
Challenges in Treating Charcot-Marie-Tooth Disease and Related Neuropathies: Current Management and Future Perspectives


Abstract
:1. Introduction
2. Current Management
2.1. Management of Pain, Fatigue, and Cramps
2.2. Rehabilitation Therapy
2.3. Surgical Treatment
2.4. General Care
3. Future Perspectives
3.1. PMP22 Downregulation in CMT1A
3.2. Gene Silencing and Gene Therapy
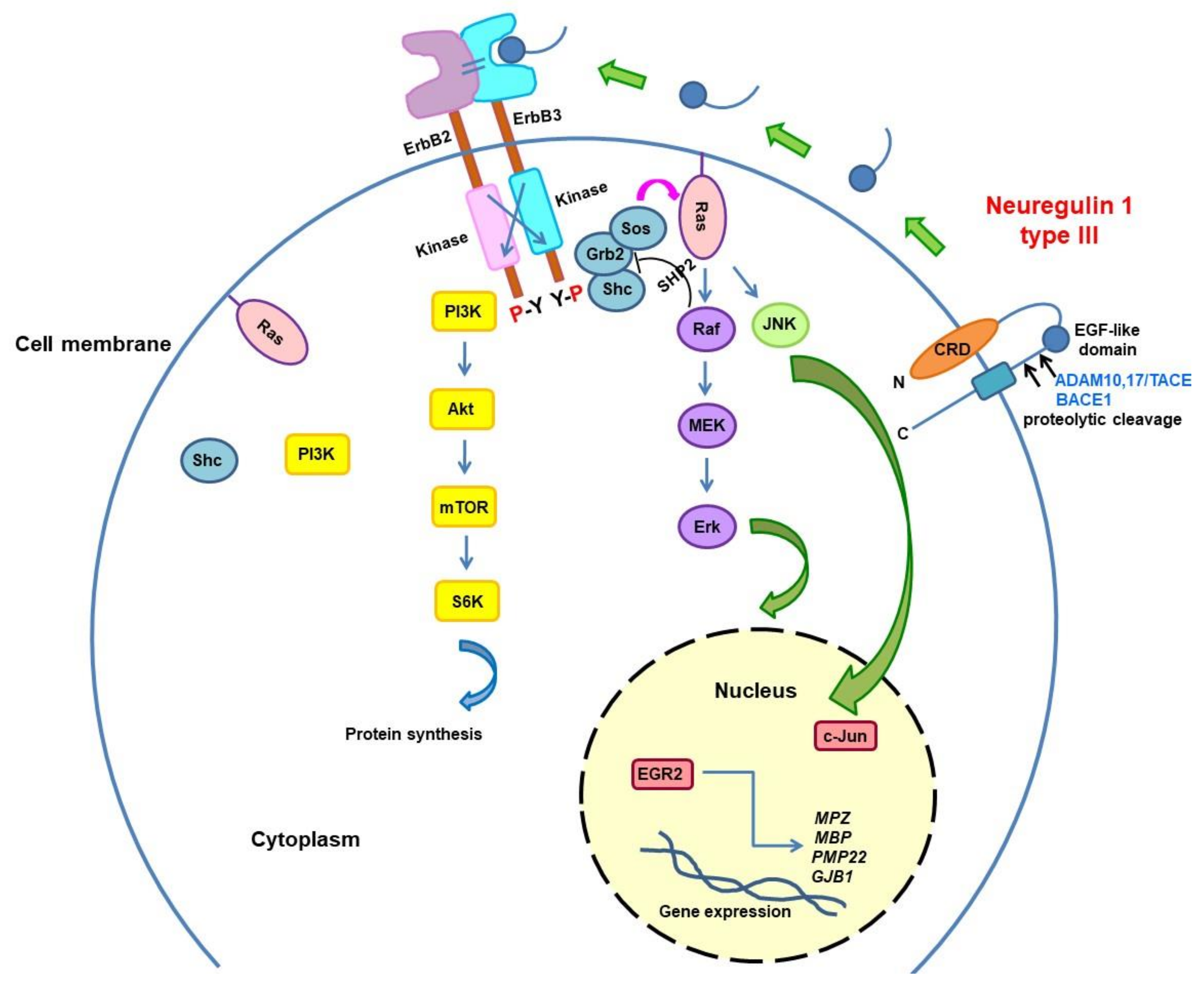
3.3. Neuregulin Pathway Modulation
3.4. Endoplasmic Reticulum (ER) Stress and Unfolded Protein Response (UPR) Activation
3.5. Axonal Degeneration and Axonal Transport
3.6. Other Approaches for Demyelinating CMT1
3.7. Correction of Metabolic Abnormalities
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pisciotta, C.; Shy, M.E. Neuropathy. Handb. Clin. Neurol. 2018, 148, 653–665. [Google Scholar] [PubMed]
- Pisciotta, C.; Saveri, P.; Pareyson, D. Updated review of therapeutic strategies for Charcot-Marie-Tooth disease and related neuropathies. Expert Rev. Neurother. 2021, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, H.; Pupe, C.; Pereira, R.; Nascimento, O.J.M. Pain in Charcot-Marie-Tooth disease: An update. Arq. Neuro-Psiquiatria 2018, 76, 273–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjelica, B.; Peric, S.; Basta, I.; Bozovic, I.; Kacar, A.; Marjanovic, A.; Ivanovic, V.; Brankovic, M.; Jankovic, M.; Novakovic, I.; et al. Neuropathic pain in patients with Charcot-Marie-Tooth type 1A. Neurol. Sci. 2020, 41, 625–630. [Google Scholar] [CrossRef]
- Carter, G.T.; Jensen, M.P.; Galer, B.S.; Kraft, G.H.; Crabtree, L.D.; Beardsley, R.M.; Abresch, R.T.; Bird, T.D. Neuropathic pain in Charcot-Marie-Tooth disease. Arch. Phys. Med. Rehabil. 1998, 79, 1560–1564. [Google Scholar] [CrossRef]
- Laurà, M.; Hutton, E.; Blake, J.; Lunn, M.P.; Fox, Z.; Pareyson, D.; Solari, A.; Radice, D.; Koltzenburg, M.; Reilly, M.M. Pain and small fiber function in charcot-marie-tooth disease type 1A. Muscle Nerve 2014, 50, 366–371. [Google Scholar] [CrossRef]
- Pazzaglia, C.; Vollono, C.; Ferraro, D.; Virdis, D.; Lupi, V.; Le Pera, D.; Tonali, P.; Padua, L.; Valeriani, M. Mechanisms of neuropathic pain in patients with Charcot-Marie-Tooth 1 A: A laser-evoked potential study. Pain 2010, 149, 379–385. [Google Scholar] [CrossRef]
- Ramchandren, S.; Jaiswal, M.; Feldman, E.; Shy, M. Effect of pain in pediatric inherited neuropathies. Neurology 2014, 82, 793–797. [Google Scholar] [CrossRef] [Green Version]
- Ribiere, C.; Bernardin, M.; Sacconi, S.; Delmont, E.; Fournier-Mehouas, M.; Rauscent, H.; Benchortane, M.; Staccini, P.; Lanteri-Minet, M.; Desnuelle, C. Pain assessment in Charcot-Marie-Tooth (CMT) disease. Ann. Phys. Rehabil. Med. 2012, 55, 160–173. [Google Scholar] [CrossRef]
- Tucker-Bartley, A.; Lemme, J.; Gomez-Morad, A.; Shah, N.; Veliu, M.; Birklein, F.; Storz, C.; Rutkove, S.; Kronn, D.; Boyce, A.M.; et al. Pain Phenotypes in Rare Musculoskeletal and Neuromuscular Diseases. Neurosci. Biobehav. Rev. 2021, 124, 267–290. [Google Scholar] [CrossRef]
- Pareyson, D.; Reilly, M.M.; Schenone, A.; Fabrizi, G.M.; Cavallaro, T.; Santoro, L.; Vita, G.; Quattrone, A.; Padua, L.; Gemignani, F.; et al. Ascorbic acid in Charcot–Marie–Tooth disease type 1A (CMT-TRIAAL and CMT-TRAUK): A double-blind randomised trial. Lancet Neurol. 2011, 10, 320–328. [Google Scholar] [CrossRef]
- Attal, N.; Bouhassira, D. Advances in the treatment of neuropathic pain. Curr. Opin. Neurol. 2021. [Google Scholar] [CrossRef]
- Callaghan, B.C.; Gallagher, G.; Fridman, V.; Feldman, E.L. Diabetic neuropathy: What does the future hold? Diabetologia 2020, 63, 891–897. [Google Scholar] [CrossRef]
- Boentert, M.; Dziewas, R.; Heidbreder, A.; Happe, S.; Kleffner, I.; Evers, S.; Young, P. Fatigue, reduced sleep quality and restless legs syndrome in Charcot-Marie-Tooth disease: A web-based survey. J. Neurol. 2010, 257, 646–652. [Google Scholar] [CrossRef] [Green Version]
- Johnson, N.E.; Heatwole, C.R.; Dilek, N.; Sowden, J.; Kirk, C.A.; Shereff, D.; Shy, M.E.; Herrmann, D.N.; Inherited Neuropathies Consortium. Quality-of-life in Charcot-Marie-Tooth disease: The patient’s perspective. Neuromuscul. Disord. 2014, 24, 1018–1023. [Google Scholar] [CrossRef] [Green Version]
- Ramdharry, G.M.; Thornhill, A.; Mein, G.; Reilly, M.M.; Marsden, J.F. Exploring the experience of fatigue in people with Charcot-Marie-Tooth disease. Neuromuscul. Disord. 2012, 22 (Suppl. 3), S208–S213. [Google Scholar] [CrossRef]
- Carter, G.T.; Han, J.J.; Mayadev, A.; Weiss, M.D. Modafinil Reduces Fatigue in Charcot-Marie-Tooth Disease Type 1A: A Case Series. Am. J. Hosp. Palliat. Med. 2006, 23, 412–416. [Google Scholar] [CrossRef]
- Moretti, A. What is the role of magnesium for skeletal muscle cramps? A Cochrane Review summary with commentary. J. Musculoskelet. Neuronal Interact. 2021, 21, 1–3. [Google Scholar]
- Young, G. Leg cramps. BMJ Clin. Evid. 2015, 2015, 1113. [Google Scholar]
- Page, P. Current concepts in muscle stretching for exercise and rehabilitation. Int. J. Sports Phys. Ther. 2012, 7, 109–119. [Google Scholar]
- Corrado, B.; Ciardi, G.; Bargigli, C. Rehabilitation Management of the Charcot-Marie-Tooth Syndrome: A Systematic Review of the Literature. Medicine 2016, 95, e3278. [Google Scholar] [CrossRef]
- Kenis-Coskun, O.; Matthews, D.J. Rehabilitation issues in Charcot-Marie-Tooth disease. J. Pediatr. Rehabil. Med. 2016, 9, 31–34. [Google Scholar] [CrossRef]
- Sman, A.D.; Hackett, D.; Fiatarone Singh, M.; Fornusek, C.; Menezes, M.P.; Burns, J. Systematic review of exercise for Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 2015, 20, 347–362. [Google Scholar] [CrossRef]
- Lindeman, E.; Leffers, P.; Reulen, J.; Spaans, F.; Drukker, J. Reduction of knee torques and leg-related functional abilities in hereditary motor and sensory neuropathy. Arch. Phys. Med. Rehabil. 1994, 75, 1201–1205. [Google Scholar] [CrossRef]
- Lindeman, E.; Leffers, P.; Spaans, F.; Drukker, J.; Reulen, J.; Kerckhoffs, M.; Köke, A. Strength training in patients with myotonic dystrophy and hereditary motor and sensory neuropathy: A randomized clinical trial. Arch. Phys. Med. Rehabil. 1995, 76, 612–620. [Google Scholar] [CrossRef]
- Lindeman, E.; Spaans, F.; Reulen, J.; Leffers, P.; Drukker, J. Progressive resistance training in neuromuscular patients. Effects on force and surface EMG. J. Electromyogr. Kinesiol. 1999, 9, 379–384. [Google Scholar] [CrossRef]
- Chetlin, R.D.; Gutmann, L.; Tarnopolsky, M.; Ullrich, I.H. Yeater, R.A. Resistance training effectiveness in patients with Charcot-Marie-Tooth disease: Recommendations for exercise prescription. Arch. Phys. Med. Rehabil. 2004, 85, 1217–1223. [Google Scholar] [CrossRef]
- Chetlin, R.D.; Gutmann, L.; Tarnopolsky, M.A.; Ullrich, I.H.; Yeater, R.A. Resistance training exercise and creatine in patients with Charcot-Marie-Tooth disease. Muscle Nerve 2004, 30, 69–76. [Google Scholar] [CrossRef]
- El Mhandi, L.; Millet, G.Y.; Calmels, P.; Richard, A.; Oullion, R.; Gautheron, V.; Féasson, L. Benefits of interval-training on fatigue and functional capacities in Charcot-Marie-Tooth disease. Muscle Nerve 2008, 37, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Matjacić, Z.; Zupan, A. Effects of dynamic balance training during standing and stepping in patients with hereditary sensory motor neuropathy. Disabil. Rehabil. 2006, 28, 1455–1459. [Google Scholar] [CrossRef]
- Ramdharry, G.M.; Pollard, A.; Anderson, C.; Laura, M.; Murphy, S.M.; Dudziec, M.; Dewar, E.L.; Hutton, E.; Grant, R.; Reilly, M.M.; et al. A pilot study of proximal strength training in Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 2014, 19, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.; Sman, A.D.; Cornett, K.; Wojciechowski, E.; Walker, T.; Menezes, M.P.; Mandarakas, M.R.; Rose, K.J.; Bray, P.; Sampaio, H.; et al. Safety and efficacy of progressive resistance exercise for Charcot-Marie-Tooth disease in children: A randomised, double-blind, sham-controlled trial. Lancet Child Adolesc. Health 2017, 1, 106–113. [Google Scholar] [CrossRef]
- Wallace, A.; Pietrusz, A.; Dewar, E.; Dudziec, M.; Jones, K.; Hennis, P.; Sterr, A.; Baio, G.; Machado, P.; Laurá, M.; et al. Community exercise is feasible for neuromuscular diseases and can improve aerobic capacity. Neurology 2019, 92, e1773–e1785. [Google Scholar] [CrossRef]
- Mori, L.; Signori, A.; Prada, V.; Pareyson, D.; Piscosquito, G.; Padua, L.; Pazzaglia, C.; Fabrizi, G.M.; Picelli, A.; Schenone, A.; et al. Treadmill training in patients affected by Charcot–Marie–Tooth neuropathy: Results of a multicenter, prospective, randomized, single-blind, controlled study. Eur. J. Neurol. 2019, 27, 280–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pazzaglia, C.; Camerota, F.; Germanotta, M.; di Sipio, E.; Celletti, C.; Padua, L. Efficacy of focal mechanic vibration treatment on balance in Charcot-Marie-Tooth 1A disease: A pilot study. J. Neurol. 2016, 263, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Zuccarino, R.; Anderson, K.M.; Shy, M.E.; Pt, J.M.W. Satisfaction with ankle foot orthoses in individuals with Charcot-Marie-Tooth disease. Muscle Nerve 2021, 63, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Laurá, M.; Singh, D.; Ramdharry, G.; Morrow, J.; Skorupinska, M.; Pareyson, D.; Burns, J.; Lewis, R.A.; Scherer, S.S.; Herrmann, D.N.; et al. Inherited Neuropathies Consortium. Prevalence and orthopedic management of foot and ankle deformities in Charcot-Marie-Tooth disease. Muscle Nerve 2018, 57, 255–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilly, M.M.; Pareyson, D.; Burns, J.; Laurá, M.; Shy, M.E.; Singh, D. ENMC CMT Foot Surgery Study Group. 221st ENMC International Workshop: Foot Surgery in Charcot-Marie-Tooth disease. 10–12 June 2016, Naarden, The Netherlands. Neuromuscul. Disord. 2017, 27, 1138–1142. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, G.B.; Gonzalez, T.; Brodsky, J.; Campbell, J.; Coetzee, C.; Conti, S.; Guyton, G.; Herrmann, D.N.; Hunt, K.; Johnson, J.; et al. A Consensus Statement on the Surgical Treatment of Charcot-Marie-Tooth Disease. Foot Ankle Int. 2020, 41, 870–880. [Google Scholar] [CrossRef]
- Horacek, O.; Mazanec, R.; Morris, C.E.; Kobesova, A. Spinal deformities in hereditary motor and sensory neuropathy: A retrospective qualitative, quantitative, genotypical, and familial analysis of 175 patients. Spine 2007, 32, 2502–2508. [Google Scholar] [CrossRef] [Green Version]
- Ibañez-Juliá, M.J.; Berzero, G.; Reyes-Botero, G.; Maisonobe, T.; Lenglet, T.; Slim, M.; Louis, S.; Balaguer, A.; Sanson, M.; Le Guern, E.; et al. Antineoplastic agents exacerbating Charcot Marie Tooth disease: Red flags to avoid permanent disability. Acta Oncol. 2017, 57, 403–411. [Google Scholar] [CrossRef]
- Laforgia, M.; Laface, C.; Calabrò, C.; Ferraiuolo, S.; Ungaro, V.; Tricarico, D.; Gadaleta, C.D.; Nardulli, P.; Ranieri, G. Peripheral Neuropathy under Oncologic Therapies: A Literature Review on Pathogenetic Mechanisms. Int. J. Mol. Sci. 2021, 22, 1980. [Google Scholar] [CrossRef]
- Lee, H.-S.; Kim, M.J.; Ko, D.S.; Jeon, E.J.; Kim, J.Y.; Kang, I.S. Preimplantation genetic diagnosis for Charcot-Marie-Tooth disease. Clin. Exp. Reprod. Med. 2013, 40, 163–168. [Google Scholar] [CrossRef]
- Pisciotta, C.; Calabrese, D.; Santoro, L.; Tramacere, I.; Manganelli, F.; Fabrizi, G.M.; Schenone, A.; Cavallaro, T.; Grandis, M.; Previtali, S.C.; et al. Pregnancy in Charcot-Marie-Tooth disease: Data from the Italian CMT national registry. Neurology 2020, 95, e3180–e3189. [Google Scholar] [CrossRef]
- Rudnik-Schöneborn, S.; Thiele, S.; Walter, M.C.; Reinecke, L.; Sereda, M.; Schöneborn, R.; Elbracht, M. Pregnancy outcome in Charcot-Marie-Tooth disease: Results of the CMT-NET cohort study in Germany. Eur. J. Neurol. 2020, 27, 1390–1396. [Google Scholar] [CrossRef]
- Passage, E.; Norreel, J.C.; Noack-Fraissignes, P.; Sanguedolce, V.; Pizant, J.; Thirion, X.; Robaglia-Schlupp, A.; Pellissier, J.F.; Fontés, M. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat. Med. 2004, 10, 396–401. [Google Scholar] [CrossRef]
- Gess, B.; Baets, J.; de Jonghe, P.; Reilly, M.M.; Pareyson, D.; Young, P. Ascorbic acid for the treatment of Charcot-Marie-Tooth disease. Cochrane Database Syst. Rev. 2015, 2015, CD011952. [Google Scholar] [CrossRef]
- Sereda, M.W.; zu Horste, G.M.; Suter, U.; Uzma, N.; Nave, K.-A. Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A). Nat. Med. 2003, 9, 1533–1537. [Google Scholar] [CrossRef]
- Meyer zu Horste, G.; Prukop, T.; Liebetanz, D.; Mobius, W.; Nave, K.A.; Sereda, M.W. Antiprogesterone therapy uncouples axonal loss from demyelination in a transgenic rat model of CMT1A neuropathy. Ann. Neurol. 2007, 61, 61–72. [Google Scholar] [CrossRef]
- Chumakov, I.; Milet, A.; Cholet, N.; Primas, G.; Boucard, A.; Pereira, Y.; Graudens, E.; Mandel, J.; Laffaire, J.; Foucquier, J.; et al. Poly-therapy with a combination of three repurposed drugs (PXT3003) down-regulates Pmp22 over-expression and improves myelination, axonal and functional parameters in models of CMT1A neuropathy. Orphanet. J. Rare Dis. 2014, 9, 201. [Google Scholar] [CrossRef] [Green Version]
- Attarian, S.; Vallat, J.M.; Magy, L.; Funalot, B.; Gonnaud, P.M.; Lacour, A.; Péréon, Y.; Dubourg, O.; Pouget, J.; Micallef, J.; et al. An ex-ploratory randomised double-blind and placebo-controlled phase 2 study of a combination of baclofen, naltrexone and sorbitol (PXT3003) in patients with Charcot-Marie-Tooth disease type 1A. Orphanet. J. Rare Dis. 2014, 9, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.T.; Damle, S.; Ikeda-Lee, K.; Kuntz, S.; Karli, I.-L.; Mohan, A.; Kim, A.; Hung, G.; Scheideler, M.A.; Scherer, S.S.; et al. PMP22 antisense oligonucleotides reverse Charcot-Marie-Tooth disease type 1A features in rodent models. J. Clin. Investig. 2017, 128, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Boutary, S.; Caillaud, M.; El Madani, M.; Vallat, J.-M.; Loisel-Duwattez, J.; Rouyer, A.; Richard, L.; Gracia, C.; Urbinati, G.; Desmaële, D.; et al. Squalenoyl siRNA PMP22 nanoparticles are effective in treating mouse models of Charcot-Marie-Tooth disease type 1 A. Commun. Biol. 2021, 4, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gautier, B.; Hajjar, H.; Soares, S.; Berthelot, J.; Deck, M.; Abbou, S.; Campbell, G.; Ceprian, M.; Gonzalez, S.; Fovet, C.M.; et al. AAV2/9-mediated silencing of PMP22 prevents the development of pathological features in a rat model of Char-cot-Marie-Tooth disease 1 A. Nat. Commun. 2021, 12, 2356. [Google Scholar] [CrossRef]
- Lee, J.-S.; Lee, J.Y.; Song, D.W.; Bae, H.S.; Doo, H.M.; Yu, H.S.; Lee, K.J.; Kim, H.K.; Hwang, H.; Kwak, G.; et al. Targeted PMP22 TATA-box editing by CRISPR/Cas9 reduces demyelinating neuropathy of Charcot-Marie-Tooth disease type 1A in mice. Nucleic Acids Res. 2019, 48, 130–140. [Google Scholar] [CrossRef]
- Lee, J.-S.; Chang, E.H.; Koo, O.J.; Jwa, D.H.; Mo, W.M.; Kwak, G.; Moon, H.W.; Park, H.T.; Bin Hong, Y.; Choi, B.-O. Pmp22 mutant allele-specific siRNA alleviates demyelinating neuropathic phenotype in vivo. Neurobiol. Dis. 2017, 100, 99–107. [Google Scholar] [CrossRef]
- Sahenk, Z.; Nagaraja, H.N.; McCracken, B.S.; King, W.M.; Freimer, M.L.; Cedarbaum, J.M.; Mendell, J.R. NT-3 promotes nerve regener-ation and sensory improvement in CMT1A mouse models and in patients. Neurology 2005, 65, 681–689. [Google Scholar] [CrossRef]
- Ozes, B.; Myers, M.; Moss, K.; Mckinney, J.; Ridgley, A.; Chen, L.; Bai, S.; Abrams, C.K.; Freidin, M.M.; Mendell, J.R.; et al. AAV1.NT-3 gene therapy for X-linked Charcot-Marie-Tooth neuropathy type 1. Gene Ther. 2021. [Google Scholar] [CrossRef]
- Sargiannidou, I.; Kagiava, A.; Bashiardes, S.; Richter, J.; Christodoulou, C.; Scherer, S.S.; Kleopa, K.A. IntraneuralGJB1gene delivery improves nerve pathology in a model of X-linked Charcot-Marie-Tooth disease. Ann. Neurol. 2015, 78, 303–316. [Google Scholar] [CrossRef]
- Kagiava, A.; Sargiannidou, I.; Theophilidis, G.; Karaiskos, C.; Richter, J.; Bashiardes, S.; Schiza, N.; Nearchou, M.; Christodoulou, C.; Scherer, S.S.; et al. Intrathecal gene therapy rescues a model of demyelinating peripheral neuropathy. Proc. Natl. Acad. Sci. USA 2016, 113, E2421–E2429. [Google Scholar] [CrossRef] [Green Version]
- Schiza, N.; Georgiou, E.; Kagiava, A.; Médard, J.-J.; Richter, J.; Tryfonos, C.; Sargiannidou, I.; Heslegrave, A.J.; Rossor, A.; Zetterberg, H.; et al. Gene replacement therapy in a model of Charcot-Marie-Tooth 4C neuropathy. Brain 2019, 142, 1227–1241. [Google Scholar] [CrossRef]
- Taveggia, C.; Zanazzi, G.; Petrylak, A.; Yano, H.; Rosenbluth, J.; Einheber, S.; Xu, X.; Esper, R.M.; Loeb, J.A.; Shrager, P.; et al. Neuregulin-1 Type III Determines the Ensheathment Fate of Axons. Neuron 2005, 47, 681–694. [Google Scholar] [CrossRef] [Green Version]
- Bolino, A.; Piguet, F.; Alberizzi, V.; Pellegatta, M.; Rivellini, C.; Guerrero-Valero, M.; Noseda, R.; Brombin, C.; Nonis, A.; D’Adamo, P.; et al. Niacin-mediated Tace activation ameliorates CMT neuropathies with focal hypermyelination. EMBO Mol. Med. 2016, 8, 1438–1454. [Google Scholar] [CrossRef]
- Scapin, C.; Ferri, C.; Pettinato, E.; Zambroni, D.; Bianchi, F.; del Carro, U.; Belin, S.; Caruso, D.; Mitro, N.; Pellegatta, M.; et al. Enhanced axonal neuregulin-1 type-III signaling ameliorates neurophysiology and hypomyelination in a Char-cot-Marie-Tooth type 1B mouse model. Hum. Mol. Genet. 2019, 28, 992–1006. [Google Scholar] [CrossRef]
- Fledrich, R.; Stassart, R.M.; Klink, A.; Rasch, L.M.; Prukop, T.; Haag, L.; Czesnik, D.; Kungl, T.; Abdelaal, T.; Keric, N.; et al. Soluble neuregulin-1 modulates disease pathogenesis in rodent models of Charcot-Marie-Tooth disease 1A. Nat. Med. 2014, 20, 1055–1061. [Google Scholar] [CrossRef]
- Bai, Y.; Wu, X.; Brennan, K.M.; Wang, D.S.; D’Antonio, M.; Moran, J.; Svaren, J.; Shy, M.E. Myelin protein zero mutations and the unfolded protein response in Charcot Marie Tooth disease type 1B. Ann. Clin. Transl. Neurol. 2018, 5, 445–455. [Google Scholar] [CrossRef]
- Patzkó, Á.; Bai, Y.; Saporta, M.A.; Katona, I.; Wu, X.; Vizzuso, D.; Feltri, M.L.; Wang, S.; Dillon, L.; Kamholz, J.; et al. Curcumin derivatives promote Schwann cell differentiation and improve neuropathy in R98C CMT1B mice. Brain 2012, 135, 3551–3566. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; La Marche, M.; Wang, D.; Mastrangelo, R.; Treins, C.; Guedat, P.; D’Antonio, M.; Shy, M. Treatment of Arg98Cys MPZ Mice In Vitro and In Vivo with IFB088. In Proceedings of the PNS 2019 Annual Meeting, Genoa, Italy, 22–25 June 2019. [Google Scholar]
- Khajavi, M.; Shiga, K.; Wiszniewski, W.; He, F.; Shaw, C.; Yan, J.; Wensel, T.; Snipes, G.J.; Lupski, J.R. Oral Curcumin Mitigates the Clinical and Neuropathologic Phenotype of the Trembler-J Mouse: A Potential Therapy for Inherited Neuropathy. Am. J. Hum. Genet. 2007, 81, 438–453. [Google Scholar] [CrossRef] [Green Version]
- Fortun, J.; Go, J.C.; Li, J.; Amici, S.A.; Dunn, W.A.; Notterpek, L. Alterations in degradative pathways and protein aggregation in a neuropathy model based on PMP22 overexpression. Neurobiol. Dis. 2006, 22, 153–164. [Google Scholar] [CrossRef]
- Lee, S.; Bazick, H.; Chittoor-Vinod, V.; Al Salihi, M.O.; Xia, G.; Notterpek, L. Elevated Peripheral Myelin Protein 22, Reduced Mitotic Potential, and Proteasome Impairment in Dermal Fibroblasts from Charcot-Marie-Tooth Disease Type 1A Patients. Am. J. Pathol. 2018, 188, 728–738. [Google Scholar] [CrossRef] [Green Version]
- D’Antonio, M.; Treins, C.; Scapin, C.; Ferri, C.; Volpi, V.; Mastrangelo, R.; Touvier, T.; Florio, F.; Bianchi, F.; del Carro, U.; et al. IFB-088 treatment improves Charcot-Marie-Tooth type 1A disease phenotype of C3-PMP22 mice. In Proceedings of the PNS 2019 Annual Meeting, Genoa, Italy, 22–25 June 2019. [Google Scholar]
- Geisler, S.; Huang, S.X.; Strickland, A.; Doan, R.A.; Summers, D.; Mao, X.; Park, J.; DiAntonio, A.; Milbrandt, J. Gene therapy targeting SARM1 blocks pathological axon degeneration in mice. J. Exp. Med. 2019, 216, 294–303. [Google Scholar] [CrossRef] [Green Version]
- D’Ydewalle, C.; Krishnan, J.; Chiheb, D.M.; van Damme, P.; Irobi, J.; Kozikowski, A.P.; van den Berghe, P.; Timmerman, V.; Robberecht, W.; van den Bosch, L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 2011, 17, 968–974. [Google Scholar] [CrossRef]
- Benoy, V.; van Helleputte, L.; Prior, R.; D’Ydewalle, C.; Haeck, W.; Geens, N.; Scheveneels, W.; Schevenels, B.; Cader, M.Z.; Talbot, K.; et al. HDAC6 is a therapeutic target in mutant GARS-induced Charcot-Marie-Tooth disease. Brain 2018, 141, 673–687. [Google Scholar] [CrossRef]
- Picci, C.; Wong, V.; Costa, C.; McKinnon, M.C.; Goldberg, D.C.; Swift, M.; Alam, N.M.; Prusky, G.T.; Shen, S.; Kozikowski, A.P.; et al. HDAC6 inhibition promotes α-tubulin acetylation and ameliorates CMT2A peripheral neuropathy in mice. Exp. Neurol. 2020, 328, 113281. [Google Scholar] [CrossRef]
- Glasser, C.E.; Gartner, M.R.; Wilson, D.; Miller, B.; Sherman, M.L.; Attie, K.M. Locally acting ACE-083 increases muscle volume in healthy volunteers. Muscle Nerve 2018, 57, 921–926. [Google Scholar] [CrossRef]
- Nobbio, L.; Sturla, L.; Fiorese, F.; Usai, C.; Basile, G.; Moreschi, I.; Benvenuto, F.; Zocchi, E.; de Flora, A.; Schenone, A.; et al. P2 × 7-mediated Increased Intracellular Calcium Causes Functional Derangement in Schwann Cells from Rats with CMT1A Neuropathy. J. Biol. Chem. 2009, 284, 23146–23158. [Google Scholar] [CrossRef] [Green Version]
- Sociali, G.; Visigalli, D.; Prukop, T.; Cervellini, I.; Mannino, E.; Venturi, C.; Bruzzone, S.; Sereda, M.W.; Schenone, A. Tolerability and efficacy study of P2 × 7 inhibition in experimental Charcot-Marie-Tooth type 1A (CMT1A) neuropathy. Neurobiol. Dis. 2016, 95, 145–157. [Google Scholar] [CrossRef]
- Keystone, E.C.; Wang, M.M.; Layton, M.; Hollis, S.; McInnes, I.B. Clinical evaluation of the efficacy of the P2 × 7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Ann. Rheum. Dis. 2012, 71, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Fledrich, R.; Abdelaal, T.; Rasch, L.; Bansal, V.; Schütza, V.; Brügger, B.; Lüchtenborg, C.; Prukop, T.; Stenzel, J.; Rahman, R.U.; et al. Tar-geting myelin lipid metabolism as a potential therapeutic strategy in a model of CMT1A neuropathy. Nat. Commun. 2018, 9, 3025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosberg, M.R.; Alvarez, S.; Klein, D.; Nielsen, F.C.; Martini, R.; Levinson, S.R.; Krarup, C.; Moldovan, M. Progression of motor axon dysfunction and ectopic Nav1.8 expression in a mouse model of Charcot-Marie-Tooth disease 1B. Neurobiol. Dis. 2016, 93, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Rosberg, M.R.; Alvarez, S.; Krarup, C.; Moldovan, M. An oral NaV1.8 blocker improves motor function in mice completely de-ficient of myelin protein P0. Neurosci. Lett. 2016, 632, 33–38. [Google Scholar] [CrossRef]
- Moldovan, M.; Pisciotta, C.; Pareyson, D.; Krarup, C. Myelin protein zero gene dose dependent axonal ion-channel dysfunction in a family with Charcot-Marie-Tooth disease. Clin. Neurophysiol. 2020, 131, 2440–2451. [Google Scholar] [CrossRef]
- Klein, D.; Patzkó, Á.; Schreiber, D.; van Hauwermeiren, A.; Baier, M.; Groh, J.; West, B.; Martini, R. Targeting the colony stimulating factor 1 receptor alleviates two forms of Charcot–Marie–Tooth disease in mice. Brain 2015, 138, 3193–3205. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Valero, M.; Grandi, F.; Cipriani, S.; Alberizzi, V.; di Guardo, R.; Chicanne, G.; Sawade, L.; Bianchi, F.; del Carro, U.; de Curtis, I.; et al. Dysregulation of myelin synthesis and actomyosin function underlies aberrant myelin in CMT4B1 neuropathy. Proc. Natl. Acad. Sci. USA 2021, 118, 2009469118. [Google Scholar] [CrossRef]
- Fridman, V.; Suriyanarayanan, S.; Novak, P.; David, W.; Macklin, E.A.; McKenna-Yasek, D.; Walsh, K.; Aziz-Bose, R.; Oaklander, A.L.; Brown, R.; et al. Randomized trial of l-serine in patients with hereditary sensory and autonomic neuropathy type 1. Neurology 2019, 92, e359–e370. [Google Scholar] [CrossRef]
- De Brouwer, A.P.; van Bokhoven, H.; Nabuurs, S.B.; Arts, W.F.; Christodoulou, J.; Duley, J. PRPS1 mutations: Four distinct syndromes and potential treatment. Am. J. Hum. Genet. 2010, 86, 506–518. [Google Scholar] [CrossRef] [Green Version]
- Cortese, A.; Zhu, Y.; Rebelo, A.P.; Negri, S.; Courel, S.; Abreu, L.; Bacon, C.J.; Bai, Y.; Bis-Brewer, D.M.; Bugiardini, E.; et al. Biallelic mu-tations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat. Genet. 2020, 52, 473–481. [Google Scholar] [CrossRef]
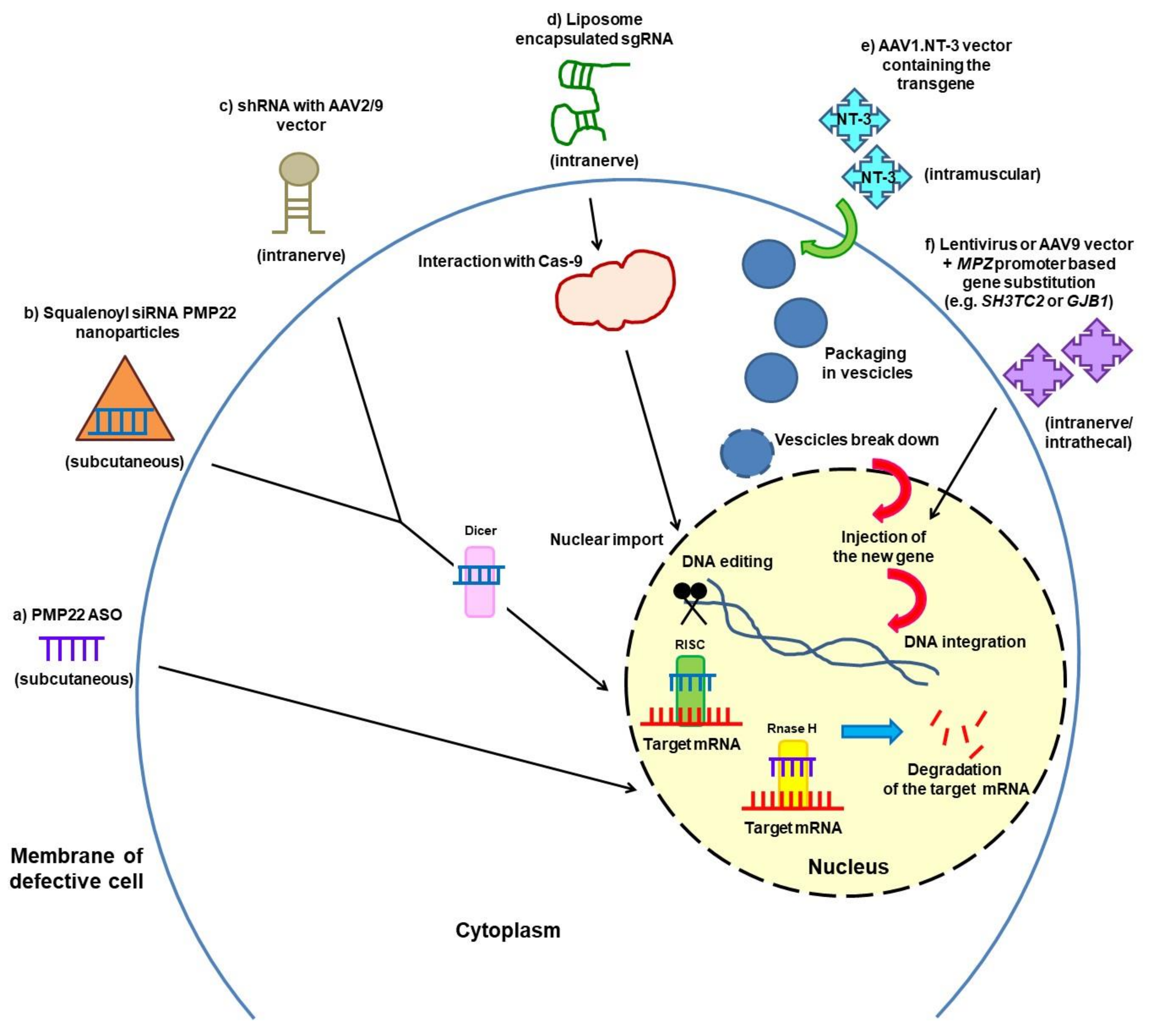

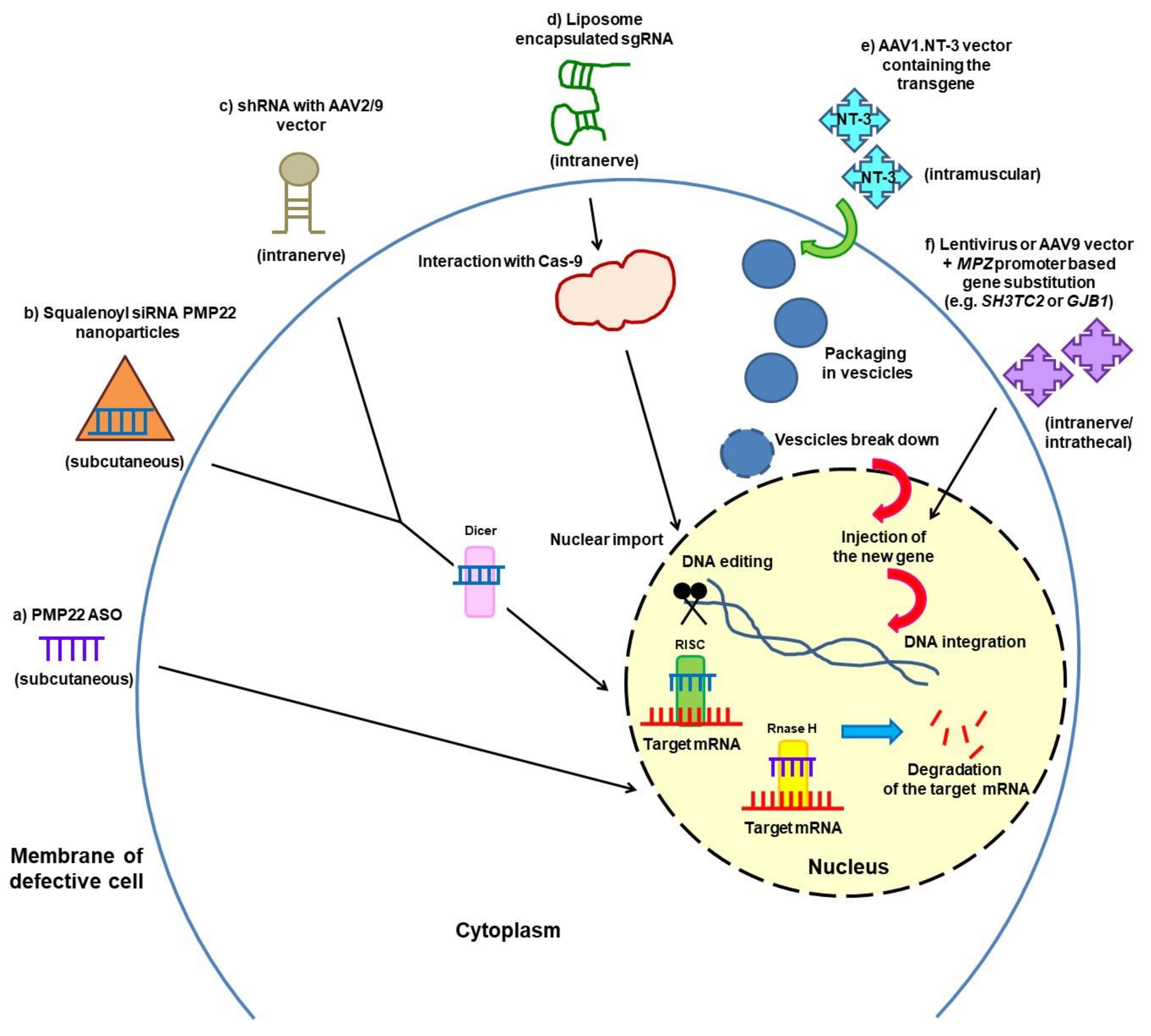{kind=link}
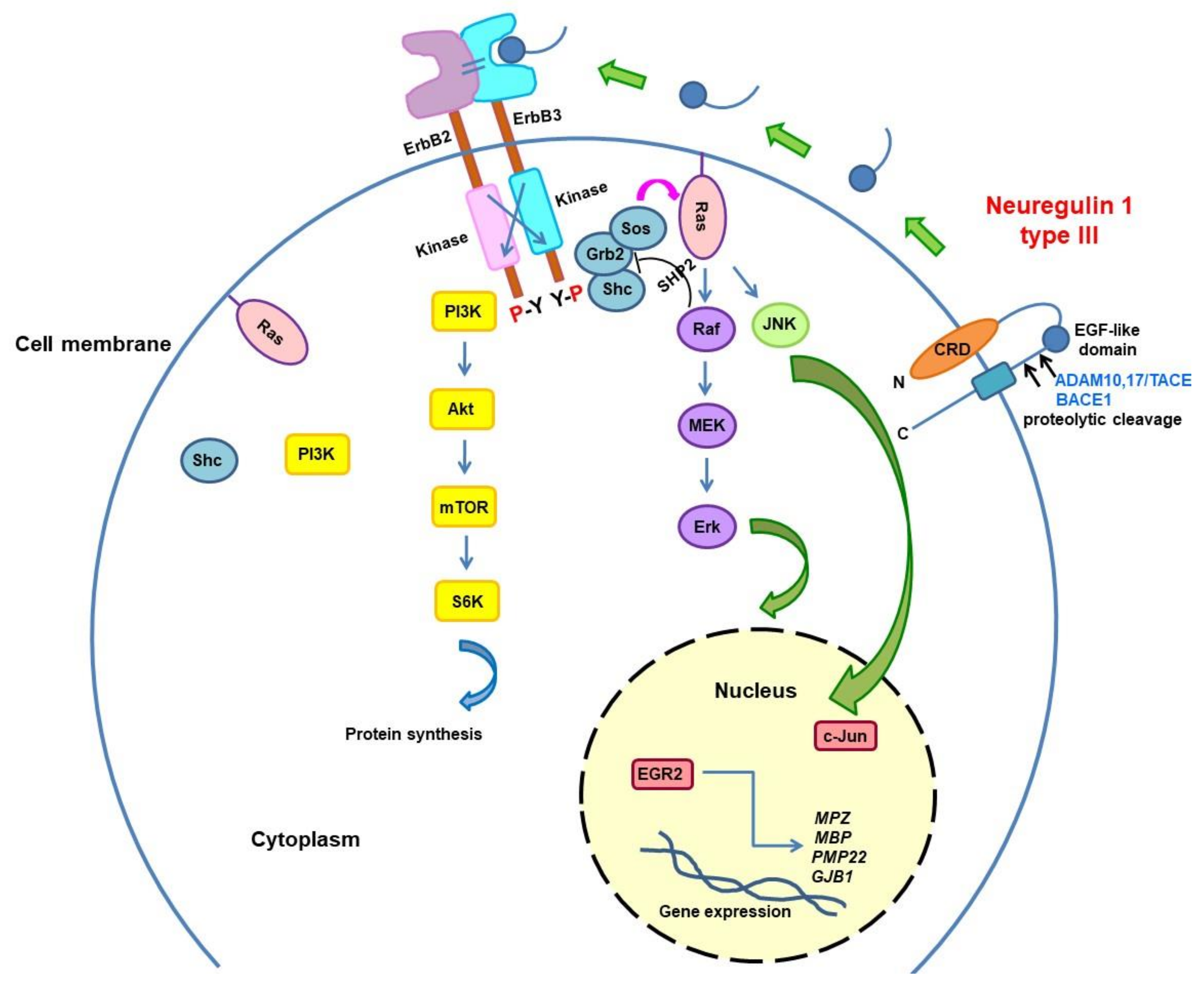{kind=link}
| CMT Type | Compound/Approach | Mechanism of Action/Aim of Therapy | Clinical Trials Status and Comments |
|---|---|---|---|
| Ascorbic acid | Reduction of PMP22 expression by reducing cAMP levels | Phase III studies concluded; all failed to meet their primary endpoints and did not show a significant effect | |
| CMT1A | Progesterone antagonists/modulators: onapristone, ulapristal | Reduction of PMP22 synthesis | Onapristone: unacceptable side effects. Ulapristal: phase II trial conducted (n = 23 out of 45 planned). Results not available |
| PXT3003 (mix of low doses of baclofen, sorbitol and naltrexone) | Inhibition of SCs proliferation and reduction of the synthesis of PMP22; baclofen, GABAB receptor modulator | PXT3003: phase II (n = 80) concluded. Phase III (n = 323) concluded but unblinding problems in the high-dose group. New Phase III requested by FDA and just started | |
| CMT1A, CMT1E | Gene silencing (ASOs, siRNAs, shRNAs, sgRNAs—CRISPR/Cas9) | Partial silencing of overexpressed (CMT1A) or mutated (CMT1E) PMP22 | Under consideration. Issues: targeting a sufficient number of SCs, obtaining a proper quantitative silencing to avoid risk of HNPP, long-life therapy |
| CMT1A, CMTX1, CMT4C | Gene therapy (e.g., AAV1-NT-3; GJB1 and SH3TC2 gene substitution) | Gene insertion (NT-3 = neurotrophic action) or (GJB1, SH3TC2) substitution | NT-3: open trial (n = 3) planned for CMT1A Gene substitution: still in preclinical phases |
| CMT1A, CMT1B, CMT4B, HNPP | Neuregulin pathways (particularly Neuregulin-1 III) | Regulation of myelin thickness | Niacin-niaspan candidate for CMT4B and HNPP? |
| CMT1A, CMT1E, CMT1B | Curcumin, sephin-1 | UPR inhibition by attenuation of the IRE1 branch | Possible clinical trial in CMT1A/CMT1B |
| CMT | FLX-787 | Activation of TRPA1 and TRPV1 channels, for cramps | Phase II (n = 120) stopped for oral intolerability in a subset of patients |
| All CMT and related neuropathies | SARM1 inhibitors | Prevention of axonal degeneration | -- |
| CMT2F, dHMN2A, CMT2A, CMT2D, dHMN5 (and others?) | HDAC6 inhibitors | Reduction of microtubules acetylation, improvement of axonal transport | Under consideration |
| CMT1, CMTX1 | ACE-083 | Action on myostatin pathway | Phase I+II (n = 62 overall) trial did not produce significant clinical improvement. Planned phase III trial abandoned |
| CMT1A | P2X7 receptor modulators (e.g., A438079) | Reduction of abnormal calcium influx into SCs | P2X 7 antagonist acceptable safety and tolerability in a previous phase II trial in rheumatoid arthritis |
| CMT1A, other CMT? | Dietary lipid supplementation | Dietary correction of defective myelin lipid biosynthesis | Trial with oral lecithin supplementation planned in Germany |
| CMT1B, other dysmyelinating CMT? | Sodium channel blockers | Blocking of Nav 1.8 channel | Lamotrigine could be a candidate compound |
| CMT1A, CMT1B, CMTX1 | CSF1R inhibitors | Decreased number and activity of macrophages in the nerve | -- |
| CMT4B1, CMT4B2 | PIKfyve enzyme inhibitors | Inhibition of PIKfyve and decrease of PI3,5P2 levels | To be considered for CMT4B1 and CMT4B2 |
| HSN1 | L-Serine | Reduction of neurotoxic deoxysphingolipids | Phase II trial (n = 18) performed, primary endpoint not reached, but underpowered trial |
| CMTX5 and allelic disorders (Arts syndrome and DFNX1) | S-adenosylmethionine (SAM) | Purine nucleotides supply | Anecdotal report |
| CMT and dHMN associated with biallelic SORD mutations | Aldose reductase inhibitors | Inhibition of aldose reductase, the enzyme converting glucose into sorbitol | Under consideration |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pisciotta, C.; Saveri, P.; Pareyson, D. Challenges in Treating Charcot-Marie-Tooth Disease and Related Neuropathies: Current Management and Future Perspectives. Brain Sci. 2021, 11, 1447. https://doi.org/10.3390/brainsci11111447
Pisciotta C, Saveri P, Pareyson D. Challenges in Treating Charcot-Marie-Tooth Disease and Related Neuropathies: Current Management and Future Perspectives. Brain Sciences. 2021; 11(11):1447. https://doi.org/10.3390/brainsci11111447
Chicago/Turabian StylePisciotta, Chiara, Paola Saveri, and Davide Pareyson. 2021. "Challenges in Treating Charcot-Marie-Tooth Disease and Related Neuropathies: Current Management and Future Perspectives" Brain Sciences 11, no. 11: 1447. https://doi.org/10.3390/brainsci11111447
APA StylePisciotta, C., Saveri, P., & Pareyson, D. (2021). Challenges in Treating Charcot-Marie-Tooth Disease and Related Neuropathies: Current Management and Future Perspectives. Brain Sciences, 11(11), 1447. https://doi.org/10.3390/brainsci11111447