Microbiome in Multiple Sclerosis: Where Are We, What We Know and Do Not Know

 ,
,  , , ,
, , ,
Abstract
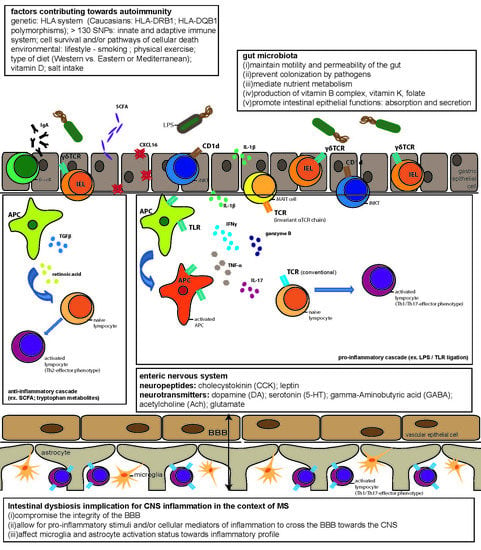
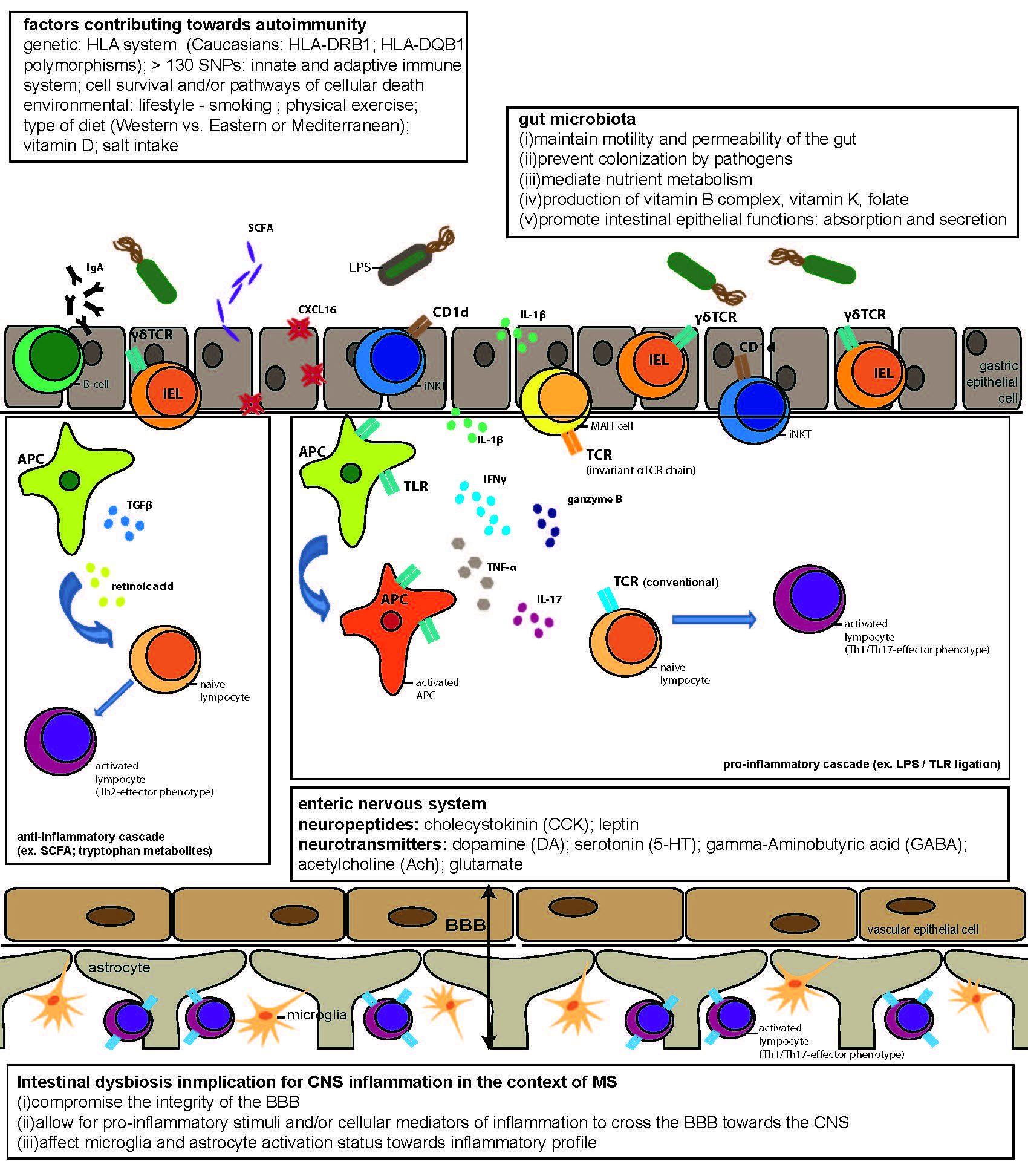{kind=link}
1. Introduction
2. The Environmental Factor in Autoimmune Disease
3. Gut Microbiota and the Role of Intestinal Dysbiosis
4. The Gut Microbiota in MS
4.1. Immunoregulation and the Gut–Brain Axis
4.2. Gut Microbiota and Innate Immunity
4.3. Gut Microbiota and Adaptive Immunity
4.4. The Role of Microbial Metabolites
4.5. The Role of Intestinal Barrier
4.6. Mechanisms of Immune-Modulation by Intestinal Microbiota—Experimental Evidence
4.7. Mechanisms of Immune-Modulation by Intestinal Microbiota—Clinical Evidence
5. Disease-Modifying Treatment (DMT) and Gut Microbiota
5.1. Interferon-β
5.2. Glatiramer Acetate
5.3. Dimethyl Fumarate
5.4. Teriflunomide
5.5. Natalizumab
5.6. Fingolimod
5.7. Alemtuzumab
6. Conclusions: Treat the Microbiome—Treat MS?
Author Contributions
Funding
Conflicts of Interest
References
- Global Health Metrics. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef]
- Grytten, N.; Torkildsen, O.; Myhr, K.M. Time trends in the incidence and prevalence of multiple sclerosis in Norway during eight decades. Acta Neurol. Scand. 2015, 132, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Rocca, M.A.; Ciccarelli, O.; De Stefano, N.; Evangelou, N.; Kappos, L.; Rovira, A.; Sastre-Garriga, J.; Tintore, M.; Frederiksen, J.L.; et al. MRI criteria for the diagnosis of multiple sclerosis: MAGNIMS consensus guidelines. Lancet Neurol. 2016, 15, 292–303. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Tintore, M.; Vidal-Jordana, A.; Sastre-Garriga, J. Treatment of multiple sclerosis—success from bench to bedside. Nat. Rev. Neurol. 2019, 15, 53–58. [Google Scholar] [CrossRef]
- Henze, T.; Rieckmann, P.; Toyka, K.V. Symptomatic treatment of multiple sclerosis. Multiple Sclerosis Therapy Consensus Group (MSTCG) of the German Multiple Sclerosis Society. Eur. Neurol. 2006, 56. [Google Scholar] [CrossRef]
- Glaser, A.; Stahmann, A.; Meissner, T.; Flachenecker, P.; Horakova, D.; Zaratin, P.; Brichetto, G.; Pugliatti, M.; Rienhoff, O.; Vukusic, S.; et al. Multiple sclerosis registries in Europe—An updated mapping survey. Mult. Scler. Relat Disord. 2019, 27, 171–178. [Google Scholar] [CrossRef]
- McKay, K.A.; Hillert, J.; Manouchehrinia, A. Long-term disability progression of pediatric-onset multiple sclerosis. Neurology 2019, 92, e2764–e2773. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, F.; Baumann, C.; Epstein, J.; Kerschen, P.; Garot, T.; Mathey, G.; Debouverie, M. Older Age at Multiple Sclerosis Onset Is an Independent Factor of Poor Prognosis: A Population-Based Cohort Study. Neuroepidemiology 2017, 48, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Patsopoulos, N.A. Genetics of Multiple Sclerosis: An Overview and New Directions. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Belbasis, L.; Bellou, V.; Evangelou, E.; Ioannidis, J.P.; Tzoulaki, I. Environmental risk factors and multiple sclerosis: An umbrella review of systematic reviews and meta-analyses. Lancet Neurol. 2015, 14, 263–273. [Google Scholar] [CrossRef]
- Zheng, C.; He, L.; Liu, L.; Zhu, J.; Jin, T. The efficacy of vitamin D in multiple sclerosis: A meta-analysis. Mult. Scler. Relat. Disord. 2018, 23, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Camara-Lemarroy, C.R.; Metz, L.M.; Yong, V.W. Focus on the gut-brain axis: Multiple sclerosis, the intestinal barrier and the microbiome. World J. Gastroenterol. 2018, 24, 4217–4223. [Google Scholar] [CrossRef] [PubMed]
- Probstel, A.K.; Baranzini, S.E. The Role of the Gut Microbiome in Multiple Sclerosis Risk and Progression: Towards Characterization of the “MS Microbiome”. Neurotherapeutics 2018, 15, 126–134. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Maloney, B. The “LEARn” (Latent Early-life Associated Regulation) model integrates environmental risk factors and the developmental basis of Alzheimer’s disease, and proposes remedial steps. Exp. Gerontol. 2010, 45, 291–296. [Google Scholar] [CrossRef]
- Xia, Z.; Steele, S.U.; Bakshi, A.; Clarkson, S.R.; White, C.C.; Schindler, M.K.; Nair, G.; Dewey, B.E.; Price, L.R.; Ohayon, J.; et al. Assessment of Early Evidence of Multiple Sclerosis in a Prospective Study of Asymptomatic High-Risk Family Members. JAMA Neurol. 2017, 74, 293–300. [Google Scholar] [CrossRef]
- Freedman, S.N.; Shahi, S.K.; Mangalam, A.K. The “Gut Feeling”: Breaking Down the Role of Gut Microbiome in Multiple Sclerosis. Neurotherapeutics 2018, 15, 109–125. [Google Scholar] [CrossRef]
- Picca, A.; Fanelli, F.; Calvani, R.; Mule, G.; Pesce, V.; Sisto, A.; Pantanelli, C.; Bernabei, R.; Landi, F.; Marzetti, E. Gut Dysbiosis and Muscle Aging: Searching for Novel Targets against Sarcopenia. Mediators Inflamm. 2018, 2018, 7026198. [Google Scholar] [CrossRef]
- Ochoa-Reparaz, J.; Magori, K.; Kasper, L.H. The chicken or the egg dilemma: Intestinal dysbiosis in multiple sclerosis. Ann. Transl. Med. 2017, 5, 145. [Google Scholar] [CrossRef]
- Blum, H.E. The human microbiome. Adv. Med. Sci. 2017, 62, 414–420. [Google Scholar] [CrossRef]
- Mowat, A.M.; Agace, W.W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Ghaisas, S.; Maher, J.; Kanthasamy, A. Gut microbiome in health and disease: Linking the microbiome-gut-brain axis and environmental factors in the pathogenesis of systemic and neurodegenerative diseases. Pharmacol. Ther. 2016, 158, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Fleck, A.K.; Schuppan, D.; Wiendl, H.; Klotz, L. Gut-CNS-Axis as Possibility to Modulate Inflammatory Disease Activity-Implications for Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 1526. [Google Scholar] [CrossRef] [PubMed]
- Vatanen, T.; Kostic, A.D.; d’Hennezel, E.; Siljander, H.; Franzosa, E.A.; Yassour, M.; Kolde, R.; Vlamakis, H.; Arthur, T.D.; Hamalainen, A.M.; et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell 2016, 165, 842–853. [Google Scholar] [CrossRef]
- Frantz, A.L.; Rogier, E.W.; Weber, C.R.; Shen, L.; Cohen, D.A.; Fenton, L.A.; Bruno, M.E.; Kaetzel, C.S. Targeted deletion of MyD88 in intestinal epithelial cells results in compromised antibacterial immunity associated with downregulation of polymeric immunoglobulin receptor, mucin-2, and antibacterial peptides. Mucosal. Immunol. 2012, 5, 501–512. [Google Scholar] [CrossRef]
- Nieuwenhuis, E.E.; Matsumoto, T.; Lindenbergh, D.; Willemsen, R.; Kaser, A.; Simons-Oosterhuis, Y.; Brugman, S.; Yamaguchi, K.; Ishikawa, H.; Aiba, Y.; et al. Cd1d-dependent regulation of bacterial colonization in the intestine of mice. J. Clin. Investig. 2009, 119, 1241–1250. [Google Scholar] [CrossRef]
- Suzuki, K.; Meek, B.; Doi, Y.; Muramatsu, M.; Chiba, T.; Honjo, T.; Fagarasan, S. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc. Natl. Acad. Sci. USA 2004, 101, 1981–1986. [Google Scholar] [CrossRef]
- Hapfelmeier, S.; Lawson, M.A.; Slack, E.; Kirundi, J.K.; Stoel, M.; Heikenwalder, M.; Cahenzli, J.; Velykoredko, Y.; Balmer, M.L.; Endt, K.; et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 2010, 328, 1705–1709. [Google Scholar] [CrossRef]
- Dias, J.; Leeansyah, E.; Sandberg, J.K. Multiple layers of heterogeneity and subset diversity in human MAIT cell responses to distinct microorganisms and to innate cytokines. Proc. Natl. Acad. Sci. USA 2017, 114, E5434–E5443. [Google Scholar] [CrossRef]
- Franchi, L.; Kamada, N.; Nakamura, Y.; Burberry, A.; Kuffa, P.; Suzuki, S.; Shaw, M.H.; Kim, Y.G.; Nunez, G. NLRC4-driven production of IL-1beta discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat. Immunol. 2012, 13, 449–456. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.H.; Kamada, N.; Kim, Y.G.; Nunez, G. Microbiota-induced IL-1beta, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J. Exp. Med. 2012, 209, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Berer, K.; Boziki, M.; Krishnamoorthy, G. Selective accumulation of pro-inflammatory T cells in the intestine contributes to the resistance to autoimmune demyelinating disease. PLoS ONE 2014, 9, e87876. [Google Scholar] [CrossRef]
- Sano, T.; Huang, W.; Hall, J.A.; Yang, Y.; Chen, A.; Gavzy, S.J.; Lee, J.Y.; Ziel, J.W.; Miraldi, E.R.; Domingos, A.I.; et al. An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell 2015, 163, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Tanoue, T.; Atarashi, K.; Honda, K. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol. 2016, 16, 295–309. [Google Scholar] [CrossRef]
- Kim, M.; Kim, C.H. Regulation of humoral immunity by gut microbial products. Gut. Microbes 2017, 8, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Haase, S.; Haghikia, A.; Wilck, N.; Muller, D.N.; Linker, R.A. Impacts of microbiome metabolites on immune regulation and autoimmunity. Immunology 2018, 154, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Camara-Lemarroy, C.R.; Metz, L.; Meddings, J.B.; Sharkey, K.A.; Wee Yong, V. The intestinal barrier in multiple sclerosis: Implications for pathophysiology and therapeutics. Brain 2018, 141, 1900–1916. [Google Scholar] [CrossRef]
- Berer, K.; Mues, M.; Koutrolos, M.; Rasbi, Z.A.; Boziki, M.; Johner, C.; Wekerle, H.; Krishnamoorthy, G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 2011, 479, 538–541. [Google Scholar] [CrossRef]
- Yokote, H.; Miyake, S.; Croxford, J.L.; Oki, S.; Mizusawa, H.; Yamamura, T. NKT cell-dependent amelioration of a mouse model of multiple sclerosis by altering gut flora. Am. J. Pathol. 2008, 173, 1714–1723. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Berer, K.; Gerdes, L.A.; Cekanaviciute, E.; Jia, X.; Xiao, L.; Xia, Z.; Liu, C.; Klotz, L.; Stauffer, U.; Baranzini, S.E.; et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 10719–10724. [Google Scholar] [CrossRef] [PubMed]
- Cekanaviciute, E.; Yoo, B.B.; Runia, T.F.; Debelius, J.W.; Singh, S.; Nelson, C.A.; Kanner, R.; Bencosme, Y.; Lee, Y.K.; Hauser, S.L.; et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl. Acad. Sci. USA 2017, 114, 10713–10718. [Google Scholar] [CrossRef] [PubMed]
- Lavasani, S.; Dzhambazov, B.; Nouri, M.; Fak, F.; Buske, S.; Molin, G.; Thorlacius, H.; Alenfall, J.; Jeppsson, B.; Westrom, B. A novel probiotic mixture exerts a therapeutic effect on experimental autoimmune encephalomyelitis mediated by IL-10 producing regulatory T cells. PLoS ONE 2010, 5, e9009. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.K.; Kim, G.C.; Kim, Y.; Hwang, W.; Jash, A.; Sahoo, A.; Kim, J.E.; Nam, J.H.; Im, S.H. Amelioration of experimental autoimmune encephalomyelitis by probiotic mixture is mediated by a shift in T helper cell immune response. Clin. Immunol. 2013, 146, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Takata, K.; Kinoshita, M.; Okuno, T.; Moriya, M.; Kohda, T.; Honorat, J.A.; Sugimoto, T.; Kumanogoh, A.; Kayama, H.; Takeda, K.; et al. The lactic acid bacterium Pediococcus acidilactici suppresses autoimmune encephalomyelitis by inducing IL-10-producing regulatory T cells. PLoS ONE 2011, 6, e27644. [Google Scholar] [CrossRef]
- Rezende, R.M.; Oliveira, R.P.; Medeiros, S.R.; Gomes-Santos, A.C.; Alves, A.C.; Loli, F.G.; Guimaraes, M.A.; Amaral, S.S.; da Cunha, A.P.; Weiner, H.L.; et al. Hsp65-producing Lactococcus lactis prevents experimental autoimmune encephalomyelitis in mice by inducing CD4+LAP+ regulatory T cells. J. Autoimmun. 2013, 40, 45–57. [Google Scholar] [CrossRef]
- Mangalam, A.; Shahi, S.K.; Luckey, D.; Karau, M.; Marietta, E.; Luo, N.; Choung, R.S.; Ju, J.; Sompallae, R.; Gibson-Corley, K.; et al. Human Gut-Derived Commensal Bacteria Suppress CNS Inflammatory and Demyelinating Disease. Cell Rep. 2017, 20, 1269–1277. [Google Scholar] [CrossRef]
- Gold, R.; Linington, C.; Lassmann, H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 2006, 129, 1953–1971. [Google Scholar] [CrossRef]
- Pollinger, B.; Krishnamoorthy, G.; Berer, K.; Lassmann, H.; Bosl, M.R.; Dunn, R.; Domingues, H.S.; Holz, A.; Kurschus, F.C.; Wekerle, H. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J. Exp. Med. 2009, 206, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, G.; Wekerle, H. EAE: An immunologist’s magic eye. Eur. J. Immunol. 2009, 39, 2031–2035. [Google Scholar] [CrossRef] [PubMed]
- Rumah, K.R.; Linden, J.; Fischetti, V.A.; Vartanian, T. Isolation of Clostridium perfringens type B in an individual at first clinical presentation of multiple sclerosis provides clues for environmental triggers of the disease. PLoS ONE 2013, 8, e76359. [Google Scholar] [CrossRef]
- Miyake, S.; Kim, S.; Suda, W.; Oshima, K.; Nakamura, M.; Matsuoka, T.; Chihara, N.; Tomita, A.; Sato, W.; Kim, S.W.; et al. Dysbiosis in the Gut Microbiota of Patients with Multiple Sclerosis, with a Striking Depletion of Species Belonging to Clostridia XIVa and IV Clusters. PLoS ONE 2015, 10, e0137429. [Google Scholar] [CrossRef] [PubMed]
- Tremlett, H.; Fadrosh, D.W.; Faruqi, A.A.; Hart, J.; Roalstad, S.; Graves, J.; Spencer, C.M.; Lynch, S.V.; Zamvil, S.S.; Waubant, E. Associations between the gut microbiota and host immune markers in pediatric multiple sclerosis and controls. BMC Neurol. 2016, 16, 182. [Google Scholar] [CrossRef]
- Tremlett, H.; Fadrosh, D.W.; Faruqi, A.A.; Zhu, F.; Hart, J.; Roalstad, S.; Graves, J.; Lynch, S.; Waubant, E. Gut microbiota in early pediatric multiple sclerosis: A case-control study. Eur. J. Neurol. 2016, 23, 1308–1321. [Google Scholar] [CrossRef] [PubMed]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef]
- Chen, J.; Chia, N.; Kalari, K.R.; Yao, J.Z.; Novotna, M.; Paz Soldan, M.M.; Luckey, D.H.; Marietta, E.V.; Jeraldo, P.R.; Chen, X.; et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci Rep. 2016, 6, 28484. [Google Scholar] [CrossRef]
- Mentis, A.A.; Dardiotis, E.; Grigoriadis, N.; Petinaki, E.; Hadjigeorgiou, G.M. Viruses and Multiple Sclerosis: From Mechanisms and Pathways to Translational Research Opportunities. Mol. Neurobiol 2017, 54, 3911–3923. [Google Scholar] [CrossRef]
- Mentis, A.A.; Dardiotis, E.; Grigoriadis, N.; Petinaki, E.; Hadjigeorgiou, G.M. Viruses and endogenous retroviruses in multiple sclerosis: From correlation to causation. Acta Neurol. Scand. 2017, 136, 606–616. [Google Scholar] [CrossRef]
- Kozhieva, M.K.; Melnikov, M.V.; Rogovsky, V.S.; Oleskin, A.V.; Kabilov, M.R.; Boyko, A.N. Gut human microbiota and multiple sclerosis. Zh Nevrol. Psikhiatr. Im S S Korsakova 2017, 117, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.; Forbes, J.D.; Zhu, F.; Bernstein, C.N.; Van Domselaar, G.; Graham, M.; Waubant, E.; Tremlett, H. The multiple sclerosis gut microbiota: A systematic review. Mult. Scler. Relat. Disord. 2020, 37, 101427. [Google Scholar] [CrossRef] [PubMed]
- Kozhieva, M.; Naumova, N.; Alikina, T.; Boyko, A.; Vlassov, V.; Kabilov, M.R. Primary progressive multiple sclerosis in a Russian cohort: Relationship with gut bacterial diversity. BMC Microbiol. 2019, 19, 309. [Google Scholar] [CrossRef]
- Riccio, P.; Rossano, R. Diet, Gut Microbiota, and Vitamins D + A in Multiple Sclerosis. Neurotherapeutics 2018, 15, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Borody, T.J.; Brandt, L.J.; Paramsothy, S. Therapeutic faecal microbiota transplantation: Current status and future developments. Curr. Opin. Gastroenterol. 2014, 30, 97–105. [Google Scholar] [CrossRef]
- Makkawi, S.; Camara-Lemarroy, C.; Metz, L. Fecal microbiota transplantation associated with 10 years of stability in a patient with SPMS. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e459. [Google Scholar] [CrossRef]
- Cammarota, G.; Ianiro, G.; Tilg, H.; Rajilic-Stojanovic, M.; Kump, P.; Satokari, R.; Sokol, H.; Arkkila, P.; Pintus, C.; Hart, A.; et al. European consensus conference on faecal microbiota transplantation in clinical practice. Gut 2017, 66, 569–580. [Google Scholar] [CrossRef]
- Chin, S.M.; Sauk, J.; Mahabamunuge, J.; Kaplan, J.L.; Hohmann, E.L.; Khalili, H. Fecal Microbiota Transplantation for Recurrent Clostridium difficile Infection in Patients with Inflammatory Bowel Disease: A Single-Center Experience. Clin. Gastroenterol. Hepatol. 2017, 15, 597–599. [Google Scholar] [CrossRef]
- Paramsothy, S.; Kamm, M.A.; Kaakoush, N.O.; Walsh, A.J.; van den Bogaerde, J.; Samuel, D.; Leong, R.W.L.; Connor, S.; Ng, W.; Paramsothy, R.; et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: A randomised placebo-controlled trial. Lancet 2017, 389, 1218–1228. [Google Scholar] [CrossRef]
- Jayasinghe, T.N.; Chiavaroli, V.; Holland, D.J.; Cutfield, W.S.; O’Sullivan, J.M. The New Era of Treatment for Obesity and Metabolic Disorders: Evidence and Expectations for Gut Microbiome Transplantation. Front. Cell Infect. Microbiol. 2016, 6, 15. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Kassam, Z.; Fagan, A.; Gavis, E.A.; Liu, E.; Cox, I.J.; Kheradman, R.; Heuman, D.; Wang, J.; Gurry, T.; et al. Fecal microbiota transplant from a rational stool donor improves hepatic encephalopathy: A randomized clinical trial. Hepatology 2017, 66, 1727–1738. [Google Scholar] [CrossRef]
- Choi, H.H.; Cho, Y.S. Fecal Microbiota Transplantation: Current Applications, Effectiveness, and Future Perspectives. Clin. Endosc. 2016, 49, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Lawson Health Research Institute. Fecal Microbial Transplantation in Relapsing Multiple Sclerosis Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT03183869 (accessed on 5 April 2020).
- Rumah, K.R.; Vartanian, T.K.; Fischetti, V.A. Oral Multiple Sclerosis Drugs Inhibit the In vitro Growth of Epsilon Toxin Producing Gut Bacterium, Clostridium perfringens. Front. Cell Infect. Microbiol. 2017, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Linden, J.R.; Ma, Y.; Zhao, B.; Harris, J.M.; Rumah, K.R.; Schaeren-Wiemers, N.; Vartanian, T. Clostridium perfringens Epsilon Toxin Causes Selective Death of Mature Oligodendrocytes and Central Nervous System Demyelination. MBio 2015, 6, e02513. [Google Scholar] [CrossRef] [PubMed]
- Katz Sand, I.; Zhu, Y.; Ntranos, A.; Clemente, J.C.; Cekanaviciute, E.; Brandstadter, R.; Crabtree-Hartman, E.; Singh, S.; Bencosme, Y.; Debelius, J.; et al. Disease-modifying therapies alter gut microbial composition in MS. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e517. [Google Scholar] [CrossRef] [PubMed]
- Giles, E.M.; Stagg, A.J. Type 1 Interferon in the Human Intestine-A Co-ordinator of the Immune Response to the Microbiota. Inflamm. Bowel Dis. 2017, 23, 524–533. [Google Scholar] [CrossRef]
- LeMessurier, K.S.; Hacker, H.; Chi, L.; Tuomanen, E.; Redecke, V. Type I interferon protects against pneumococcal invasive disease by inhibiting bacterial transmigration across the lung. PLoS Pathog. 2013, 9, e1003727. [Google Scholar] [CrossRef]
- Nakahashi-Oda, C.; Udayanga, K.G.; Nakamura, Y.; Nakazawa, Y.; Totsuka, N.; Miki, H.; Iino, S.; Tahara-Hanaoka, S.; Honda, S.; Shibuya, K.; et al. Apoptotic epithelial cells control the abundance of Treg cells at barrier surfaces. Nat. Immunol. 2016, 17, 441–450. [Google Scholar] [CrossRef]
- Castillo-Alvarez, F.; Perez-Matute, P.; Oteo, J.A.; Marzo-Sola, M.E. The influence of interferon beta-1b on gut microbiota composition in patients with multiple sclerosis. Neurologia 2018. [Google Scholar] [CrossRef]
- Aharoni, R.; Sonego, H.; Brenner, O.; Eilam, R.; Arnon, R. The therapeutic effect of glatiramer acetate in a murine model of inflammatory bowel disease is mediated by anti-inflammatory T-cells. Immunol. Lett. 2007, 112, 110–119. [Google Scholar] [CrossRef]
- Yablecovitch, D.; Shabat-Simon, M.; Aharoni, R.; Eilam, R.; Brenner, O.; Arnon, R. Beneficial effect of glatiramer acetate treatment on syndecan-1 expression in dextran sodium sulfate colitis. J. Pharmacol. Exp. Ther. 2011, 337, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Shahi, S.K.; Freedman, S.N.; Murra, A.C.; Zarei, K.; Sompallae, R.; Gibson-Corley, K.N.; Karandikar, N.J.; Murray, J.A.; Mangalam, A.K. Prevotella histicola, A Human Gut Commensal, Is as Potent as COPAXONE(R) in an Animal Model of Multiple Sclerosis. Front. Immunol. 2019, 10, 462. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Waubant, E.; Chehoud, C.; Kuczynski, J.; DeSantis, T.Z.; Warrington, J.; Venkatesan, A.; Fraser, C.M.; Mowry, E.M. Gut microbiota in multiple sclerosis: Possible influence of immunomodulators. J. Investig. Med. 2015, 63, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Wu, Y.; Xie, F.; Du, K.; Wang, Y.; Shi, L.; Ji, L.; Liu, T.; Ma, X. Dimethyl fumarate reduces the risk of mycotoxins via improving intestinal barrier and microbiota. Oncotarget 2017, 8, 44625–44638. [Google Scholar] [CrossRef] [PubMed]
- Pitarokoili, K.; Bachir, H.; Sgodzai, M.; Gruter, T.; Haupeltshofer, S.; Duscha, A.; Pedreiturria, X.; Motte, J.; Gold, R. Induction of Regulatory Properties in the Intestinal Immune System by Dimethyl Fumarate in Lewis Rat Experimental Autoimmune Neuritis. Front. Immunol. 2019, 10, 2132. [Google Scholar] [CrossRef]
- Ochoa-Reparaz, J.; Colpitts, S.L.; Kircher, C.; Kasper, E.J.; Telesford, K.M.; Begum-Haque, S.; Pant, A.; Kasper, L.H. Induction of gut regulatory CD39(+) T cells by teriflunomide protects against EAE. Neurol Neuroimmunol. Neuroinflamm. 2016, 3, e291. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Bryant, R.V.; Travis, S. Interfering with leukocyte trafficking in Crohn’s disease. Best Pract. Res. Clin. Gastroenterol. 2019, 38–39, 101617. [Google Scholar] [CrossRef]
- Kunisawa, J.; Kurashima, Y.; Higuchi, M.; Gohda, M.; Ishikawa, I.; Ogahara, I.; Kim, N.; Shimizu, M.; Kiyono, H. Sphingosine 1-phosphate dependence in the regulation of lymphocyte trafficking to the gut epithelium. J. Exp. Med. 2007, 204, 2335–2348. [Google Scholar] [CrossRef]
- Deguchi, Y.; Andoh, A.; Yagi, Y.; Bamba, S.; Inatomi, O.; Tsujikawa, T.; Fujiyama, Y. The S1P receptor modulator FTY720 prevents the development of experimental colitis in mice. Oncol. Rep. 2006, 16, 699–703. [Google Scholar] [CrossRef]
- Huang, Y.; Mao, K.; Chen, X.; Sun, M.A.; Kawabe, T.; Li, W.; Usher, N.; Zhu, J.; Urban, J.F., Jr.; Paul, W.E.; et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science 2018, 359, 114–119. [Google Scholar] [CrossRef]
- Vidal-Martinez, G.; Vargas-Medrano, J.; Gil-Tommee, C.; Medina, D.; Garza, N.T.; Yang, B.; Segura-Ulate, I.; Dominguez, S.J.; Perez, R.G. FTY720/Fingolimod Reduces Synucleinopathy and Improves Gut Motility in A53T Mice: CONTRIBUTIONS OF PRO-BRAIN-DERIVED NEUROTROPHIC FACTOR (PRO-BDNF) AND MATURE BDNF. J. Biol. Chem. 2016, 291, 20811–20821. [Google Scholar] [CrossRef] [PubMed]
- Bonitz, J.A.; Son, J.Y.; Chandler, B.; Tomaio, J.N.; Qin, Y.; Prescott, L.M.; Feketeova, E.; Deitch, E.A. A sphingosine-1 phosphate agonist (FTY720) limits trauma/hemorrhagic shock-induced multiple organ dysfunction syndrome. Shock 2014, 42, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.G.; Liu, F.; Verin, A.D.; Birukova, A.; Dechert, M.A.; Gerthoffer, W.T.; Bamberg, J.R.; English, D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Investig. 2001, 108, 689–701. [Google Scholar] [CrossRef]
- Wang, X.; Maruvada, R.; Morris, A.J.; Liu, J.O.; Wolfgang, M.J.; Baek, D.J.; Bittman, R.; Kim, K.S. Sphingosine 1-Phosphate Activation of EGFR As a Novel Target for Meningitic Escherichia coli Penetration of the Blood-Brain Barrier. PLoS Pathog. 2016, 12, e1005926. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Orengo, L.; Daniels, B.P.; Dorsey, D.; Basak, S.A.; Grajales-Reyes, J.G.; McCandless, E.E.; Piccio, L.; Schmidt, R.E.; Cross, A.H.; Crosby, S.D.; et al. Enhanced sphingosine-1-phosphate receptor 2 expression underlies female CNS autoimmunity susceptibility. J. Clin. Investig. 2014, 124, 2571–2584. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Gardell, S.E.; Herr, D.R.; Rivera, R.; Lee, C.W.; Noguchi, K.; Teo, S.T.; Yung, Y.C.; Lu, M.; Kennedy, G.; et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 751–756. [Google Scholar] [CrossRef]
- Colombo, E.; Di Dario, M.; Capitolo, E.; Chaabane, L.; Newcombe, J.; Martino, G.; Farina, C. Fingolimod may support neuroprotection via blockade of astrocyte nitric oxide. Ann. Neurol. 2014, 76, 325–337. [Google Scholar] [CrossRef]
- Gohda, M.; Kunisawa, J.; Miura, F.; Kagiyama, Y.; Kurashima, Y.; Higuchi, M.; Ishikawa, I.; Ogahara, I.; Kiyono, H. Sphingosine 1-phosphate regulates the egress of IgA plasmablasts from Peyer’s patches for intestinal IgA responses. J. Immunol. 2008, 180, 5335–5343. [Google Scholar] [CrossRef]
- Kunisawa, J.; Kurashima, Y.; Gohda, M.; Higuchi, M.; Ishikawa, I.; Miura, F.; Ogahara, I.; Kiyono, H. Sphingosine 1-phosphate regulates peritoneal B-cell trafficking for subsequent intestinal IgA production. Blood 2007, 109, 3749–3756. [Google Scholar] [CrossRef]
- Coles, A.J.; Cohen, J.A.; Fox, E.J.; Giovannoni, G.; Hartung, H.P.; Havrdova, E.; Schippling, S.; Selmaj, K.W.; Traboulsee, A.; Compston, D.A.S.; et al. Alemtuzumab CARE-MS II 5-year follow-up: Efficacy and safety findings. Neurology 2017, 89, 1117–1126. [Google Scholar] [CrossRef]
- Havrdova, E.; Horakova, D.; Kovarova, I. Alemtuzumab in the treatment of multiple sclerosis: Key clinical trial results and considerations for use. Ther. Adv. Neurol. Disord. 2015, 8, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Holmoy, T.; Fevang, B.; Olsen, D.B.; Spigset, O.; Bo, L. Adverse events with fatal outcome associated with alemtuzumab treatment in multiple sclerosis. BMC Res. Notes 2019, 12, 497. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Giovannoni, G.; Schmierer, K. Marked neutropenia: Significant but rare in people with multiple sclerosis after alemtuzumab treatment. Mult. Scler. Relat. Disord. 2017, 18, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Vijiaratnam, N.; Rath, L.; Xu, S.S.; Skibina, O. Pancolitis a novel early complication of Alemtuzumab for MS treatment. Mult. Scler. Relat. Disord. 2016, 7, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.L.; Lyu, Y.Q.; Jiang, H.T.; Shan, T.; Zhang, J.B.; Li, Q.R.; Li, J.S. Effect of alemtuzumab on intestinal intraepithelial lymphocytes and intestinal barrier function in cynomolgus model. Chin. Med. J. (Engl.) 2015, 128, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, Q.; Wang, C.; Jiang, S.; Li, N.; Li, J. The response of intestinal stem cells and epithelium after alemtuzumab administration. Cell Mol. Immunol. 2011, 8, 325–332. [Google Scholar] [CrossRef]
- Qu, L.; Li, Q.; Jiang, H.; Gu, L.; Zhang, Q.; Wang, C.; Li, J. Effect of anti-mouse CD52 monoclonal antibody on mouse intestinal intraepithelial lymphocytes. Transplantation 2009, 88, 766–772. [Google Scholar] [CrossRef]
- Li, Q.R.; Wang, C.Y.; Tang, C.; He, Q.; Li, N.; Li, J.S. Reciprocal interaction between intestinal microbiota and mucosal lymphocyte in cynomolgus monkeys after alemtuzumab treatment. Am. J. Transplant. 2013, 13, 899–910. [Google Scholar] [CrossRef]
- Metz, L.M.; Li, D.K.B.; Traboulsee, A.L.; Duquette, P.; Eliasziw, M.; Cerchiaro, G.; Greenfield, J.; Riddehough, A.; Yeung, M.; Kremenchutzky, M.; et al. Trial of Minocycline in a Clinically Isolated Syndrome of Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 2122–2133. [Google Scholar] [CrossRef]
- Mentis, A.A.; Pantelidi, K.; Dardiotis, E.; Hadjigeorgiou, G.M.; Petinaki, E. Precision Medicine and Global Health: The Good, the Bad, and the Ugly. Front. Med. (Lausanne) 2018, 5, 67. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boziki, M.K.; Kesidou, E.; Theotokis, P.; Mentis, A.-F.A.; Karafoulidou, E.; Melnikov, M.; Sviridova, A.; Rogovski, V.; Boyko, A.; Grigoriadis, N. Microbiome in Multiple Sclerosis: Where Are We, What We Know and Do Not Know. Brain Sci. 2020, 10, 234. https://doi.org/10.3390/brainsci10040234
Boziki MK, Kesidou E, Theotokis P, Mentis A-FA, Karafoulidou E, Melnikov M, Sviridova A, Rogovski V, Boyko A, Grigoriadis N. Microbiome in Multiple Sclerosis: Where Are We, What We Know and Do Not Know. Brain Sciences. 2020; 10(4):234. https://doi.org/10.3390/brainsci10040234
Chicago/Turabian StyleBoziki, Marina Kleopatra, Evangelia Kesidou, Paschalis Theotokis, Alexios-Fotios A. Mentis, Eleni Karafoulidou, Mikhail Melnikov, Anastasia Sviridova, Vladimir Rogovski, Alexey Boyko, and Nikolaos Grigoriadis. 2020. "Microbiome in Multiple Sclerosis: Where Are We, What We Know and Do Not Know" Brain Sciences 10, no. 4: 234. https://doi.org/10.3390/brainsci10040234
APA StyleBoziki, M. K., Kesidou, E., Theotokis, P., Mentis, A.-F. A., Karafoulidou, E., Melnikov, M., Sviridova, A., Rogovski, V., Boyko, A., & Grigoriadis, N. (2020). Microbiome in Multiple Sclerosis: Where Are We, What We Know and Do Not Know. Brain Sciences, 10(4), 234. https://doi.org/10.3390/brainsci10040234