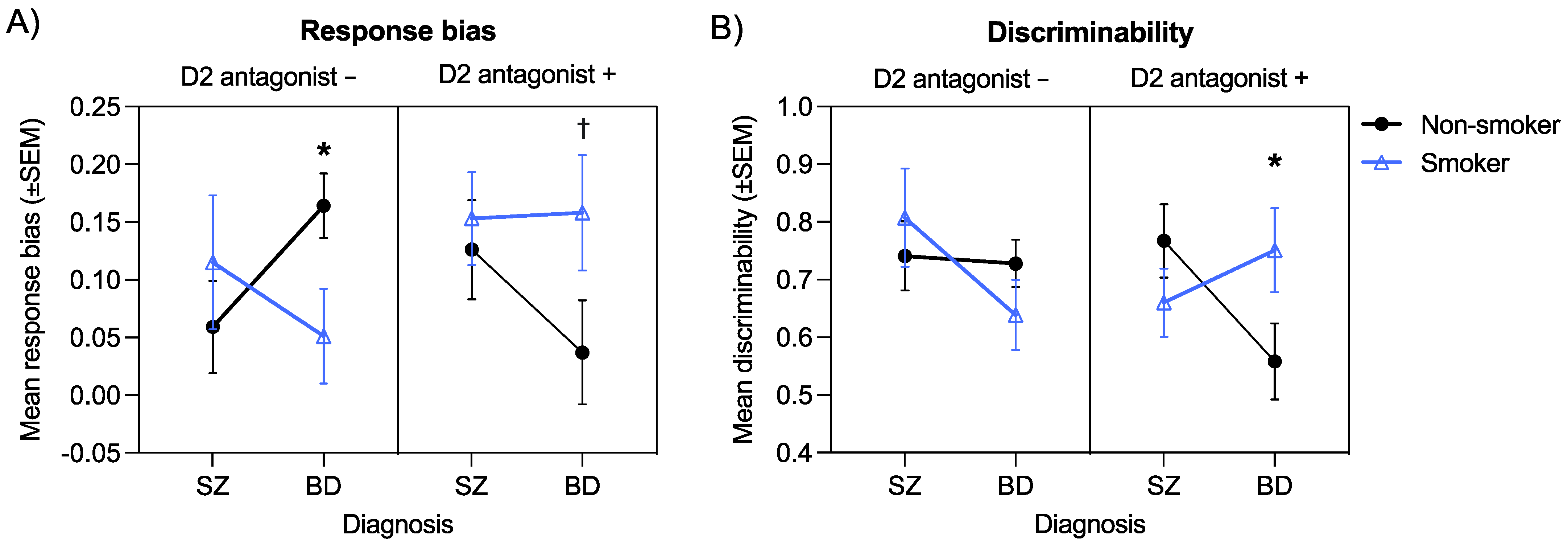
3.7. Interactive Effects of Diagnosis, Smoking, and D2 Antagonism on Response Bias
3.8. Interactive Effects of Diagnosis, Smoking, and D2 Antagonism on Discriminability
3.9. Evaluation of Potential Confounding Factors
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sterzer, P.; Adams, R.; Fletcher, P.; Frith, C.; Lawrie, S.M.; Muckli, L.; Petrovic, P.; Uhlhaas, P.; Voss, M.; Corlett, P.R. The predictive coding account of psychosis. Biol. Psychiatry 2018, 84, 634–643. [Google Scholar] [CrossRef]
- Horga, G.; Abi-Dargham, A. An integrative framework for perceptual disturbances in psychosis. Nat. Rev. Neurosci. 2019, 20, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Millard, S.J.; Bearden, C.E.; Karlsgodt, K.H.; Sharpe, M.J. The prediction-error hypothesis of schizophrenia: New data point to circuit-specific changes in dopamine activity. Neuropsychopharmacology 2021. epub ahead of print. [Google Scholar] [CrossRef]
- Whitton, A.E.; Treadway, M.T.; Pizzagalli, D.A. Reward processing dysfunction in major depression, bipolar disorder and schizophrenia. Curr. Opin. Psychiatry 2015, 28, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Strauss, G.P.; Waltz, J.A.; Gold, J.M. A review of reward processing and motivational impairment in schizophrenia. Schizophr. Bull. 2014, 40, S107–S116. [Google Scholar] [CrossRef]
- Whitton, A.E.; Lewandowski, K.E. Reward processing and social functioning in psychosis. In Social Cognition in Psychosis; Lewandowski, K.E., Moustafa, A.A., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 177–200. [Google Scholar]
- Radua, J.; Schmidt, A.; Borgwardt, S.; Heinz, A.; Schlagenhauf, F.; McGuire, P.; Fusar-Poli, P. Ventral striatal activation during reward processing in psychosis: A neurofunctional meta-analysis. JAMA Psychiatry 2015, 72, 1243–1251. [Google Scholar] [CrossRef]
- McCutcheon, R.; Beck, K.; Jauhar, S.; Howes, O.D. Defining the locus of dopaminergic dysfunction in schizophrenia: A meta-analysis and test of the mesolimbic hypothesis. Schizophr. Bull. 2018, 44, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Maia, T.V.; Frank, M.J. An integrative perspective on the role of dopamine in schizophrenia. Biol. Psychiatry 2017, 81, 52–66. [Google Scholar] [CrossRef]
- Waltz, J.A.; Frank, M.J.; Robinson, B.M.; Gold, J.M. Selective reinforcement learning deficits in schizophrenia support predictions from computational models of striatal-cortical dysfunction. Biol. Psychiatry 2007, 62, 756–764. [Google Scholar] [CrossRef]
- Heerey, E.A.; Bell-Warren, K.R.; Gold, J.M. Decision-making impairments in the context of intact reward sensitivity in schizophrenia. Biol. Psychiatry 2008, 64, 62–69. [Google Scholar] [CrossRef]
- Gold, J.M.; Waltz, J.A.; Prentice, K.J.; Morris, S.E.; Heerey, E.A. Reward processing in schizophrenia: A deficit in the representation of value. Schizophr. Bull. 2008, 34, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Murray, G.; Cheng, F.; Clark, L.; Barnett, J.H.; Blackwell, A.D.; Fletcher, P.C.; Robbins, T.W.; Bullmore, E.T.; Jones, P.B. Reinforcement and reversal learning in first-episode psychosis. Schizophr. Bull. 2008, 34, 848–855. [Google Scholar] [CrossRef]
- Weiler, J.A.; Bellebaum, C.; Brüne, M.; Juckel, G.; Daum, I. Impairment of probabilistic reward-based learning in schizophrenia. Neuropsychology 2009, 23, 571. [Google Scholar] [CrossRef] [PubMed]
- Herbener, E.S. Impairment in long-term retention of preference conditioning in schizophrenia. Biol. Psychiatry 2009, 65, 1086–1090. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barch, D.M.; Carter, C.S.; Gold, J.M.; Johnson, S.L.; Kring, A.M.; MacDonald, A.W.; Pizzagalli, D.A.; Ragland, J.D.; Silverstein, S.M.; Strauss, M.E. Explicit and implicit reinforcement learning across the psychosis spectrum. J. Abnorm. Psychol. 2017, 126, 694–711. [Google Scholar] [CrossRef] [PubMed]
- Strauss, G.P.; Thaler, N.S.; Matveeva, T.M.; Vogel, S.J.; Sutton, G.P.; Lee, B.G.; Allen, D.N. Predicting psychosis across diagnostic boundaries: Behavioral and computational modeling evidence for impaired reinforcement learning in schizophrenia and bipolar disorder with a history of psychosis. J. Abnorm. Psychol. 2015, 124, 697. [Google Scholar] [CrossRef]
- Ford, J.M.; Mathalon, D.H. Corollary discharge dysfunction in schizophrenia: Can it explain auditory hallucinations? Int. J. Psychophysiol. 2005, 58, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Waters, F.; Allen, P.; Aleman, A.; Fernyhough, C.; Woodward, T.S.; Badcock, J.C.; Barkus, E.; Johns, L.; Varese, F.; Menon, M. Auditory hallucinations in schizophrenia and nonschizophrenia populations: A review and integrated model of cognitive mechanisms. Schizophr. Bull. 2012, 38, 683–693. [Google Scholar] [CrossRef]
- Smith, D.M.; Grant, B.; Fisher, D.J.; Borracci, G.; Labelle, A.; Knott, V.J. Auditory verbal hallucinations in schizophrenia correlate with P50 gating. Clin. Neurophysiol. 2013, 124, 1329–1335. [Google Scholar] [CrossRef]
- Thoma, R.J.; Meier, A.; Houck, J.; Clark, V.P.; Lewine, J.D.; Turner, J.; Calhoun, V.; Stephen, J. Diminished auditory sensory gating during active auditory verbal hallucinations. Schizophr. Res. 2017, 188, 125–131. [Google Scholar] [CrossRef]
- Faugere, M.; Micoulaud-Franchi, J.-A.; Boyer, L.; Cermolacce, M.; Richieri, R.; Faget, C.; Vion-Dury, J.; Lancon, C. Does sensory gating have a protective effect against hallucinatory behavior in schizophrenia? Clin. Neurophysiol. 2016, 127, 1746–1748. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.H.; Schulze, K.; Rijsdijk, F.; Picchioni, M.; Ettinger, U.; Bramon, E.; Freedman, R.; Murray, R.M.; Sham, P. Heritability and reliability of P300, P50 and duration mismatch negativity. Behav. Genet. 2006, 36, 845–857. [Google Scholar] [CrossRef]
- Atagun, M.I.; Drukker, M.; Hall, M.-H.; Altun, I.K.; Tatli, S.Z.; Guloksuz, S.; van Os, J.; van Amelsvoort, T. Meta-analysis of auditory P50 sensory gating in schizophrenia and bipolar disorder. Psychiatry Res. Neuroimaging 2020, 300, 111078. [Google Scholar] [CrossRef]
- Toyomaki, A.; Hashimoto, N.; Kako, Y.; Tomimatsu, Y.; Koyama, T.; Kusumi, I. Different P50 sensory gating measures reflect different cognitive dysfunctions in schizophrenia. Schizophr. Res. Cogn. 2015, 2, 166–169. [Google Scholar] [CrossRef]
- Hamilton, H.K.; Williams, T.J.; Ventura, J.; Jasperse, L.J.; Owens, E.M.; Miller, G.A.; Subotnik, K.L.; Nuechterlein, K.H.; Yee, C.M. Clinical and cognitive significance of auditory sensory processing deficits in schizophrenia. Am. J. Psychiatry 2018, 175, 275–283. [Google Scholar] [CrossRef]
- Xia, L.; Wang, D.; Wang, J.; Xu, H.; Huo, L.; Tian, Y.; Dai, Q.; Wei, S.; Wang, W.; Zhang, G. Association of cognitive and P50 suppression deficits in chronic patients with schizophrenia. Clin. Neurophysiol. 2020, 131, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Erwin, R.; Turetsky, B.; Moberg, P.; Gur, R.; Gur, R. P50 abnormalities in schizophrenia: Relationship to clinical and neuropsychological indices of attention. Schizophr. Res. 1998, 33, 157–167. [Google Scholar] [CrossRef]
- Light, G.A.; Geyer, M.A.; Clementz, B.A.; Cadenhead, K.S.; Braff, D.L. Normal P50 suppression in schizophrenia patients treated with atypical antipsychotic medications. Am. J. Psychiatry 2000, 157, 767–771. [Google Scholar] [CrossRef]
- Louchart-de la Chapelle, S.; Levillain, D.; Menard, J.-F.; Van der Elst, A.; Allio, G.; Haouzir, S.; Dollfus, S.; Campion, D.; Thibaut, F. P50 inhibitory gating deficit is correlated with the negative symptomatology of schizophrenia. Psychiatry Res. 2005, 136, 27–34. [Google Scholar] [CrossRef]
- Adler, L.E.; Waldo, M.C.; Tatcher, A.; Cawthra, E.; Baker, N.; Freedman, R. Lack of relationship of auditory gating defects to negative symptoms in schizophrenia. Schizophr. Res. 1990, 3, 131–138. [Google Scholar] [CrossRef]
- Cromwell, H.C.; Mears, R.P.; Wan, L.; Boutros, N.N. Sensory gating: A translational effort from basic to clinical science. Clin. EEG Neurosci. 2008, 39, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Bramon, E.; Rabe-Hesketh, S.; Sham, P.; Murray, R.M.; Frangou, S. Meta-analysis of the P300 and P50 waveforms in schizophrenia. Schizophr. Res. 2004, 70, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.K.; Hall, M.-H.; McDonald, C.; Marshall, N.; Walshe, M.; Murray, R.M.; Bramon, E. P50 auditory evoked potential suppression in bipolar disorder patients with psychotic features and their unaffected relatives. Biol. Psychiatry 2007, 62, 121–128. [Google Scholar] [CrossRef]
- Grill-Spector, K.; Henson, R.; Martin, A. Repetition and the brain: Neural models of stimulus-specific effects. Trends Cogn. Sci. 2006, 10, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Martens, U.; Gruber, T. Sharpening and formation: Two distinct neuronal mechanisms of repetition priming. Eur. J. Neurosci. 2012, 36, 2989–2995. [Google Scholar] [CrossRef]
- Summerfield, C.; Trittschuh, E.H.; Monti, J.M.; Mesulam, M.-M.; Egner, T. Neural repetition suppression reflects fulfilled perceptual expectations. Nat. Neurosci. 2008, 11, 1004–1006. [Google Scholar] [CrossRef]
- Summerfield, C.; Wyart, V.; Mareike Johnen, V.; De Gardelle, V. Human scalp electroencephalography reveals that repetition suppression varies with expectation. Front. Hum. Neurosci. 2011, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Mayrhauser, L.; Bergmann, J.; Crone, J.; Kronbichler, M. Neural repetition suppression: Evidence for perceptual expectation in object-selective regions. Front. Hum. Neurosci. 2014, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Livesey, E.J. Prediction-based attenuation as a general property of learning in neural circuits. J. Exp. Psychol. Anim. Learn. Cogn. 2021, 47, 14. [Google Scholar] [CrossRef] [PubMed]
- Whitton, A.E.; Green, A.I.; Pizzagalli, D.A.; Roth, R.M.; Williams, J.M.; Brunette, M.F. Potent dopamine D2 antagonists block the reward-enhancing effects of nicotine in smokers with schizophrenia. Schizophr. Bull. 2019, 45, 1300–1308. [Google Scholar] [CrossRef]
- Adler, L.E.; Hoffer, L.D.; Wiser, A.; Freedman, R. Normalization of auditory physiology by cigarette smoking in schizophrenic patients. Am. J. Psychiatry 1993, 150, 1856–1861. [Google Scholar]
- George, T.P.; Krystal, J.H. Comorbidity of psychiatric and substance abuse disorders. Curr. Opin. Psychiatry 2000, 13, 327–331. [Google Scholar] [CrossRef]
- Dani, J.A.; De Biasi, M. Cellular mechanisms of nicotine addiction. Pharmacol. Biochem. Behav. 2001, 70, 439–446. [Google Scholar] [CrossRef]
- Laviolette, S.R.; Van Der Kooy, D. The neurobiology of nicotine addiction: Bridging the gap from molecules to behaviour. Nat. Rev. Neurosci. 2004, 5, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.; Adler, L.E.; Bickford, P.; Byerley, W.; Coon, H.; Cullum, C.M.; Griffith, J.M.; Harris, J.G.; Leonard, S.; Miller, C. Schizophrenia and nicotinic receptors. Harv. Rev. Psychiatry 1994, 2, 179–192. [Google Scholar] [CrossRef]
- Gil, S.M.; Metherate, R. Enhanced sensory–cognitive processing by activation of nicotinic acetylcholine receptors. Nicotine Tob. Res. 2019, 21, 377–382. [Google Scholar] [CrossRef]
- Menegas, W.; Babayan, B.M.; Uchida, N.; Watabe-Uchida, M. Opposite initialization to novel cues in dopamine signaling in ventral and posterior striatum in mice. eLife 2017, 6, e21886. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, M.J.; Chang, C.Y.; Liu, M.A.; Batcherlor, H.M.; Mueller, L.E.; Jones, J.L.; Niv, Y.; Schoenbaum, G. Dopamine transients are sufficient and necessary for acquisition of model-based associations. Nat. Neurosci. 2017, 20, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, M.J.; Batchelor, H.M.; Mueller, L.E.; Chang, C.Y.; Maes, E.J.P.; Niv, Y.; Schoenbaum, G. Dopamine transients do not act as model-free prediction errors during associative learning. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.H.; Anttila, V.; Won, H.; Feng, Y.-C.A.; Rosenthal, J.; Zhu, Z.; Tucker-Drob, E.M.; Nivard, M.G.; Grotzinger, A.D.; Pasthuma, D. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 2019, 179, 1469–1482.e11. [Google Scholar] [CrossRef]
- Cheng, C.-H.; Chan, P.-Y.S.; Liu, C.-Y.; Hsu, S.-C. Auditory sensory gating in patients with bipolar disorders: A meta-analysis. J. Affect. Disord. 2016, 203, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Heffner, J.L.; Strawn, J.R.; DelBello, M.P.; Strakowski, S.M.; Anthenelli, R.M. The co-occurrence of cigarette smoking and bipolar disorder: Phenomenology and treatment considerations. Bipolar Disord. 2011, 13, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Alloy, L.B.; Nusslock, R. Future directions for understanding adolescent bipolar spectrum disorders: A reward hypersensitivity perspective. J. Clin. Child Adolesc. Psychol. 2019, 48, 669–683. [Google Scholar] [CrossRef]
- Nusslock, R.; Alloy, L.B. Reward processing and mood-related symptoms: An RDoC and translational neuroscience perspective. J. Affect. Disord. 2017, 216, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Smucny, J.; Tully, L.M.; Howell, A.M.; Lesh, T.A.; Johnson, S.L.; O’Reilly, R.G.; Minzenberg, M.J.; Ursu, S.; Yoon, J.H.; Niendam, T.A. Schizophrenia and bipolar disorder are associated with opposite brain reward anticipation-associated response. Neuropsychopharmacology 2021, 46, 1152–1160. [Google Scholar] [CrossRef]
- First, M.B.; Spitzer, R.L.; Gibbon, M.; Williams, J.B. (Eds.) Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Patient Edition (SCID-I/P); Biometrics Research New York State Psychiatric Institute: New York, NY, USA, 2002. [Google Scholar]
- Uttl, B. North American Adult Reading Test: Age norms, reliability, and validity. J. Clin. Exp. Neuropsychol. 2002, 24, 1123–1137. [Google Scholar] [CrossRef] [PubMed]
- Kay, S.R.; Fiszbein, A.; Opler, L.A. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr. Bull. 1987, 13, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Young, R.C.; Biggs, J.T.; Ziegler, V.E.; Meyer, D.A. A rating scale for mania: Reliability, validity and sensitivity. Br. J. Psychiatry 1978, 133, 429–435. [Google Scholar] [CrossRef]
- Montgomery, S.A.; Åsberg, M. A new depression scale designed to be sensitive to change. Br. J. Psychiatry 1979, 134, 382–389. [Google Scholar] [CrossRef]
- Watson, D.; Weber, K.; Assenheimer, J.S.; Clark, L.A.; Strauss, M.E.; McCormick, R.A. Testing a tripartite model: I. Evaluating the convergent and discriminant validity of anxiety and depression symptom scales. J. Abnorm. Psychol. 1995, 104, 3–14. [Google Scholar] [CrossRef]
- Barker, S.; Barron, N.; McFarland, B.H.; Bigelow, D.A. A community ability scale for chronically mentally ill consumers: Part I. Reliability and validity. Community Ment. Health J. 1994, 30, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, K.E.; Cohen, B.M.; Keshavan, M.S.; Sperry, S.H.; Öngür, D. Neuropsychological functioning predicts community outcomes in affective and non-affective psychoses: A 6-month follow-up. Schizophr. Res. 2013, 148, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Baldessarini, R.J. Chemotherapy in Psychiatry: Pharmacological Basis of Treatments for Major Mental Illness, 3rd ed.; Springer: New York, NY, USA, 2013. [Google Scholar]
- Hall, M.H.; Chen, C.-Y.; Cohen, B.M.; Spencer, K.M.; Levy, D.L.; Ongur, D.; Smoller, J.W. Genomewide association analyses of electrophysiological endophenotypes for schizophrenia and psychotic bipolar disorders: A preliminary report. Am. J. Med. Genet. B Neuropsychiatry Genet. 2015, 168, 151–161. [Google Scholar] [CrossRef]
- Hall, M.H.; Levy, D.L.; Salisbury, D.F.; Haddad, S.; Gallagher, P.; Lohan, M.; Cohen, B.; Ongur, D.; Smoller, J.W. Neurophysiologic effect of GWAS derived schizophrenia and bipolar risk variants. Am. J. Med. Genet. B Neuropsychiatry Genet. 2014, 165, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.-H.; Taylor, G.; Salisbury, D.F.; Levy, D.L. Sensory gating event–related potentials and oscillations in schizophrenia patients and their unaffected relatives. Schizophr. Bull. 2011, 37, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Nagamoto, H.T.; Adler, L.E.; Waldo, M.C.; Freedman, R. Sensory gating in schizophrenics and normal controls: Effects of changing stimulation interval. Biol. Psychiatry 1989, 25, 549–561. [Google Scholar] [CrossRef]
- Clementz, B.A.; Geyer, M.A.; Braff, D.L. P50 suppression among schizophrenia and normal comparison subjects: A methodological analysis. Biol. Psychiatry 1997, 41, 1035–1044. [Google Scholar] [CrossRef]
- Pizzagalli, D.A.; Jahn, A.L.; O’Shea, J.P. Toward an objective characterization of an anhedonic phenotype: A signal-detection approach. Biol. Psychiatry 2005, 57, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Pizzagalli, D.A.; Iosifescu, D.; Hallett, L.A.; Ratner, K.G.; Fava, M. Reduced hedonic capacity in major depressive disorder: Evidence from a probabilistic reward task. J Psychiatr. Res. 2008, 43, 76–87. [Google Scholar] [CrossRef]
- Whitton, A.E.; Rabinovich, N.E.; Lindt, J.D.; Pergadia, M.L.; Pizzagalli, D.A.; Gilbert, D.G. Genetic and depressive traits moderate the reward-enhancing effects of acute nicotine in young light smokers. Nicotine Tob. Res. 2021, 23, 1779–1786. [Google Scholar] [CrossRef] [PubMed]
- Macmillan, N.; Creelman, C. Detection Theory: A User’s Guide; Cambridge University Press: Cambridge, UK, 1991. [Google Scholar]
- Hautus, M.J. Corrections for extreme proportions and their biasing effects on estimated values of d′. Behav. Res. Method Instrum. Comput. 1995, 27, 46–51. [Google Scholar] [CrossRef]
- Perkins, K. Baseline-dependency of nicotine effects: A review. Behav. Pharmacol. 1999, 10, 597–615. [Google Scholar] [CrossRef] [PubMed]
- Newhouse, P.A.; Potter, A.; Singh, A. Effects of nicotinic stimulation on cognitive performance. Curr. Opin. Pharmacol. 2004, 4, 36–46. [Google Scholar] [CrossRef] [PubMed]
- De la Salle, S.; Smith, D.; Choueiry, J.; Impey, D.; Philippe, T.; Dort, H.; Millar, A.; Albert, P.; Knott, V. Effects of COMT genotype on sensory gating and its modulation by nicotine: Differences in low and high P50 suppressors. Neuroscience 2013, 241, 147–156. [Google Scholar] [CrossRef][Green Version]
- Millar, A.; Smith, D.; Choueiry, J.; Fisher, D.; Albert, P.; Knott, V. The moderating role of the dopamine transporter 1 gene on P50 sensory gating and its modulation by nicotine. Neuroscience 2011, 180, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Kumari, V.; Postma, P. Nicotine use in schizophrenia: The self medication hypotheses. Neurosci. Biobehav. Rev. 2005, 29, 1021–1034. [Google Scholar] [CrossRef] [PubMed]
- Boggs, D.L.; Surti, T.S.; Esterlis, I.; Pittman, B.; Cosgrove, K.; Sewell, R.A.; Ranganathan, M.; D’Souza, D.C. Minimal effects of prolonged smoking abstinence or resumption on cognitive performance challenge the “self-medication” hypothesis in schizophrenia. Schizophr. Res. 2018, 194, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Hahn, B.; Harvey, A.N.; Concheiro-Guisan, M.; Huestis, M.A.; Holcomb, H.H.; Gold, J.M. A test of the cognitive self-medication hypothesis of tobacco smoking in schizophrenia. Biol. Psychiatry 2013, 74, 436–443. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Control | Schizophrenia | Bipolar Disorder | |||
|---|---|---|---|---|---|
| Non-Smoker (n = 129) | Non-Smoker (n = 45) | Smoker (n = 34) | Non-Smoker (n = 66) | Smoker (n = 38) | |
| Age | 32.02 (12.13) | 43.44 (13.45) | 41.65 (12.49) | 40.14 (13.46) | 41.47 (13.27) |
| Female, N (%) | 81 (62.8) | 17 (37.8) | 6 (17.6) | 37 (56.1) | 19 (50.0) |
| Yrs. Educ. | 15.96 (2.08) | 14.86 (2.05) | 13.94 (1.94) | 15.68 (1.96) | 14.69 (1.86) |
| CPZ equiv. | - | 460.23 (446.00) | 661.79 (595.40) | 161.39 (240.96) | 317.63 (368.41) |
| D2 antag., N (%) a | - | 20 (46.5) | 23 (67.6) | 18 (27.3) | 15 (40.5) |
| PANSS Negative | - | 10.57 (9.90) | 10.26 (8.58) | 7.49 (5.78) | 8.71 (6.51) |
| PANSS Positive | - | 10.95 (9.42) | 14.94 (10.90) | 10.24 (8.88) | 15.34 (10.38) |
| PANSS General | - | 20.93 (16.88) | 23.91 (16.43) | 21.98 (15.88) | 25.00 (14.43) |
| YMRS | - | 5.59 (7.99) | 10.15 (10.10) | 8.25 (12.61) | 12.23 (13.89) |
| MADRS | - | 11.23 (12.21) | 12.15 (12.67) | 10.98 (12.43) | 10.91 (10.22) |
| MASQ GDA | - | 21.71 (8.44) | 22.03 (8.42) | 20.77 (8.22) | 20.75 (5.66) |
| MASQ GDD | - | 27.69 (11.85) | 24.29 (11.41) | 25.36 (11.07) | 26.58 (10.64) |
| MASQ AA | - | 27.67 (11.10) | 28.41 (11.29) | 25.53 (9.43) | 24.31 (7.64) |
| MASQ AD | - | 66.84 (16.28) | 62.09 (18.80) | 60.65 (17.43) | 65.50 (16.82) |
| MCAS | - | 27.31 (22.29) | 29.76 (21.27) | 33.95 (22.99) | 32.78 (22.14) |
| Years smoking | - | - | 21.38 (13.72) | - | 19.29 (13.68) |
| Cigarettes/day | - | - | 10.62 (14.88) | - | 10.73 (12.90) |
| Control | Schizophrenia | Bipolar Disorder | |||
|---|---|---|---|---|---|
| Non-Smoker (n = 129) | Non-Smoker (n = 45) | Smoker (n = 34) | Non-Smoker (n = 66) | Smoker (n = 38) | |
| Sensory gating, M (SD) | |||||
| P50 ratio | 37.48 (25.84) | 80.73 (43.37) | 63.37 (34.97) | 60.91 (28.60) | 77.45 (46.01) |
| P50 S1 amp | 3.32 (1.56) | 2.48 (1.05) | 2.66 (1.66) | 3.20 (1.27) | 2.79 (1.47) |
| P50 S2 amp | 1.20 (0.91) | 1.85 (1.06) | 1.49 (0.82) | 1.84 (0.93) | 1.92 (1.42) |
| Response bias, M (SD) | |||||
| Block 1 | 0.10 (0.25) | 0.09 (0.26) | 0.13 (0.20) | 0.06 (0.21) | 0.06 (0.20) |
| Block 2 | 0.14 (0.24) | 0.09 (0.30) | 0.16 (0.24) | 0.16 (0.27) | 0.08 (0.19) |
| Block 3 | 0.14 (0.27) | 0.12 (0.29) | 0.14 (0.26) | 0.16 (0.27) | 0.15 (0.27) |
| Total | 0.12 (0.19) | 0.09 (0.21) | 0.14 (0.19) | 0.13 (0.18) | 0.09 (0.18) |
| Discriminability, M (SD) | |||||
| Block 1 | 0.89 (0.29) | 0.73 (0.33) | 0.64 (0.28) | 0.65 (0.25) | 0.65 (0.29) |
| Block 2 | 0.88 (0.29) | 0.76 (0.38) | 0.74 (0.37) | 0.72 (0.34) | 0.70 (0.29) |
| Block 3 | 0.89 (0.30) | 0.74 (0.36) | 0.75 (0.39) | 0.67 (0.25) | 0.69 (0.29) |
| Total | 0.87 (0.25) | 0.73 (0.32) | 0.69 (0.29) | 0.67 (0.23) | 0.67 (0.25) |
| P50 Ratio | Response Bias | Discriminability | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| df | F | p | ηp2 | df | F | p | ηp2 | df | F | p | ηp2 | |
| Between-subjects effects | ||||||||||||
| Diagnosis | 1 | 0.15 | 0.70 | 0.001 | 1 | 0.12 | 0.73 | 0.001 | 1 | 2.71 | 0.10 | 0.02 |
| Smoking | 1 | 0.003 | 0.96 | <0.001 | 1 | 0.54 | 0.46 | 0.003 | 1 | 0.12 | 0.73 | 0.001 |
| D2 antag. | 1 | 0.78 | 0.38 | 0.005 | 1 | 0.48 | 0.49 | 0.003 | 1 | 0.97 | 0.33 | 0.006 |
| Diagnosis × Smoking | 1 | 6.12 | 0.01 | 0.03 | 1 | 0.36 | 0.55 | 0.002 | 1 | 0.64 | 0.43 | 0.004 |
| Diagnosis × D2 antag. | 1 | 2.05 | 0.16 | 0.01 | 1 | 1.04 | 0.31 | 0.006 | 1 | 0.12 | 0.73 | 0.001 |
| Smoking × D2 antag. | 1 | 0.002 | 0.97 | <0.001 | 1 | 2.69 | 0.10 | 0.02 | 1 | 0.36 | 0.55 | 0.002 |
| Diagnosis × Smoking × D2 antag. | 1 | 0.74 | 0.39 | 0.004 | 1 | 4.50 | 0.04 | 0.03 | 1 | 6.25 | 0.01 | 0.04 |
| Error | 172 | 172 | 170 a | |||||||||
| Within-subjects effects | ||||||||||||
| Block | 2 | 2.74 | 0.07 | 0.02 | 2 | 5.90 | 0.003 | 0.03 | ||||
| Diagnosis × Block | 2 | 0.58 | 0.56 | 0.003 | 2 | 0.73 | 0.48 | 0.004 | ||||
| Smoking × Block | 2 | 0.05 | 0.95 | <0.001 | 2 | 1.84 | 0.16 | 0.01 | ||||
| D2 antag. × Block | 2 | 1.98 | 0.14 | 0.01 | 2 | 1.25 | 0.29 | 0.007 | ||||
| Diagnosis × Smoking × Block | 2 | 0.72 | 0.49 | 0.004 | 2 | 1.01 | 0.35 | 0.006 | ||||
| Diagnosis × D2 antag. × Block | 2 | 0.31 | 0.73 | 0.002 | 2 | 0.15 | 0.86 | 0.001 | ||||
| Smoking × D2 antag. × Block | 2 | 0.88 | 0.42 | 0.005 | 2 | 1.74 | 0.18 | 0.01 | ||||
| Diagnosis × Smoking × D2 antag. × Block | 2 | 1.13 | 0.35 | 0.007 | 2 | 1.86 | 0.16 | 0.01 | ||||
| Error (Block) | 344 | 340 | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whitton, A.E.; Lewandowski, K.E.; Hall, M.-H. Smoking as a Common Modulator of Sensory Gating and Reward Learning in Individuals with Psychotic Disorders. Brain Sci. 2021, 11, 1581. https://doi.org/10.3390/brainsci11121581
Whitton AE, Lewandowski KE, Hall M-H. Smoking as a Common Modulator of Sensory Gating and Reward Learning in Individuals with Psychotic Disorders. Brain Sciences. 2021; 11(12):1581. https://doi.org/10.3390/brainsci11121581
Chicago/Turabian StyleWhitton, Alexis E., Kathryn E. Lewandowski, and Mei-Hua Hall. 2021. "Smoking as a Common Modulator of Sensory Gating and Reward Learning in Individuals with Psychotic Disorders" Brain Sciences 11, no. 12: 1581. https://doi.org/10.3390/brainsci11121581
APA StyleWhitton, A. E., Lewandowski, K. E., & Hall, M.-H. (2021). Smoking as a Common Modulator of Sensory Gating and Reward Learning in Individuals with Psychotic Disorders. Brain Sciences, 11(12), 1581. https://doi.org/10.3390/brainsci11121581