Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review



Abstract
1. Introduction
1.1. Alzheimer’s Disease
1.2. Parkinson’s Disease
1.3. Amyotrophic Lateral Sclerosis
1.4. Huntington’s Disease
1.5. Spinocerebellar Ataxias
1.6. Transmissible Spongiform Encephalopathy
1.7. Multiple Sclerosis
1.8. Protein Aggregation in Neurodegeneration
1.9. Post-Translational Modifications and NDDs
2. Methods
2.1. Search Strategy
2.2. Eligibility Criteria
2.3. Data Acquisition and Analysis
3. Results
3.1. Alzheimer’s Disease
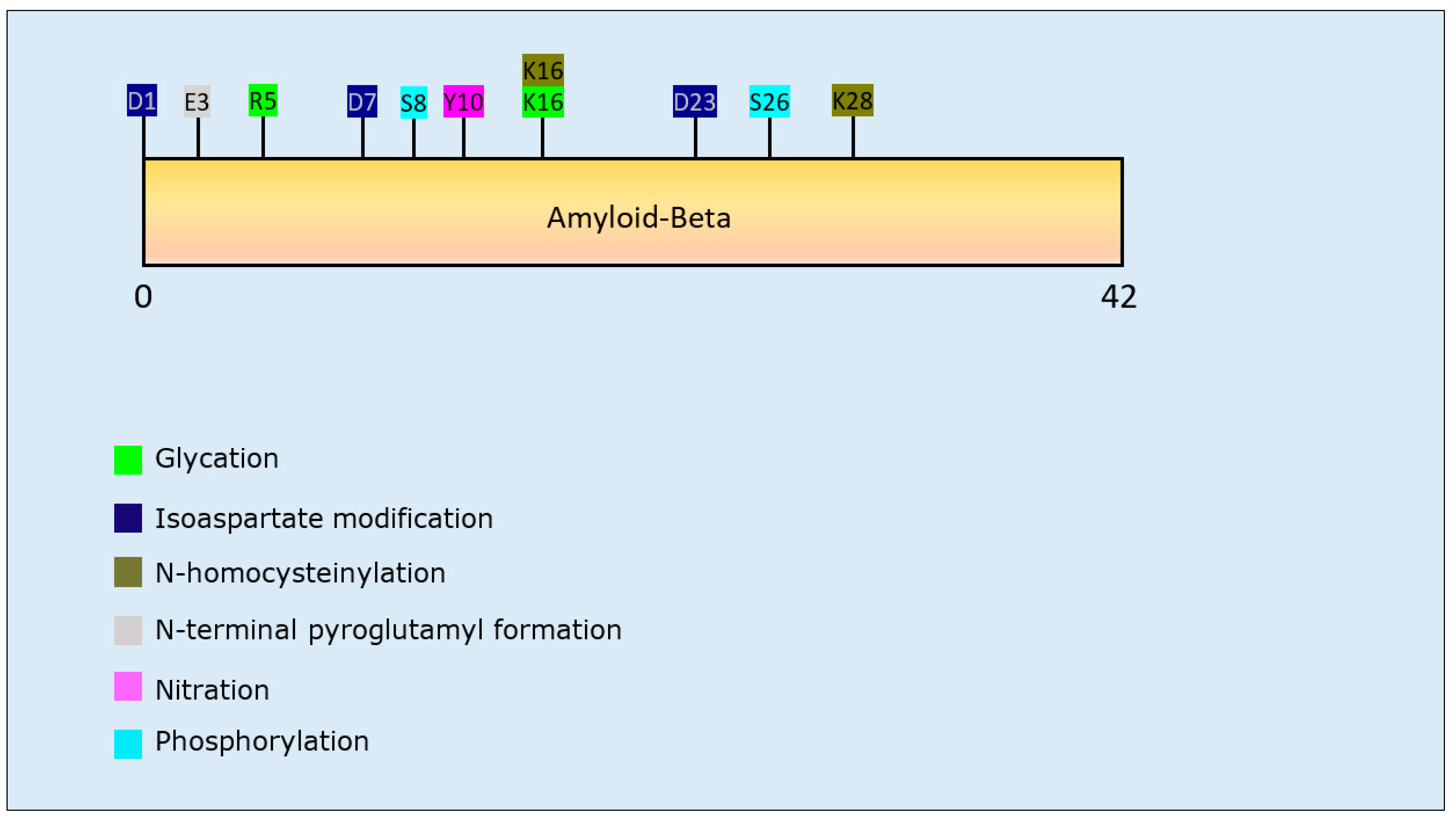
3.1.1. Aβ PTMs and Propensity for Aggregation
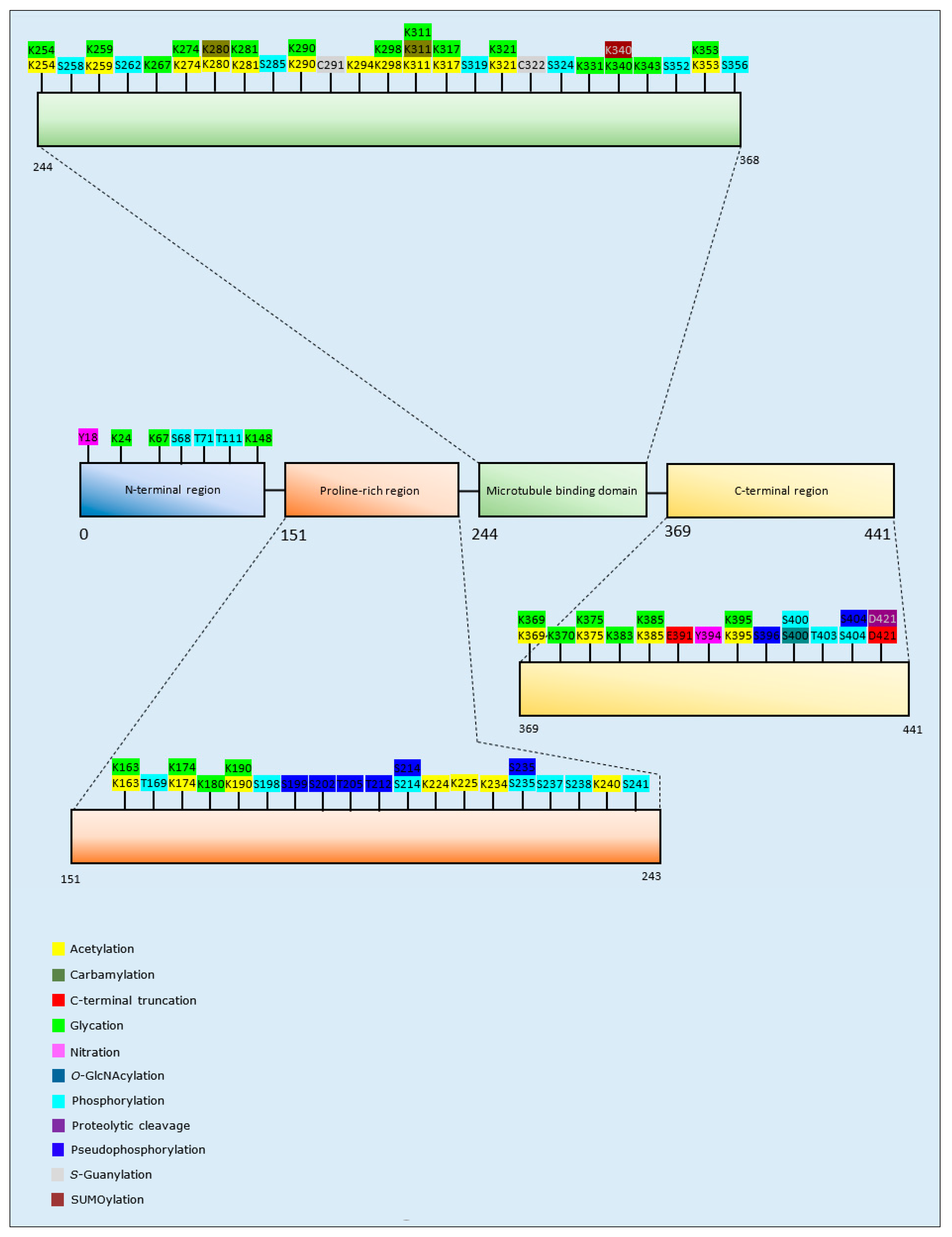
3.1.2. Tau PTMs and Propensity for Aggregation
3.2. Parkinson’s Disease
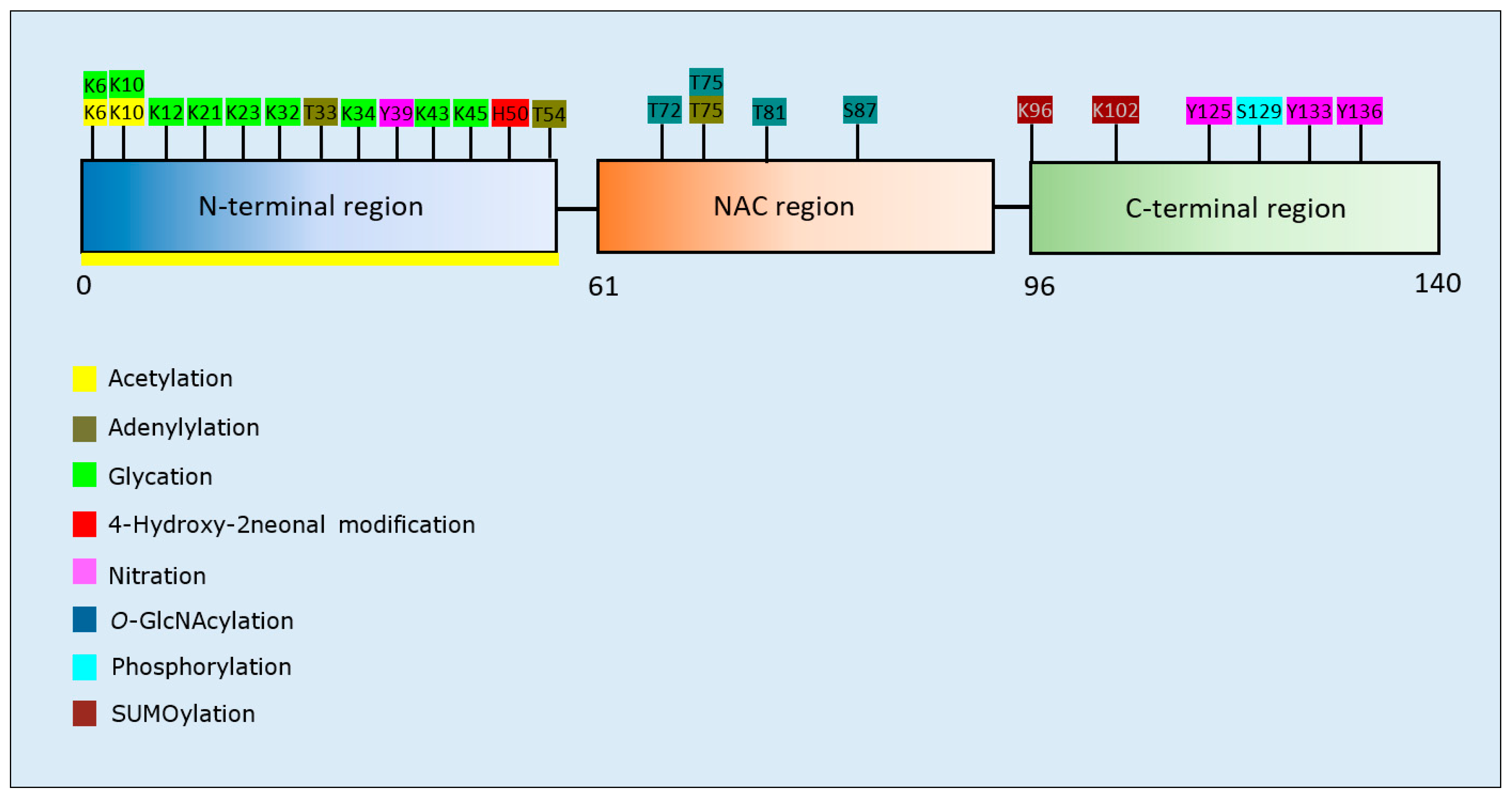
α-Synuclein PTMs and Propensity for Aggregation
3.3. Amyotrophic Lateral Sclerosis
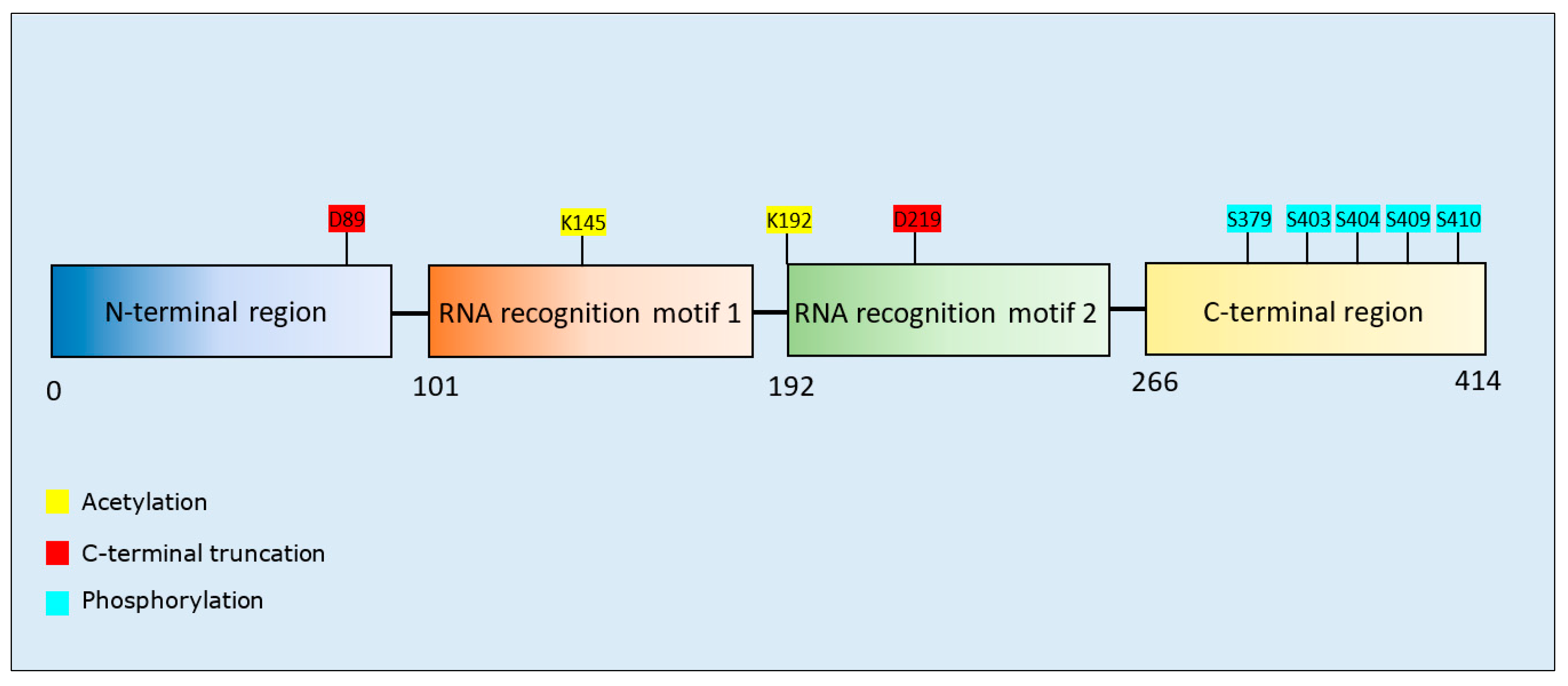
3.3.1. TAR DNA-Binding Protein 43 PTMs and Propensity for Aggregation
3.3.2. SOD1 PTMs and Propensity for Aggregation
3.4. Huntington’s Disease
Htt PTMs and Propensity for Aggregation
3.5. Spinocerebellar Ataxias
Ataxins PTMs and Propensity for Aggregation
3.6. Transmissible Spongiform Encephalopathies
Prion Protein PTMs and Propensity for Aggregation
3.7. Multiple Sclerosis
4. Discussion
4.1. Study Limitations
4.2. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical Neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [PubMed]
- Dugger, B.N.; Dickson, D.W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. (Ed.) Solving the puzzle of neurodegeneration. In The Molecular and Cellular Basis of Neurodegenerative Diseases: Underlying Mechanisms; Elsevier: London, UK, 2018. [Google Scholar]
- Sullivan, R.; Yau, W.Y.; O’Connor, E.; Houlden, H. Spinocerebellar ataxia: An update. J. Neurol. 2019, 266, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; Zubair, H.; Pursell, S.; Shahab, M. Neurodegenerative diseases: Regenerative mechanisms and novel therapeutic approaches. Brain Sci. 2018, 8, 177. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Wan, L.; Xu, K.; Chen, Z.; Tang, B.; Jiang, H. Roles of post-translational modifications in spinocerebellar ataxias. Front. Cell. Neurosci. 2018, 12, 290. [Google Scholar] [CrossRef]
- David, M.A.; Tayebi, M. Detection of protein aggregates in brain and cerebrospinal fluid derived from multiple sclerosis patients. Front. Neurol. 2014, 5, 251. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Blokhuis, A.M.; Groen, E.J.N.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef]
- Ugalde, C.L.; Finkelstein, D.I.; Lawson, V.A.; Hill, A.F. Pathogenic mechanisms of prion protein, amyloid-β and α-synuclein misfolding: The prion concept and neurotoxicity of protein oligomers. J. Neurochem. 2016, 139, 162–180. [Google Scholar] [CrossRef]
- Schattling, B.; Engler, J.B.; Volkmann, C.; Rothammer, N.; Woo, M.S.; Petersen, M.; Winkler, I.; Kaufmann, M.; Rosenkranz, S.C.; Fejtova, A.; et al. Bassoon proteinopathy drives neurodegeneration in multiple sclerosis. Nat. Neurosci. 2019, 22, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, J.; Banati, R.B.; Cuzner, M.L.; Kreutzberg, G.W.; Newcombe, J. Amyloid precursor protein (APP) expression in multiple sclerosis lesions. Glia 1995, 15, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Coarelli, G.; Brice, A.; Durr, A. Recent advances in understanding dominant spinocerebellar ataxias from clinical and genetic points of view. F1000Res 2018, 7, Rev-1781. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, T.; Watanabe, A.; Saito, Y.; Murayama, S.; Mann, D.M.A.; Yamazaki, M.; Ravid, R.; Morishima-Kawashima, M.; Nagashima, K.; Ihara, Y. Visualization of newly deposited tau in neurofibrillary tangles and neuropil threads. J. Neuropathol. Exp. Neurol. 2005, 64, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Yoshida, S.; Sugeno, N.; Kobayashi, J.; Aoki, M. DnaJ/Hsp40 family and parkinson’s disease. Front. Neurosci. 2018, 11, 743. [Google Scholar] [CrossRef] [PubMed]
- Pihlstrøm, L.; Wiethoff, S.; Houlden, H.; Kovacs, G.G.; Alafuzoff, I. Genetics of neurodegenerative diseases: An overview. Handb. Clin. Neurol. 2017, 145, 309–323. [Google Scholar]
- Wirths, O.; Bayer, T.A. Neuron Loss in Transgenic Mouse Models of Alzheimer’s Disease. Int. J. Alzheimers Dis. 2010, 12, 723782. [Google Scholar] [CrossRef]
- Sánchez, A.; Milà, M.; Castellví-Bel, S.; Rosich, M.; Jiménez, D.; Badenas, C.; Estivill, X. Maternal transmission in sporadic Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 1997, 62, 535–537. [Google Scholar] [CrossRef][Green Version]
- Sim, V. Prion disease. In BMJ Best Practice; BMJ Publishing Group: London, UK, 2018; Available online: https://bestpractice.bmj.com/topics/en-gb/484/pdf/484.pdf (accessed on 20 September 2019).
- Steenhof, M.; Stenager, E.; Nielsen, N.M.; Kyvik, K.; Möller, S.; Hertz, J.M. Familial multiple sclerosis patients have a shorter delay in diagnosis than sporadic cases. Mult. Scler. Relat. Discord. 2019, 32, 97–102. [Google Scholar] [CrossRef]
- Kraft, S.; Furtado, S.; Ranawaya, R.; Parboosingh, J.; Bleoo, S.; McElligott, K.; Bridge, P.; Spacey, S.; Das, S.; Suchowersky, O. Adult onset spinocerebellar ataxia in a Canadian movement disorders clinic. Can. J. Neurol. Sci. 2005, 32, 450–458. [Google Scholar] [CrossRef]
- Chen, C.; Dong, X.P. Epidemiological characteristics of human prion diseases. Infect. Dis. Poverty 2016, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Niclis, J.C.; Pinar, A.; Haynes, J.M.; Alsanie, W.; Jenny, R.; Dottori, M.; Cram, D.S. Characterization of forebrain neurons derived from late-onset Huntington’s disease human embryonic stem cell lines. Front. Cell. Neurosci. 2013, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- GBD 2016 Motor Neuron Disease Collaborators. Global, regional, and national burden of motor neuron diseases 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 1083–1097. [Google Scholar] [CrossRef]
- GBD 2016 Multiple Sclerosis Collaborators. Global, regional, and national burden of multiple sclerosis 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 269–285. [Google Scholar] [CrossRef]
- GBD 2016 Dementia Collaborators. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural. Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Burré, J. The synaptic function of α-Synuclein. J. Park. Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimer’s Dement. J. Alzheimers Dis. 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Wortmann, M. Dementia: A global health priority - highlights from an ADI and World Health Organization report. Alzheimers Res. Ther. 2012, 4, 40. [Google Scholar] [CrossRef]
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158. [Google Scholar] [CrossRef]
- Hane, F.T.; Robinson, M.; Lee, B.Y.; Bai, O.; Leonenko, Z.; Albert, M.S. Recent progress in Alzheimer’s disease research, Part 3: Diagnosis and treatment. J. Alzheimers Dis. 2017, 57, 645–665. [Google Scholar] [CrossRef] [PubMed]
- Perl, D.P. Neuropathology of Alzheimer’s disease. Mt. Sinai. J. Med. 2010, 77, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Gouras, G.K.; Olsson, T.T.; Hansson, O. β-Amyloid peptides and amyloid plaques in Alzheimer’s disease. Neurotherapeutics 2015, 12, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Brothers, H.M.; Gosztyla, M.L.; Robinson, S.R. The physiological roles of amyloid-β peptide hint at new ways to treat Alzheimer’s disease. Front. Ageing Nurosci. 2018, 10, 118. [Google Scholar] [CrossRef]
- Pearson, H.A.; Peers, C. Physiological roles for amyloid beta peptides. J. Physiol. 2006, 575, 5–10. [Google Scholar] [CrossRef]
- Lührs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Döbeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid-beta (1–42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef]
- Medina, M.; Hernández, F.; Avila, J. New features about tau function and dysfunction. Biomolecules 2016, 6, 21. [Google Scholar] [CrossRef]
- Jouanne, M.; Rault, S.; Voisin-Chiret, A.S. Tau protein aggregation in Alzheimer’s disease: An attractive target for the development of novel therapeutic agents. Eur. J. Med. Chem. 2017, 139, 153–167. [Google Scholar] [CrossRef]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Pakkenberg, H.; Brody, H. The number of nerve cells in the substantia nigra in paralysis agitans. Acta Neuropathol. 1965, 5, 320–324. [Google Scholar] [CrossRef] [PubMed]
- DeMaagd, G.; Philip, A. Parkinson’s disease and its management: Part 1: Disease entity, risk factors, pathophysiology, clinical presentation, and diagnosis. P T 2015, 40, 504–532. [Google Scholar] [PubMed]
- Spillanti, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Venda, L.L.; Cragg, S.J.; Buchman, V.L.; Wade-Martins, R. α-Synuclein and dopamine at the crossroads of Parkinson’s disease. Trends Neurosci. 2010, 33, 559–568. [Google Scholar] [CrossRef]
- Cookson, M.R. alpha-Synuclein and neuronal cell death. Mol. Neurodegener. 2009, 4, 9. [Google Scholar] [CrossRef]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar]
- Oskarsson, B.; Gendron, T.F.; Staff, N.P. Amyotrophic lateral sclerosis: An update for 2018. Mayo Clin. Proc. 2018, 93, 1617–1628. [Google Scholar] [CrossRef]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef]
- Hergesheimer, R.C.; Chami, A.A.; de Assis, D.R.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Lanznaster, D.; Blasco, H. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: A resolution in sight? Brain 2019, 142, 1176–1194. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.M.A.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Highley, J.R.; Kirby, J.; Jansweijer, J.A.; Webb, P.S.; Hewamadduma, C.A.; Heath, P.R.; Higginbottom, A.; Raman, R.; Ferraiuolo, L.; Cooper-Knock, J.; et al. Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol. Appl. Neurobiol. 2014, 40, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Rakhit, R.; Chakrabartty, A. Structure, folding, and misfolding of Cu,Zn superoxide dismutase in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 1025–1037. [Google Scholar] [CrossRef]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease. Int. J. Mol. Sci. 2018, 19, 1345. [Google Scholar] [CrossRef]
- Medinas, D.B.; Rozas, P.; Martínez Traub, F.; Woehlbier, U.; Brown, R.H.; Bosco, D.A.; Hetz, C. Endoplasmic reticulum stress leads to accumulation of wild-type SOD1 aggregates associated with sporadic amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 8209. [Google Scholar] [CrossRef]
- Myers, R.H. Huntington’s disease genetics. NeuroRx 2004, 1, 255–262. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Huang, W.J.; Chen, W.W.; Zhang, X. Huntington’s disease: Molecular basis of pathology and status of current therapeutic approaches. Exp. Ther. Med. 2016, 12, 1951–1956. [Google Scholar] [CrossRef]
- Roos, R.A. Huntington’s disease: A clinical review. Orphanet J Rare Dis 2010, 5, 40. [Google Scholar] [CrossRef]
- Nasir, J.; Floresco, S.B.; O’Kusky, J.R.; Diewert, V.M.; Richman, J.M.; Zeisler, J.; Borowski, A.; Marth, J.D.; Phillips, A.G.; Hayden, M.R. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 1995, 81, 811–823. [Google Scholar] [CrossRef]
- McKinstry, S.U.; Karadeniz, Y.B.; Worthington, A.K.; Hayrapetyan, V.Y.; Ozlu, M.I.; Serafin-Molina, K.; Risher, W.C.; Ustunkaya, T.; Dragatsis, I.; Zeitlin, S.; et al. Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J. Neurosci. 2014, 34, 9455–9472. [Google Scholar] [CrossRef] [PubMed]
- Finkbeiner, S. Huntington’s disease. Cold Spring Harb Perspect Biol. 2011, 3, a007476. [Google Scholar] [CrossRef] [PubMed]
- Kim, M. Beta conformation of polyglutamine track revealed by a crystal structure of Huntingtin N-terminal region with insertion of three histidine residues. Prion 2013, 7, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Arrasate, M.; Finkbeiner, S. Protein aggregates in Huntington’s disease. Exp. Neurol. 2012, 238, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, I.; Mahlke, C.; Yuan, J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 2003, 421, 373–379. [Google Scholar] [CrossRef]
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef]
- Ramachandra, N.B.; Kusuma, L. An understanding of spinocerebellar ataxia. Indian J. Med. Res. 2015, 141, 148–150. [Google Scholar] [CrossRef]
- Paulson, H.L. The spinocerebellar ataxias. J. Neuroophthalmol. 2009, 29, 227–237. [Google Scholar] [CrossRef]
- Rabinovici, G.D.; Wang, P.N.; Levin, J.; Cook, L.; Pravdin, M.; Davis, J.; DeArmond, S.J.; Barbaro, N.M.; Martindale, J.; Miller, B.L.; et al. First symptom in sporadic Creutzfeldt–Jakob disease. Neurology 2006, 66, 286. [Google Scholar] [CrossRef]
- Jeffrey, M.; Scott, J.; Williams, A.; Fraser, H. Ultrastructural features of spongiform encephalopathy transmitted to mice from three species of bovidae. Acta Neuropathol. 1992, 84, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Hirsch, N. Prion diseases. Anaesth. Inten. Care Med. 2013, 14, 407–409. [Google Scholar] [CrossRef]
- Collins, S.J.; Sanchez-Juan, P.; Masters, C.L.; Klug, G.M.; van Duijn, C.; Poleggi, A.; Pocchiari, M.; Almonti, S.; Cuadrado-Corrales, N.; de Pedro-Cuesta, J.; et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt–Jakob disease. Brain 2006, 129, 2278–2287. [Google Scholar] [CrossRef]
- Moya, K.L.; Salès, N.; Hässig, R.; Créminon, C.; Grassi, J.; Di Giamberardino, L. Immunolocalization of the cellular prion protein in normal brain. Microsc. Res. Tech. 2000, 50, 58–65. [Google Scholar] [CrossRef]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Morantes, C.Y.; Wille, H. The structure of human prions: From biology to structural models-considerations and pitfalls. Viruses 2014, 6, 3875–3892. [Google Scholar] [CrossRef] [PubMed]
- Bueler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef]
- Ambadi Thody, S.; Mathew, M.K.; Udgaonkar, J.B. Mechanism of aggregation and membrane interactions of mammalian prion protein. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1927–1935. [Google Scholar] [CrossRef]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Budka, H. Molecular pathology of human prion diseases. Int. J. Mol. Sci. 2009, 10, 976–999. [Google Scholar] [CrossRef]
- Laurent, M. Autocatalytic processes in cooperative mechanisms of prion diseases. FEBS Lett. 1997, 407, 1–6. [Google Scholar] [CrossRef]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple sclerosis: Pathogenesis, symptoms, diagnoses and cell-based therapy. Cell J. 2017, 19, 1–10. [Google Scholar] [PubMed]
- Dutta, R.; Trapp, B.D. Relapsing and progressive forms of multiple sclerosis: Insights from pathology. Curr. Opin. Neurol. 2014, 27, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Altrock, W.D.; tom Dieck, S.; Sokolov, M.; Meyer, A.C.; Sigler, A.; Brakebusch, C.; Fässler, R.; Richter, K.; Boeckers, T.M.; Potschka, H. Functional inactivation of a fraction of excitatory synapses in mice deficient for the active zone protein bassoon. Neuron 2003, 37, 787–800. [Google Scholar] [CrossRef]
- Takalo, M.; Salminen, A.; Soininen, H.; Hiltunen, M.; Haapasalo, A. Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2013, 2, 1–14. [Google Scholar]
- Lim, J.; Yue, Z. Neuronal aggregates: Formation, clearance, and spreading. Dev. Cell 2015, 32, 491–501. [Google Scholar] [CrossRef]
- Uniprot Modified Residues in Proteins. Available online: http://www.uniprot.org/docs/ptmlist.txt (accessed on 15 March 2020).
- Xu, H.; Wang, Y.; Lin, S.; Deng, W.; Peng, D.; Cui, Q.; Xue, Y. PTMD: A database of human disease-associated Post-translational Modifications. Genom. Proteom. Bioinform. 2018, 16, 244–251. [Google Scholar] [CrossRef]
- Lee, K.W.; Chen, W.; Junn, E.; Im, J.Y.; Grosso, H.; Sonsalla, P.K.; Feng, X.; Ray, N.; Fernandez, J.R.; Chao, Y.; et al. Enhanced phosphatase activity attenuates α-synucleinopathy in a mouse model. J. Neurosci. 2011, 31, 6963–6971. [Google Scholar] [CrossRef]
- Lindstedt, P.R.; Aprile, F.A.; Matos, M.J.; Perni, M.; Bertoldo, J.B.; Bernardim, B.; Peter, Q.; Jiménez-Osés, G.; Knowles, T.P.J.; Dobson, C.M.; et al. Enhancement of the anti-aggregation activity of a molecular chaperone using a rationally designed post-translational modification. ACS Cent. Sci. 2019, 5, 1417–1424. [Google Scholar] [CrossRef]
- Xue, C.; Lin, T.Y.; Chang, D.; Guo, Z. Thioflavin T as an amyloid dye: Fibril quantification, optimal concentration and effect on aggregation. R. Soc. Open Sci. 2017, 4, 160696. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. BMJ 2009, 339, b2535. [Google Scholar] [CrossRef]
- Fossati, S.; Todd, K.; Sotolongo, K.; Ghiso, J.; Rostagno, A. Differential contribution of isoaspartate post-translational modifications to the fibrillization and toxic properties of amyloid β and the Asn23 Iowa mutation. Biochem. J. 2013, 456, 347–360. [Google Scholar] [CrossRef]
- Shimizu, T.; Fukuda, H.; Murayama, S.; Izumiyama, N.; Shirasawa, T. Isoaspartate formation at position 23 of amyloid beta peptide enhanced fibril formation and deposited onto senile plaques and vascular amyloids in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Lauber, T.; Schaupp, M.; Manhart, S.; Scheel, E.; Böhm, G.; Demuth, H.U. On the Seeding and Oligomerization of pGlu-Amyloid Peptides (in vitro). Biochemistry 2006, 45, 12393–12399. [Google Scholar] [CrossRef]
- Jamasbi, E.; Separovic, F.; Hossain, M.A.; Ciccotosto, G.D. Phosphorylation of a full length amyloid-β peptide modulates its amyloid aggregation, cell binding and neurotoxic properties. Mol. Biosyst. 2017, 13, 1545–1551. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Rezaei-Ghaleh, N.; Terwel, D.; Thal, D.R.; Richard, M.; Hoch, M.; Mc Donald, J.M.; Wüllner, U.; Glebov, K.; Heneka, M.T.; et al. Extracellular phosphorylation of the amyloid β-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer’s disease. EMBO J. 2011, 30, 2255–2265. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Wirths, O.; Stüber, K.; Wunderlich, P.; Koch, P.; Theil, S.; Rezaei-Ghaleh, N.; Zweckstetter, M.; Bayer, T.A.; Brüstle, O.; et al. Phosphorylation of the amyloid β-peptide at Ser26 stabilizes oligomeric assembly and increases neurotoxicity. Acta Neuropathol. 2016, 131, 525–537. [Google Scholar] [CrossRef]
- Khodadadi, S.; Riazi, G.H.; Ahmadian, S.; Hoveizi, E.; Karima, O.; Aryapour, H. Effect of N-homocysteinylation on physicochemical and cytotoxic properties of amyloid β-peptide. FEBS Lett. 2012, 586, 127–131. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, P.; Li, H.; Gao, Z. Nitration of Y10 in Aβ1–40: Is it a compensatory reaction against oxidative/nitrative stress and Aβ aggregation? Chem. Res. Toxicol. 2015, 28, 401–407. [Google Scholar] [CrossRef]
- Guivernau, B.; Bonet, J.; Valls-Comamala, V.; Bosch-Morató, M.; Godoy, J.A.; Inestrosa, N.C.; Perálvarez-Marín, A.; Fernández-Busquets, X.; Andreu, D.; Oliva, B.; et al. Amyloid-β peptide nitrotyrosination stabilizes oligomers and enhances NMDAR-Mediated Toxicity. J. Neurosci. 2016, 36, 11693–11703. [Google Scholar] [CrossRef]
- Kummer Markus, P.; Hermes, M.; Delekarte, A.; Hammerschmidt, T.; Kumar, S.; Terwel, D.; Walter, J.; Pape, H.C.; König, S.; Roeber, S.; et al. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron 2011, 71, 833–844. [Google Scholar] [CrossRef]
- Emendato, A.; Milordini, G.; Zacco, E.; Sicorello, A.; Dal Piaz, F.; Guerrini, R.; Thorogate, R.; Picone, D.; Pastore, A. Glycation affects fibril formation of Aβ peptides. J. Biol. Chem. 2018, 293, 13100–13111. [Google Scholar] [CrossRef]
- Trzeciakiewicz, H.; Tseng, J.H.; Wander, C.M.; Madden, V.; Tripathy, A.; Yuan, C.X.; Cohen, T.J. A dual pathogenic mechanism links tau acetylation to sporadic tauopathy. Sci. Rep. 2017, 7, 44102. [Google Scholar] [CrossRef]
- Haj-Yahya, M.; Lashuel, H.A. Protein semisynthesis provides access to tau disease-associated post-translational modifications (PTMs) and paves the way to deciphering the tau ptm code in health and diseased states. J. Am. Chem. Soc. 2018, 140, 6611–6621. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef]
- Ferreon, J.C.; Jain, A.; Choi, K.J.; Tsoi, P.S.; MacKenzie, K.R.; Jung, S.Y.; Ferreon, A.C. Acetylation disfavors tau phase separation. Int. J. Mol. Sci. 2018, 19, 1360. [Google Scholar] [CrossRef] [PubMed]
- Kamah, A.; Huvent, I.; Cantrelle, F.X.; Qi, H.; Lippens, G.; Landrieu, I.; Smet-Nocca, C. Nuclear magnetic resonance analysis of the acetylation pattern of the neuronal tau protein. Biochemistry 2014, 53, 3020. [Google Scholar] [CrossRef] [PubMed]
- Carlomagno, Y.; Chung, D.E.C.; Yue, M.; Castanedes-Casey, M.; Madden, B.J.; Dunmore, J.; Tong, J.; DeTure, M.; Dickson, D.W.; Petrucelli, L.; et al. An acetylation-phosphorylation switch that regulates tau aggregation propensity and function. J. Biol. Chem. 2017, 292, 15277–15286. [Google Scholar] [CrossRef] [PubMed]
- Guru KrishnaKumar, V.; Baweja, L.; Ralhan, K.; Gupta, S. Carbamylation promotes amyloidogenesis and induces structural changes in Tau-core hexapeptide fibrils. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2590–2604. [Google Scholar] [CrossRef]
- Yin, H.; Kuret, J. C-terminal truncation modulates both nucleation and extension phases of τ fibrillization. FEBS Lett. 2006, 580, 211–215. [Google Scholar] [CrossRef]
- Liu, K.; Liu, Y.; Li, L.; Qin, P.; Iqbal, J.; Deng, Y.; Qing, H. Glycation alter the process of Tau phosphorylation to change Tau isoforms aggregation property. Biochim. Biophys. Acta 2016, 1862, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Mead, E.; Kestoras, D.; Gibson, Y.; Hamilton, L.; Goodson, R.; Jones, S.; Eversden, S.; Davies, P.; O’Neill, M.; Hutton, M.; et al. Halting of caspase activity protects tau from MC1-Conformational change and aggregation. J. Alzheimers Dis. 2016, 54, 1521–1538. [Google Scholar] [CrossRef] [PubMed]
- Necula, M.; Kuret, J. Pseudophosphorylation and glycation of tau protein enhance but do not trigger fibrillization in vitro. J. Biol. Chem. 2004, 279, 49694–49703. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Kim, S.; Schafer, K.N.; Kuret, J. Pseudophosphorylation of tau protein directly modulates its aggregation kinetics. Biochim. Biophys. Acta 2011, 1814, 388–395. [Google Scholar] [CrossRef]
- Luo, H.B.; Xia, Y.Y.; Shu, X.J.; Liu, Z.C.; Feng, Y.; Liu, X.H.; Yu, G.; Yin, G.; Xiong, Y.S.; Zeng, K.; et al. SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc. Natl. Acad. Sci. USA 2014, 111, 16586–16591. [Google Scholar] [CrossRef] [PubMed]
- Yuzwa, S.A.; Cheung, A.H.; Okon, M.; McIntosh, L.P.; Vocadlo, D.J. O-GlcNAc modification of tau directly inhibits its aggregation without perturbing the conformational properties of tau monomers. J. Mol. Biol. 2014, 426, 1736–1752. [Google Scholar] [CrossRef]
- Yoshitake, J.; Soeda, Y.; Ida, T.; Sumioka, A.; Yoshikawa, M.; Matsushita, K.; Akaike, T.; Takashima, A. Modification of Tau by 8-Nitroguanosine 3’,5’-Cyclic Monophosphate (8-Nitro-cGMP): Effects of nitric oxide-linked chemical modification on tau aggregation. J. Biol. Chem. 2016, 291, 22714–22720. [Google Scholar] [CrossRef]
- Funk, K.E.; Thomas, S.N.; Schafer, K.N.; Cooper, G.L.; Liao, Z.; Clark, D.J.; Yang, A.J.; Kuret, J. Lysine methylation is an endogenous post-translational modification of tau protein in human brain and a modulator of aggregation propensity. Biochem. J. 2014, 462, 77–88. [Google Scholar] [CrossRef]
- Reynolds, M.R.; Berry, R.W.; Binder, L.I. Site-Specific nitration differentially influences τ assembly in vitro. Biochemistry 2005, 44, 13997–14009. [Google Scholar] [CrossRef]
- Bartels, T.; Kim, N.C.; Luth, E.S.; Selkoe, D.J. N-alpha-acetylation of α-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS ONE 2014, 9, e103727. [Google Scholar] [CrossRef]
- Kang, L.; Moriarty, G.M.; Woods, L.A.; Ashcroft, A.E.; Radford, S.E.; Baum, J. N-terminal acetylation of α-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. 2012, 21, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Bu, B.; Tong, X.; Li, D.; Hu, Y.; He, W.; Zhao, C.; Hu, R.; Li, X.; Shao, Y.; Liu, C.; et al. N-Terminal Acetylation Preserves α-Synuclein from Oligomerization by Blocking Intermolecular Hydrogen Bonds. ACS Chem. Neurosci. 2017, 8, 2145–2151. [Google Scholar] [CrossRef]
- de Oliveira, R.M.; Vicente, M.H.; Francelle, L.; Pinho, R.; Szegö, É.M.; Martinho, R.; Munari, F.; Lázaro, D.F.; Moniot, S.; Guerreiro, P.; et al. The mechanism of sirtuin 2-mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease. PLoS Biol. 2017, 15, e2000374. [Google Scholar] [CrossRef] [PubMed]
- Birol, M.; Wojcik, S.P.; Miranker, A.D.; Rhoades, E. Identification of N-linked glycans as specific mediators of neuronal uptake of acetylated α-Synuclein. PLoS Biol. 2019, 17, e3000318. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; Dutta, S.; Camara, A.; Chandran, A.; Koller, A.; Watson, B.G.; Sengupta, R.; Ysselstein, D.; Montenegro, P.; Cannon, J.; et al. Alpha-Synuclein is a target of fic-mediated adenylylation/ampylation: Possible implications for parkinson’s disease. J. Mol. Biol. 2019, 431, 2266–2282. [Google Scholar] [CrossRef] [PubMed]
- Vicente, M.H.; Szego, E.M.; Oliveira, L.M.A.; Breda, C.; Darendelioglu, E.; de Oliveira, R.M.; Ferreira, D.G.; Gomes, M.A.; Rott, R.; Oliveira, M.; et al. Glycation potentiates alpha-synuclein-associated neurodegeneration in synucleinopathies. Brain 2017, 140, 1399–1419. [Google Scholar] [CrossRef]
- Krumova, P.; Meulmeester, E.; Garrido, M.; Tirard, M.; Hsiao, H.H.; Bossis, G.; Urlaub, H.; Zweckstetter, M.; Kügler, S.; Melchior, F.; et al. Sumoylation inhibits alpha-synuclein aggregation and toxicity. J. Cell Biol. 2011, 194, 49–60. [Google Scholar] [CrossRef]
- Xiang, W.; Schlachetzki, J.C.M.; Helling, S.; Bussmann, J.C.; Berlinghof, M.; Schäffer, T.E.; Marcus, K.; Winkler, J.; Klucken, J.; Becker, C.M. Oxidative stress-induced posttranslational modifications of alpha-synuclein: Specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol. Cell. Neurosci. 2013, 54, 71–83. [Google Scholar] [CrossRef]
- Xiang, W.; Menges, S.; Schlachetzki, J.C.; Meixner, H.; Hoffmann, A.C.; Schlötzer-Schrehardt, U.; Becker, C.M.; Winkler, J.; Klucken, J. Posttranslational modification and mutation of histidine 50 trigger alpha synuclein aggregation and toxicity. Mol. Neurodegener. 2015, 10, 8. [Google Scholar] [CrossRef]
- Qin, Z.; Hu, D.; Han, S.; Reaney, S.; Di Monte, D.; Fink, A. Effect of 4-hydroxy-2-neonal modification on alpha-synuclein aggregation. J. Biol. Chem. 2007, 282, 5862–5870. [Google Scholar] [CrossRef]
- Levine, P.M.; Galesic, A.; Balana, A.T.; Mahul-Mellier, A.L.; Navarro, M.X.; De Leon, C.A.; Lashuel, H.A.; Pratt, M.R. α-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. PNAS USA 2019, 116, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lei, H.; Chen, Y.; Ma, Y.T.; Jiang, F.; Tan, J.; Zhang, Y.; Li, J.D. Enzymatic O-GlcNAcylation of alpha-synuclein reduces aggregation and increases SDS-resistant soluble oligomers. Neurosci. Lett. 2017, 655, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Marotta, N.P.; Lin, Y.H.; Lewis, Y.E.; Ambroso, M.R.; Zaro, B.W.; Roth, M.T.; Arnold, D.B.; Langen, R.; Pratt, M.R. O-GlcNAc modification blocks the aggregation and toxicity of the protein α-synuclein associated with Parkinson’s disease. Nat. Chem. 2015, 7, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Samuel, F.; Flavin, W.P.; Iqbal, S.; Pacelli, C.; Sri Renganathan, S.D.; Trudeau, L.E.; Campbell, E.M.; Fraser, P.E.; Tandon, A. Effects of Serine 129 Phosphorylation on α-Synuclein Aggregation, Membrane Association, and Internalization. J. Biol. Chem. 2016, 291, 4374–4385. [Google Scholar] [CrossRef]
- Burai, R.; Ait-Bouziad, N.; Chiki, A.; Lashuel, H.A. Elucidating the role of site-specific nitration of α-synuclein in the pathogenesis of parkinson’s disease via protein semisynthesis and mutagenesis. J. Am. Chem. Soc. 2015, 137, 5041–5052. [Google Scholar] [CrossRef]
- Liu, Y.; Qiang, M.; Wei, Y.; He, R. A novel molecular mechanism for nitrated {alpha}-synuclein-induced cell death. J. Mol. Cell. Biol. 2011, 3, 239–249. [Google Scholar] [CrossRef]
- Hodara, R.; Norris, E.H.; Giasson, B.I.; Mishizen-Eberz, A.J.; Lynch, D.R.; Lee, V.M.; Ischiropoulos, H. Functional consequences of alpha-synuclein tyrosine nitration: Diminished binding to lipid vesicles and increased fibril formation. J. Biol. Chem. 2004, 279, 47746–47753. [Google Scholar] [CrossRef]
- Souza, J.M.; Giasson, B.I.; Chen, Q.; Lee, V.M.; Ischiropoulos, H. Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 2000, 275, 18344–18349. [Google Scholar] [CrossRef]
- Wang, P.; Wander, CM.; Yuan, C.X.; Bereman, M.S.; Cohen, T.J. Acetylation-induced TDP-43 pathology is suppressed by an HSF1-dependent chaperone program. Nat. Commun. 2017, 8, 82. [Google Scholar] [CrossRef]
- Cohen, T.J.; Hwang, A.W.; Restrepo, C.R.; Yuan, C.X.; Trojanowski, J.Q.; Lee, V.M.Y. An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 2015, 6, 5845. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Xu, Y.F.; Cook, C.; Gendron, T.F.; Roettges, P.; Link, C.D.; Lin, W.L.; Tong, J.; Castanedes-Casey, M.; Ash, P.; et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 7607–7612. [Google Scholar] [CrossRef] [PubMed]
- Carlomagno, Y.; Zhang, Y.; Davis, M.; Lin, W.L.; Cook, C.; Dunmore, J.; Tay, W.; Menkosky, K.; Cao, X.; Petrucelli, L.; et al. Casein kinase II induced polymerization of soluble TDP-43 into filaments is inhibited by heat shock proteins. PLoS ONE 2014, 9, e90452. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Arai, T.; Nonaka, T.; Kametani, F.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.; Morita, M.; et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 2008, 64, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Brady, O.A.; Meng, P.; Zheng, Y.; Mao, Y.; Hu, F. Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J. Neurochem. 2011, 116, 248–259. [Google Scholar] [CrossRef]
- Li, H.Y.; Yeh, P.A.; Chiu, H.C.; Tang, C.Y.; Tu, B.P. Hyperphosphorylation as a defense mechanism to reduce TDP-43 aggregation. PLoS ONE 2011, 6, e23075. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Sivalingam, V.; Bharathi, V.; Girdhar, A.; Patel, B.K. The amyloidogenicity of a C-terminal region of TDP-43 implicated in Amyotrophic Lateral Sclerosis can be affected by anions, acetylation and homodimerization. Biochimie 2018, 150, 76–87. [Google Scholar] [CrossRef]
- Yang, C.; Tan, W.; Whittle, C.; Qui, L.; Cao, L.; Akbarian, S.; Xu, Z. The C-terminal TDP-43 fragments have a high aggregation propensity and harm neurons by a dominant-negative mechanism. PLoS ONE 2011, 5, e15878. [Google Scholar] [CrossRef]
- Rasouli, S.; Abdolvahabi, A.; Croom, C.M.; Plewman, D.L.; Shi, Y.; Ayers, J.I.; Shaw, B.F. Lysine acylation in superoxide dismutase-1 electrostatically inhibits formation of fibrils with prion-like seeding. J. Biol. Chem. 2017, 292, 19366–19380. [Google Scholar] [CrossRef]
- Niikura, T.; Kita, Y.; Abe, Y. SUMO3 modification accelerates the aggregation of ALS-linked SOD1 mutants. PLoS ONE 2014, 9, e101080. [Google Scholar] [CrossRef]
- Fei, E.; Jia, N.; Yan, M.; Ying, Z.; Sun, Q.; Wang, H.; Zhang, T.; Ma, X.; Ding, H.; Yao, X.; et al. SUMO-1 modification increases human SOD1 stability and aggregation. Biochem. Biophys. Res. Commun. 2006, 347, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Chaibva, M.; Jawahery, S.; Pilkington Albert, W.; Arndt James, R.; Sarver, O.; Valentine, S.; Matysiak, S.; Legleiter, J. Acetylation within the first 17 residues of huntingtin exon 1 alters aggregation and lipid binding. Biophys. J. 2016, 111, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Chiki, A.; DeGuire, S.M.; Ruggeri, F.S.; Sanfelice, D.; Ansaloni, A.; Wang, Z.M.; Cendrowska, U.; Burai, R.; Vieweg, S.; Pastore, A.; et al. Mutant exon1 huntingtin aggregation is regulated by T3 phosphorylation-induced structural changes and crosstalk between T3 phosphorylation and acetylation at K6. Angew. Chem. Int. Ed. Engl. 2017, 56, 5202–5207. [Google Scholar] [CrossRef] [PubMed]
- Ansaloni, A.; Wang, Z.M.; Jeong, J.S.; Ruggeri, F.S.; Dietler, G.; Lashuel, H.A. One-Pot semisynthesis of Exon 1 of the huntingtin protein: New tools for elucidating the role of posttranslational modifications in the pathogenesis of huntington’s disease. Angew. Chem. Int. Ed. Engl. 2014, 53, 1928–1933. [Google Scholar] [CrossRef]
- Cariulo, C.; Azzollini, L.; Verani, M.; Martufi, P.; Boggio, R.; Chiki, A.; Deguire, S.M.; Cherubini, M.; Gines, S.; Marsh, J.L.; et al. Phosphorylation of huntingtin at residue T3 is decreased in Huntington’s disease and modulates mutant huntingtin protein conformation. Proc. Natl. Acad. Sci. USA 2017, 114, E10809. [Google Scholar] [CrossRef]
- DeGuire, S.M.; Ruggeri, F.S.; Fares, M.B.; Chiki, A.; Cendrowska, U.; Dietler, G.; Lashuel, H.A. N-terminal Huntingtin (Htt) phosphorylation is a molecular switch regulating Htt aggregation, helical conformation, internalization, and nuclear targeting. J. Biol. Chem. 2018, 293, 18540–18558. [Google Scholar] [CrossRef]
- Gu, X.; Greiner, E.R.; Mishra, R.; Kodali, R.; Osmand, A.; Finkbeiner, S.; Steffan, J.S.; Thompson, L.M.; Wetzel, R.; Yang, X.W. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron 2009, 64, 828–840. [Google Scholar] [CrossRef]
- Lunkes, A.; Lindenberg, K.S.; Ben-Haïem, L.; Weber, C.; Devys, D.; Landwehrmeyer, G.B.; Mandel, J.L.; Trottier, Y. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell 2002, 10, 259–269. [Google Scholar] [CrossRef]
- Raspe, M.; Gillis, J.; Krol, H.; Krom, S.; Bosch, K.; van Veen, H.; Reits, E. Mimicking proteasomal release of polyglutamine peptides initiates aggregation and toxicity. J. Cell Sci. 2009, 122, 3262–3271. [Google Scholar] [CrossRef]
- Ryu, J.; Cho, S.; Park, B.C.; Lee, D.H. Oxidative stress-enhanced SUMOylation and aggregation of ataxin-1: Implication of JNK pathway. Biochem. Biophys. Res. Commun. 2010, 393, 280–285. [Google Scholar] [CrossRef]
- Haacke, A.; Broadley, S.A.; Boteva, R.; Tzvetkov, N.; Hartl, F.U.; Breuer, P. Proteolytic cleavage of polyglutamine-expanded ataxin-3 is critical for aggregation and sequestration of non-expanded ataxin-3. Hum. Mol. Genet. 2006, 15, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Dear, D.V.; Young, D.S.; Kazlauskaite, J.; Meersman, F.; Oxley, D.; Webster, J.; Pinheiro, T.J.; Gill, A.C.; Bronstein, I.; Lowe, C.R. Effects of post-translational modifications on prion protein aggregation and the propagation of scrapie-like characteristics in vitro. Biochim. Biophys. Acta 2007, 1774, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, P.N.; Robertson, C.; Jodoin, J.; Paudel, H.; Booth, S.A.; LeBlanc, A.C. Phosphorylation of prion protein at serine 43 induces prion protein conformational change. J. Neurosci. 2009, 29, 8743–8751. [Google Scholar] [CrossRef] [PubMed]
- Moro, M.L.; Phillips, A.S.; Gaimster, K.; Paul, C.; Mudher, A.; Nicoll, J.A.R.; Boche, D. Pyroglutamate and Isoaspartate modified Amyloid-Beta in ageing and Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Vigneswara, V.; Cass, S.; Wayne, D.; Bolt, E.L.; Ray, D.E.; Carter, W.G. Molecular ageing of alpha- and Beta-synucleins: Protein damage and repair mechanisms. PLoS ONE 2013, 8, E61442. [Google Scholar] [CrossRef] [PubMed]
- Vigneswara, V.; Lowenson, J.D.; Powell, C.D.; Thakur, M.; Bailey, K.; Clarke, S.; Ray, D.E.; Carter, W.G. Proteomic identification of novel substrates of a protein isoaspartyl methyltransferase repair enzyme. J. Biol. Chem. 2006, 281, 32619–32629. [Google Scholar] [CrossRef]
- Neddens, J.; Temmel, M.; Flunkert, S.; Kerschbaumer, B.; Hoeller, C.; Loeffler, T.; Niederkofler, V.; Daum, G.; Attems, J.; Hutter-Paier, B. Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 52. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Helwig, M.; Klinkenberg, M.; Rusconi, R.; Musgrove, R.E.; Majbour, N.K.; El-Agnaf, O.M.; Ulusoy, A.; Di Monte, D.A. Brain propagation of transduced alpha-synuclein involves non-fibrillar protein species and is enhanced in alpha-synuclein null mice. Brain 2016, 139, 856–870. [Google Scholar] [CrossRef]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef]
- Saito, Y.; Kawashima, A.; Ruberu, N.N.; Fujiwara, H.; Koyama, S.; Sawabe, M.; Arai, T.; Nagura, H.; Yamanouchi, H.; Hasegawa, M.; et al. Accumulation of phosphorylated α-synuclein in aging human brain. J. Neuropathol. Exp. Neurol. 2003, 62, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Dangoumau, A.; Marouillat, S.; Burlaud Gaillard, J.; Uzbekov, R.; Veyrat-Durebex, C.; Blasco, H.; Arnoult, C.; Corcia, P.; Andres, C.R.; Vourc’h, P. Inhibition of Pathogenic mutant SOD1 aggregation in cultured motor neuronal cells by prevention of its sumoylation on lysine 75. Neurodegener. Dis. 2016, 16, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Bowie, L.E.; Maiuri, T.; Alpaugh, M.; Gabriel, M.; Arbez, N.; Galleguillos, D.; Hung, C.L.K.; Patel, S.; Xia, J.; Hertz, N.T.; et al. N6-Furfuryladenine is protective in Huntington’s disease models by signaling huntingtin phosphorylation. Proc. Natl. Acad. Sci. USA 2018, 115, E7081–E7090. [Google Scholar] [CrossRef] [PubMed]
- Herrera, F.; Branco-Santos, J.; Outeiro, T. Threonine 3 regulates Serine 13/16 phosphorylation in the huntingtin exon 1. Matters Sel. 2019. [Google Scholar] [CrossRef]
- Verma, M.; Vats, A.; Taneja, V. Toxic species in amyloid disorders: Oligomers or mature fibrils. Ann. Indian Acad. Neurol. 2015, 18, 138–145. [Google Scholar] [PubMed]
- Celej María, S.; Sarroukh, R.; Goormaghtigh, E.; Fidelio Gerardo, D.; Ruysschaert, J.M.; Raussens, V. Toxic prefibrillar α-synuclein amyloid oligomers adopt a distinctive antiparallel β-sheet structure. Biochem. J. 2012, 443, 719–726. [Google Scholar] [CrossRef]
- Cerf, E.; Sarroukh, R.; Tamamizu-Kato, S.; Breydo, L.; Derclaye, S.; Dufrene, Y.F.; Narayanaswami, V.; Goormaghtigh, E.; Ruysschaert, J.M.; Raussens, V. Antiparallel beta-sheet: A signature structure of the oligomeric amyloid beta-peptide. Biochem. J. 2009, 421, 415–423. [Google Scholar] [CrossRef]
- Simmons, L.K.; May, P.C.; Tomaselli, K.J.; Rydel, R.E.; Fuson, K.S.; Brigham, E.F.; Wright, S.; Lieberburg, I.; Becker, G.W.; Brems, D.N.; et al. Secondary structure of amyloid beta peptide correlates with neurotoxic activity in vitro. Mol. Pharmacol. 1994, 45, 373–379. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NDD | Commonly Mutated Proteins | Primary Region of Damage | Compartment of Aggregate Deposition | Aggregate-Forming Proteins | Global Prevalence | Sporadic Cases | Familial Cases |
|---|---|---|---|---|---|---|---|
| AD | APP, presenilins | Cortex, hippocampus | Extracellular, intracytoplasmic | Aβ (plaques), tau (tangles) | 593:100,000 | >98% | <2% |
| PD | α-synuclein, LRRK2 | Substantia nigra, cortex | Intracytoplasmic | α-synuclein (Lewy bodies) | 1–2:1000 | >90% | <10% |
| HD | Htt | Striatum, basal ganglia | Intranuclear, intracytoplasmic | Htt | 1:10,000 | 3% | 97% |
| ALS | TDP-43, SOD1, c9orf72 | Spinal motor neurons, motor cortex | Intracytoplasmic | SOD1, TDP-43 | 5:100,000 | 90–95% | 5–10% |
| MS | - | Basal ganglia, brainstem | Intracytoplasmic, extracellular | Aβ, tau, APP, bassoon protein | 30.1:100,000 | 80–90% | 10–20% |
| SCAs | ATX1, ATX2, ATX3, CACNA1A, ATX7, TBP, ATN1 | Cerebellum, brainstem | Intranuclear | Atrophin-1, ataxins | 3:100,000 | No data | No data |
| TSEs | PrP | Cortex, brainstem, thalamus, cerebellum | Extracellular | PrP | 1–2:1,000,000 | 85–90% | 10–15% |
| Post-Translational Modification | Residues Modified | Author & Year | Aggregation | Formation of Oligomers | Formation of Fibrillar Aggregates | Formation of Amorphous Aggregates |
|---|---|---|---|---|---|---|
| Glycation | R5, K16 | Emendato et al., 2018 [105] | Decrease | - | Decrease | - |
| Isoaspartate formation | D23 | Shimizu et al., 2002 [96] | Increase | - | Increase | - |
| D1, D7, D23 | Fossati et al., 2013 [95] | Increase | Increase | Increase | - | |
| N-Homocysteinylation | K16, K28 | Khodadadi et al., 2012 [101] | Decrease | Increase | Decrease | - |
| N-terminal pyroglutamylation | N-terminal E3 | Schilling et al., 2006 [97] | Increase | Increase | Increase | - |
| Nitration | Y10 | Kummer et al., 2011 [104] | Increase | Increase | Increase | - |
| Y10 | Zhao et al., 2015 [102] | Decrease | Decrease | Decrease | - | |
| Y10 | Guivernau et al., 2016 [103] | Decrease | Increase | Decrease | - | |
| Phosphorylation | S8 | Jamasbi et al., 2017 [98] | Increase | - | Increase | - |
| S8 | Kumar et al., 2011 [99] | Increase | Increase | Increase | - | |
| S26 | Kumar et al., 2016 [100] | Decrease | Increase | Decrease | - |
| Post-Translational Modification | Isoform and Residues Modified | Author & Year | Aggregation | Formation of Oligomers | Formation of Fibrillar Aggregates | Formation of Amorphous Aggregates |
|---|---|---|---|---|---|---|
| Acetylation | 4R2N: K280/K281 | Trzeciakiewicz et al., 2017 [106] | Increase | - | Increase | - |
| 4R2N: K163/K174/K190/K224/K234/K240/K254/K280/K281/K290/K311/K375/K385/K395 | Ferreon et al., 2018 [109] | Decrease | - | Decrease | - | |
| 4R0N: K321, K259/K290/K321/K353, K290/K321, K274 | Carlomagno et al., 2017 [111] | Decrease | - | Decrease | - | |
| 4R2N: K163, K174, K224, K225, K234, K240, K259, K274, K280, K290/K321, K294, K298, K317, K353, K369 | Kamah et al., 2014 [110] | Decrease | - | Decrease | - | |
| 4R2N: K280 | Haj-Yahya and Lashuel, 2018 [107] | Increase | Increase | Decrease | - | |
| 4R, Tau-K18: K163/K280/K281/K369 | Cohen et al., 2011 [108] | Increase | - | Increase | - | |
| Carbamylation | 4R2N: K311, K280, K311/K280 | KrishnaKumar et al., 2018 [112] | Increase | - | Increase | - |
| C-terminal Truncation | 4R2N: D421, E391 | Yin and Kuret, 2006 [113] | Increase | - | Increase | - |
| Glycation | 4R2N: K67, K148, K163, K180, K190, K259, K267, K274, K281, K290, K298, K311, K317, K321, K331, K340, K343, K353, K369, K370, K375, K383, K385, K395 | Liu et al., 2016 [114] | Increase | - | Increase | - |
| 3R2N: K24, K163, K174, K180, K190, K254, K259, K267, K311, K343, K353, K369, K385 | Liu et al., 2016 [114] | Increase | - | Increase | - | |
| Methylation | 4R2N: Multiple residues* | Funk et al., 2014 [121] | Decrease | - | Decrease | - |
| Nitration | 4R2N: Y18, Y394 | Reynolds et al., 2005 [122] | Decrease | - | Decrease | - |
| O-GlcNAcylation | 4R2N: S400 | Yuzwa et al., 2014 [119] | Decrease | - | Decrease | - |
| Phosphorylation | 4R2N: S68, T169, S214, S262, S285, S319, S356, T403 | Liu et al., 2016 [114] | Increase | - | Increase | - |
| 3R1N: T71 | Liu et al., 2016 [114] | Increase | - | Increase | - | |
| 4R0N: T111, S198, S214, S237, S238, S241, S258, S324, S352, S356, S400, S404 | Liu et al., 2016 [114] | Decrease | - | Decrease | - | |
| 3R2N: S235, S237, S324 | Liu et al., 2016 [114] | Decrease | - | Decrease | - | |
| Proteolytic cleavage | 4R2N: D421 | Mead et al., 2016 [115] | Increase | - | - | Increase |
| Pseudo-phosphorylation | 4R2N: S199, S199/S202/T205, T212, S214, T212/S214, S396/S404, | Necula and Kuret, 2004 [116] | Increase | - | Increase | - |
| 4R2N: S235 | Necula and Kuret, 2004 [116] | Decrease | - | Decrease | - | |
| 4R2N: T212 | Chang et al., 2011 [117] | Increase | - | Increase | - | |
| S-Guanylation | 3R2N: C291 | Yoshitake et al., 2016 [120] | Decrease | Decrease | Decrease | - |
| 4R2N: C291, C322 | Yoshitake et al., 2016 [120] | Decrease | Decrease | Decrease | - | |
| SUMOylation | 4R2N: K340 | Luo et al., 2014 [118] | Increase | - | - | - |
| Post-Translational Modification | Residues Modified | Author & Year | Aggregation | Formation of Oligomers | Formation of Fibrillar Aggregates | Formation of Amorphous Aggregates |
|---|---|---|---|---|---|---|
| Acetylation | N-terminus | Bartels et al., 2014 [123] | Decrease | - | Decrease | - |
| N-terminus | Kang et al., 2012 [124] | Decrease | - | Decrease | - | |
| N- terminus | Bu et al., 2017 [125] | Decrease | Decrease | Decrease | - | |
| N-terminus | Birol et al., 2019 [127] | Increase | - | Increase | - | |
| N-terminus, K6, K10 | Oliveira et al., 2017 [126] | Decrease | Decrease | Decrease | - | |
| Adenylylation | T33, T54, T75 | Sanyal et al., 2019 [128] | Decrease | - | Decrease | - |
| Glycation | K6, K10, K12, K21, K23, K32, K34, K43, K45 | Vicente et al., 2017 [129] | Increase | Increase | Decrease | Increase |
| 4-Hydroxy-2-neonalModification | H50, and other Lys residues | Qin et al., 2006 [133] | Decrease | Increase | Decrease | - |
| H50, and other Lys residues | Xiang et al., 2013 [131] | Decrease | Increase | Decrease | - | |
| H50 | Xiang et al., 2015 [132] | - | Increase | - | - | |
| Nitration | Y39, Y125, Y133/Y136 | Burai et al., 2015 [139] | Decrease | Increase | Decrease | Increase |
| Y39, Y125, Y133, Y136 | Liu et al., 2011 [140] | - | Increase | - | Increase | |
| Y39, Y125, Y133, Y136 | Hodara et al., 2004 [141] | Increase | - | - | - | |
| Y39, Y125, Y133, Y136 | Souza et al., 2000 [142] | Increase | Increase | - | - | |
| Y39/Y125/Y133/136 | Xiang et al., 2013 [131] | Decrease | Increase | Decrease | - | |
| O-GlcNAcylation | T72 | Levine et al., 2019 [134] | Decrease | - | Decrease | - |
| T75 | Levine et al., 2019 [134] | Decrease | - | Decrease | - | |
| T81 | Levine et al., 2019 [134] | Decrease | Increase | Decrease | - | |
| S87 | Levine et al., 2019 [134] | Decrease | Increase | Decrease | - | |
| T72/T75/T81 | Levine et al., 2019 [134] | Decrease | - | Decrease | - | |
| Muliple sites * | Zhang et al., 2017 [135] | Decrease | Increase | Decrease | - | |
| T72 | Marotta et al., 2015 [136] | Decrease | Decrease | Decrease | - | |
| Phosphorylation | S129 | Fujiwara et al., 2002 [137] | Increase | Increase | Increase | - |
| S129 | Samuel et al., 2016 [138] | Increase | - | Increase | - | |
| SUMOylation | K96, K102 | Krumova et al., 2011 [130] | Decrease | Increase | Decrease | - |
| Post-Translational Modification | Residues Modified | Author & Year | Aggregation | Formation of Oligomers | Formation of Fibrillar Aggregates | Formation of Amorphous Aggregates |
|---|---|---|---|---|---|---|
| Acetylation | K145 | Wang et al., 2017 [143] | Increase | - | - | - |
| K145, K192 | Cohen et al., 2015 [144] | Increase | - | - | - | |
| C-terminal fragmentation | D89, D219 | Zhang et al., 2009 [145] | Increase | - | - | - |
| Phosphorylation | S379, S403, S404, S409, S410, S403/S404, S409/S410, S379/S403/S404, S379/S409/S410, S403/S404/S409/S410 | Li et al., 2011 [149] | Decrease | - | - | - |
| S409/S410 | Carlomagno et al., 2014 [146] | Increase | - | Increase | - | |
| S379, S403/404, S409, S410, S409/S410 | Hasegawa et al., 2008 [147] | Increase | Increase | Increase | - | |
| S409/S410 | Brady et al., 2011 [148] | Decrease | - | - | - |
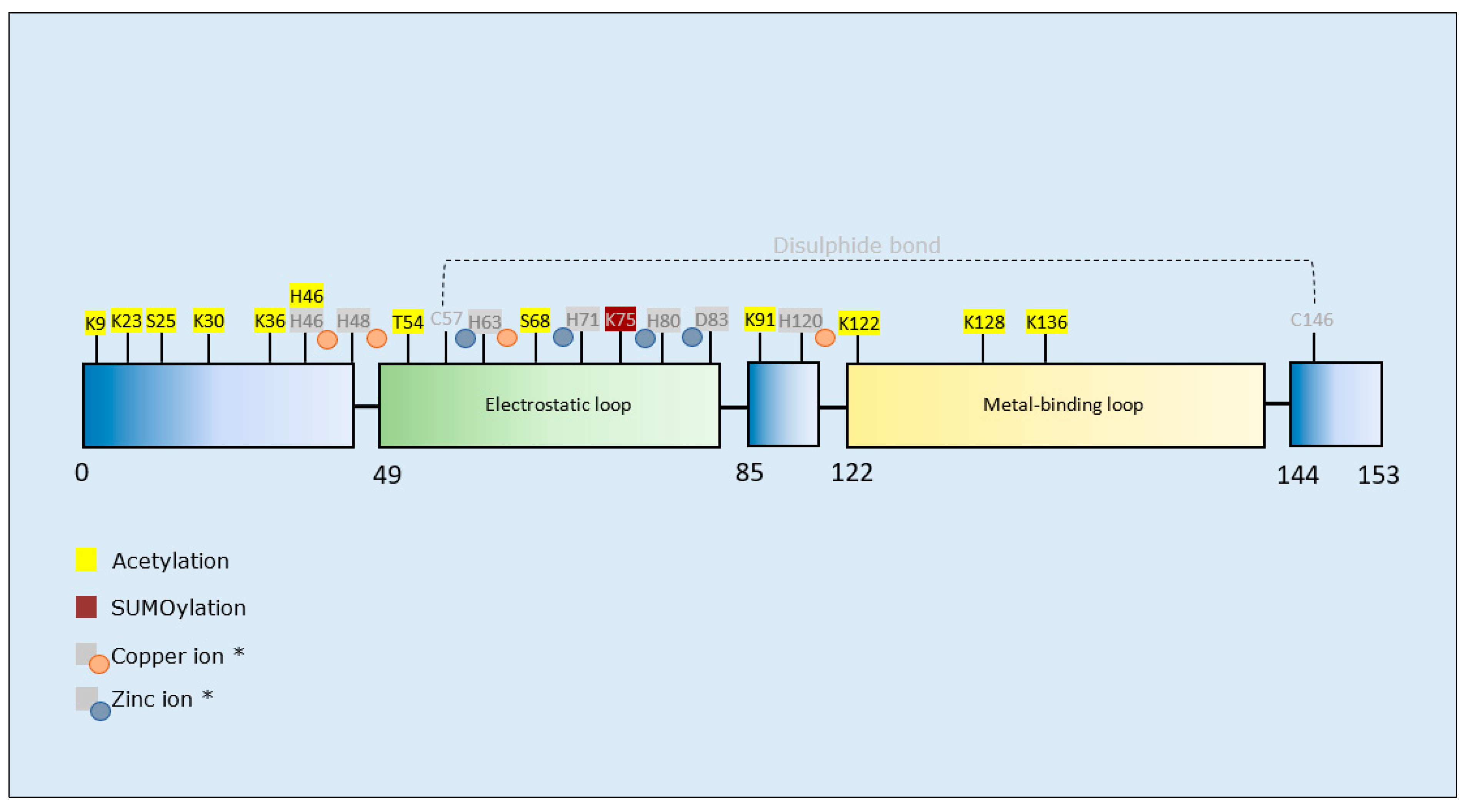
| Post-Translational Modification | Residues Modified | Author & Year | Aggregation | Formation of Oligomers | Formation of Fibrillar Aggregates | Formation of Amorphous Aggregates |
|---|---|---|---|---|---|---|
| Acetylation | K23/K30/K36/H46/K91/K122/K128/K136 (BP *) | Rasouli et al., 2017 [152] | - | - | Decrease | - |
| K36/K128 (BT *) | Rasouli et al., 2017 [152] | - | - | Decrease | Increase | |
| K23/S25/K30/K36/K91/K122/K128/K136 (PM *) | Rasouli et al., 2017 [152] | - | - | Decrease | Increase | |
| K9 (CA*) | Rasouli et al., 2017 [152] | - | - | Increase | - | |
| K9/K23/K30/K36/K91/K122/K136/T54/S68 (GA *) | Rasouli et al., 2017 [152] | - | - | Decrease | Increase | |
| K23/S25/K30/K36/K122/K128/K136 (SA *) | Rasouli et al., 2017 [152] | - | - | Increase | - | |
| SUMOylation | K75 (SUMO3) † | Niikura et al., 2014 [153] | Increase | - | - | - |
| K75 (SUMO1) ‡ | Fei et al., 2006 [154] | Increase | - | - | - |
| Post-Translational Modification | Residues Modified | Author & Year | Aggregation | Formation of Oligomers | Formation of Fibrillar Aggregates | Formation of Amorphous Aggregates |
|---|---|---|---|---|---|---|
| Acetylation | K6, K9, K15 ‡ | Chaibva et al., 2016 [155] | Decrease | Decrease | - | |
| Phosphorylation | T3 * | Chiki et al., 2017 [156] | Decrease | - | Decrease | - |
| T3 | Ansaloni et al., 2014 [157] | Decrease | Decrease | Decrease | - | |
| T3 * | Cariulo et al., 2017 [158] | Decrease | - | - | - | |
| S13, S16, S13/S16 † | DeGuire et al., 2018 [159] | Decrease | Increase | Decrease | - | |
| Pseudo-phosphorylation | S13, S16 † | DeGuire et al., 2018 [159] | Decrease | Increase | Decrease | - |
| S13/S16 * | Gu et al., 2009 [160] | Decrease | - | Decrease | - | |
| Proteolytic Cleavage | Cleavage site between residue 104 and 114 * | Lunkes et al., 2002 [161] | Increase | - | - | - |
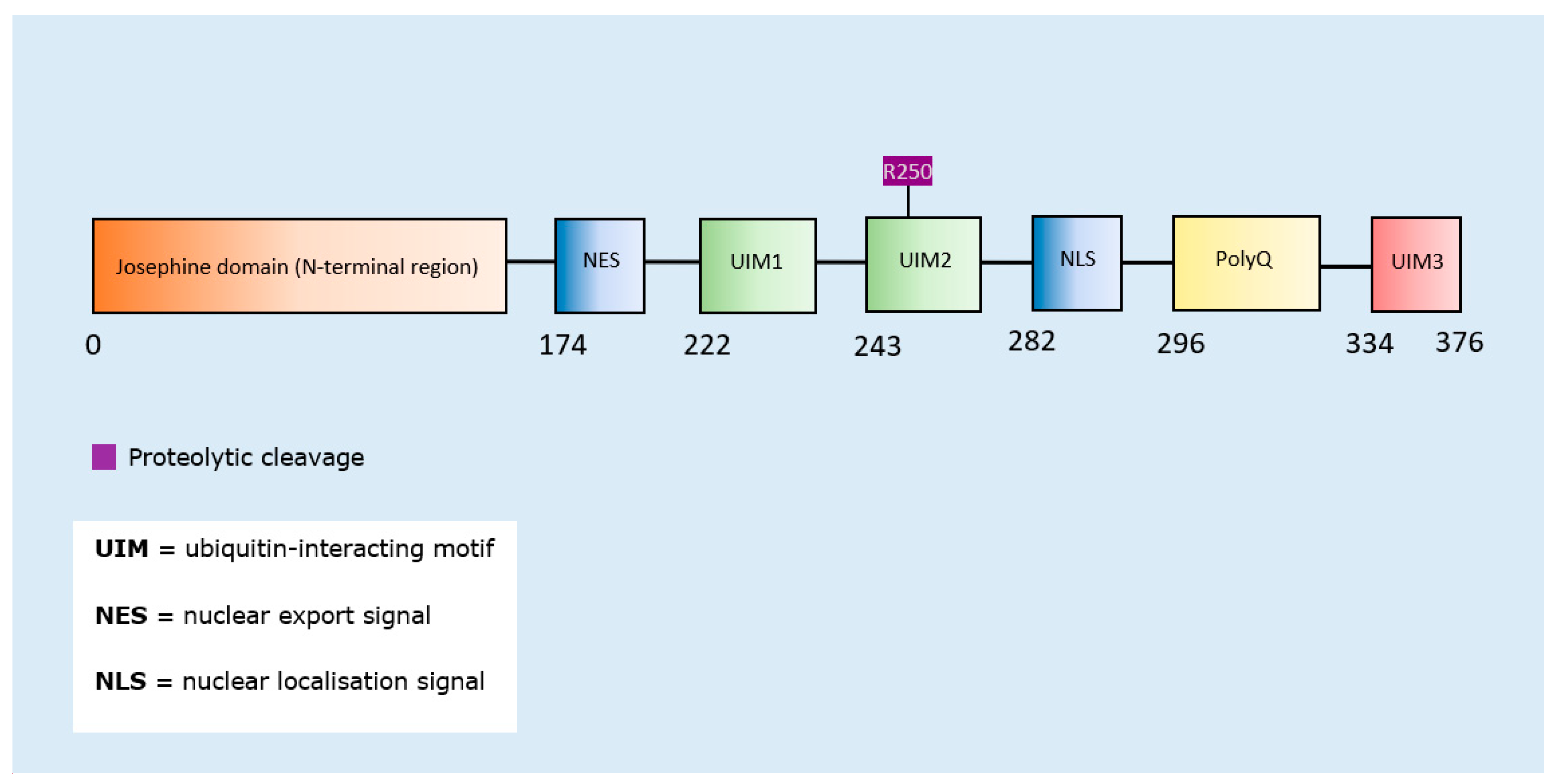
| Post-Translational Modification | Residues Modified | Author & Year | Aggregation | Formation of Oligomers | Formation of Fibrillar Aggregates | Formation of Amorphous Aggregates |
|---|---|---|---|---|---|---|
| SUMOylation (ataxin-1) | Multiple residues | Ryu et al., 2010 [163] | Increase | - | - | - |
| Proteolytic Cleavage (ataxin-3) | N-terminal region cleavage near residue 250 | Haacke et al., 2006 [164] | Increase | - | - | - |
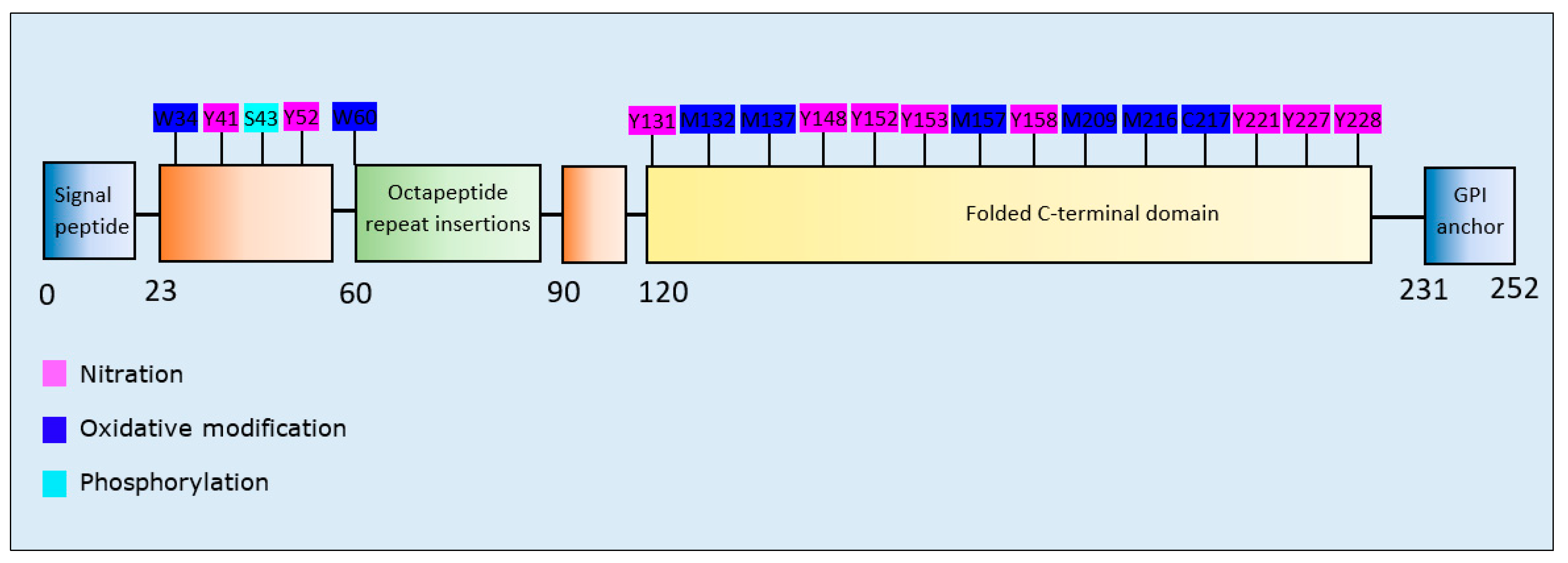
| Post-Translational Modification | Residues Modified | Author & Year | Aggregation | Formation of Oligomers | Formation of Fibrillar Aggregates | Formation of Amorphous Aggregates |
|---|---|---|---|---|---|---|
| Oxidative modification and Nitration | Oxidation: W34, W60, M157, M209, M216, C217, M132/M137 | Dear et al., 2007 [165] | Increase | - | Increase | Increase |
| Nitration: Y41, Y41/Y52, Y131, Y148, Y152, Y153, Y158, Y221, Y227/Y228 | Increase | - | Increase | Increase | ||
| Phosphorylation | S43 | Giannopoulos et al., 2009 [166] | Increase | - | Increase | Decrease |
| Protein | PTM | Residues | Schematic Representation of Modification | Suggested Pharmacological Intervention |
|---|---|---|---|---|
| Aβ | Isoaspartate modification | D1, D7, D23 |  | Inhibition |
| Aβ | Phosphorylation | S8 |  | Inhibition |
| Aβ | Phosphorylation | S26 |  | Enhancement |
| αS | Acetylation | K6, K10, N-terminal region |  | Enhancement |
| αS | HNE modification | H50 |  | Inhibition |
| αS | O-GlcNacylation | T72, T75, T81, S87 |  | Enhancement |
| αS | Phosphorylation | S129 |  | Inhibition |
| TDP-43 | Acetylation | K145, K192 |  | Inhibition |
| SOD1 | SUMOylation | K75 |  | Inhibition |
| HTT | Phosphorylation | T3, S13, S16 |  | Enhancement |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaffert, L.-N.; Carter, W.G. Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sci. 2020, 10, 232. https://doi.org/10.3390/brainsci10040232
Schaffert L-N, Carter WG. Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sciences. 2020; 10(4):232. https://doi.org/10.3390/brainsci10040232
Chicago/Turabian StyleSchaffert, Larissa-Nele, and Wayne G. Carter. 2020. "Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review" Brain Sciences 10, no. 4: 232. https://doi.org/10.3390/brainsci10040232
APA StyleSchaffert, L.-N., & Carter, W. G. (2020). Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sciences, 10(4), 232. https://doi.org/10.3390/brainsci10040232