Downregulation of Astrocytic Kir4.1 Potassium Channels Is Associated with Hippocampal Neuronal Hyperexcitability in Type 2 Diabetic Mice

 ,
,  and
and 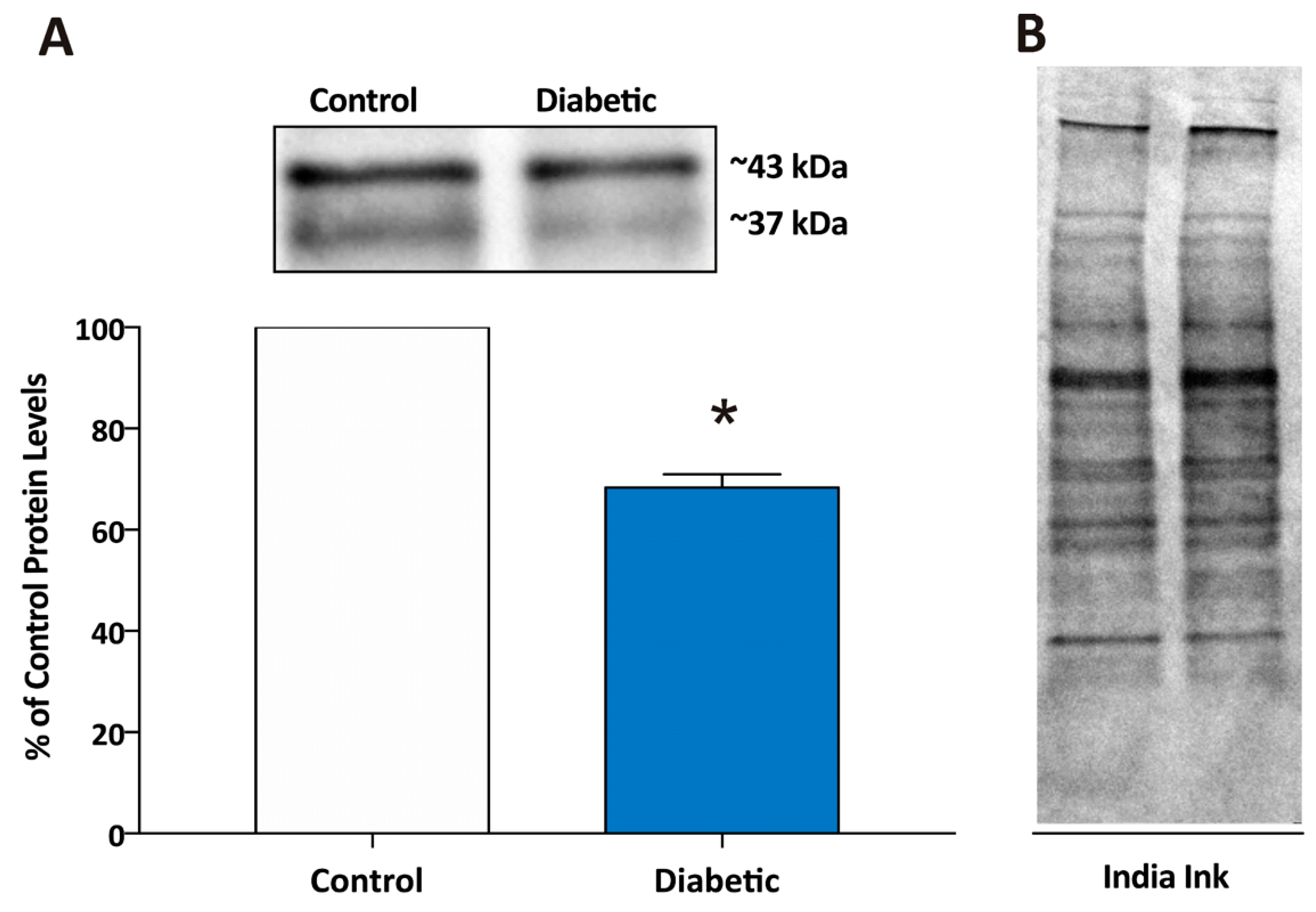{kind=link}
{kind=link}
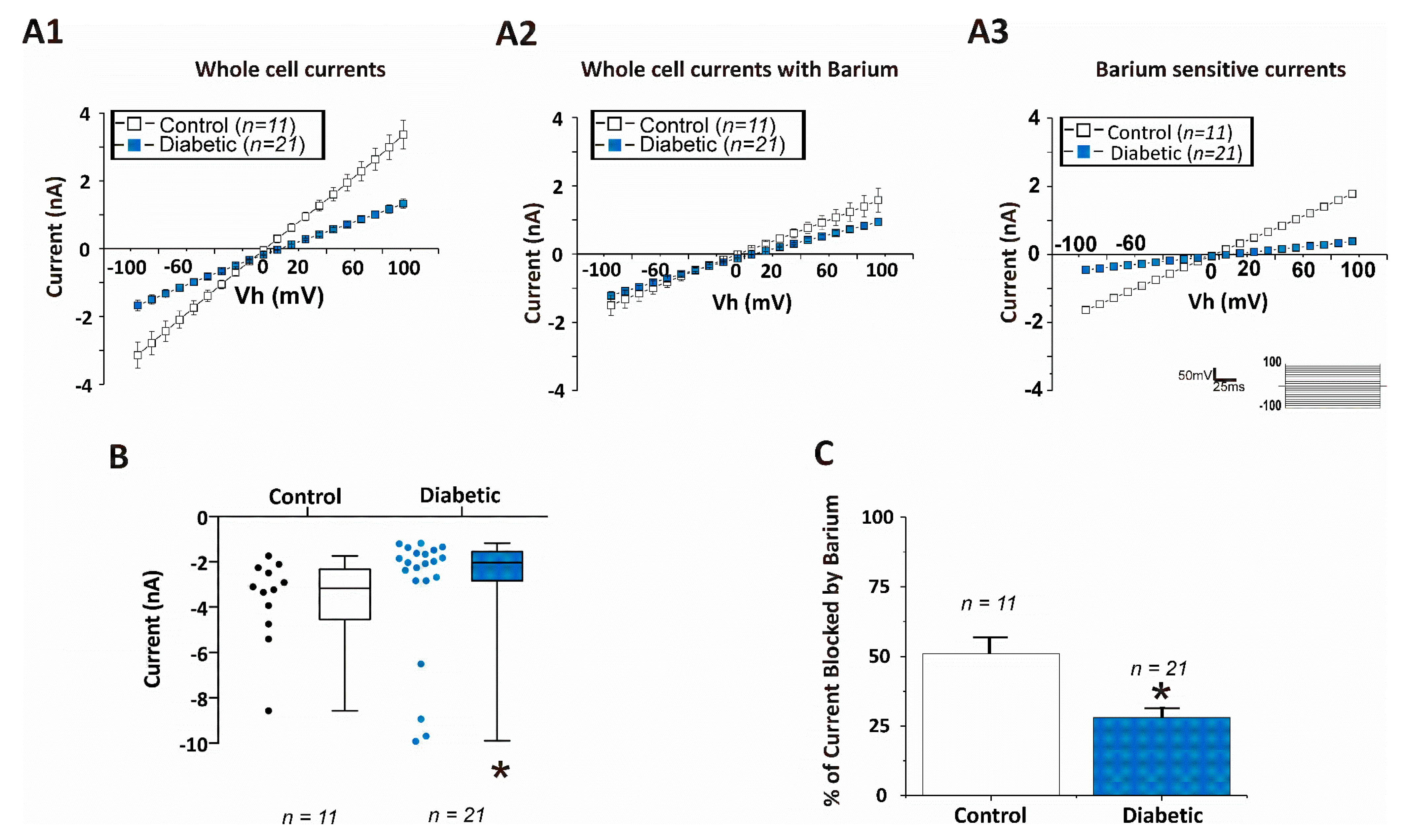{kind=link}
{kind=link}
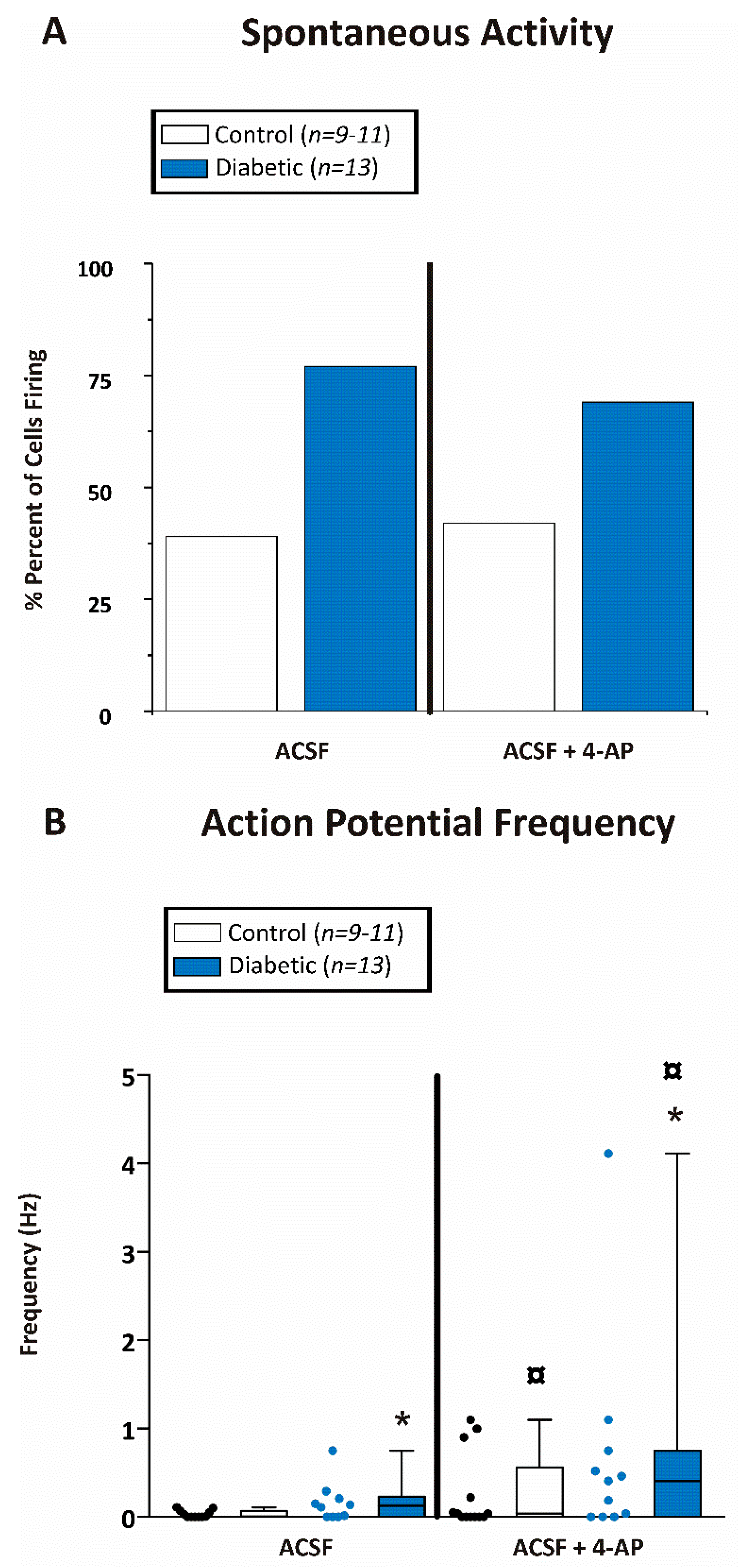{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Blood Glucose Measurement
2.3. SDS-PAGE and Western Blotting Analysis
2.4. Electrophysiological Recording from Neurons and Astrocytes in Brain Slices
2.4.1. Selection and Recording from Astrocytes
2.4.2. Recording from Pyramidal Neurons
Statistical Methodology
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE Official Report: A Practical Clinical Definition of Epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Shorvon, S.D. The Etiologic Classification of Epilepsy. Epilepsia 2011, 52, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, X. Association between Seizures and Diabetes Mellitus: A Comprehensive Review of Literature. Curr. Diabetes Rev. 2013, 9, 350–354. [Google Scholar]
- Younes, S.; Cherif, Y.; Aissi, M.; Alaya, W.; Berriche, O.; Boughammoura, A.; Frih-Ayed, M.; Zantour, B.; Habib Sfar, M. Seizures and Movement Disorders Induced by Hyperglycemia without Ketosis in Elderly. Iran J. Neurol. 2014, 13, 172–176. [Google Scholar] [PubMed]
- Grant, C.; Warlow, C. Focal Epilepsy in Diabetic Non-Ketotic Hyperglycaemia. Br. Med. J. (Clin. Res. Ed.) 1985, 290, 1204–1205. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-L.; Chang, Y.-H.; Sun, Y.; Li, C.-Y. A Population-Based Study of Epilepsy Incidence in Association with Type 2 Diabetes and Severe Hypoglycaemia. Diabetes Res. Clin. Pract. 2018, 140, 97–106. [Google Scholar] [CrossRef]
- Brennan, M.R.; Whitehouse, F.W. Case Study: Seizures and Hypoglycemia. Clin. Diabetes 2012, 30, 23–24. [Google Scholar] [CrossRef]
- Di Mario, U.; Morano, S.; Valle, E.; Pozzessere, G. Electrophysiological Alterations of the Central Nervous System in Diabetes Mellitus. Diabetes Metab. Rev. 1995, 11, 259–277. [Google Scholar] [CrossRef]
- De Leon-Morales, L.V.D.; Jauregui-Renaud, K.; Garay-Sevilla, M.E.; Hernandez-Prado, J.; Malacara-Hernandez, J.M. Auditory Impairment in Patients with Type 2 Diabetes Mellitus. Arch. Med. Res. 2005, 36, 507–510. [Google Scholar] [CrossRef]
- Kamal, A.; Biessels, G.J.; Gispen, W.H.; Ramakers, G.M. Synaptic Transmission Changes in the Pyramidal Cells of the Hippocampus in Streptozotocin-Induced Diabetes Mellitus in Rats. Brain Res. 2006, 1073–1074, 276–280. [Google Scholar] [CrossRef]
- Butt, A.M.; Kalsi, A. Inwardly Rectifying Potassium Channels (Kir) in Central Nervous System Glia: A Special Role for Kir4.1 in Glial Functions. J. Cell. Mol. Med. 2006, 10, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Kucheryavykh, Y.V.; Kucheryavykh, L.Y.; Nichols, C.G.; Maldonado, H.M.; Baksi, K.; Reichenbach, A.; Skatchkov, S.N.; Eaton, M.J. Downregulation of Kir4.1 Inward Rectifying Potassium Channel Subunits by RNAi Impairs Potassium Transfer and Glutamate Uptake by Cultured Cortical Astrocytes. Glia 2007, 55, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Djukic, B.; Casper, K.B.; Philpot, B.D.; Chin, L.S.; McCarthy, K.D. Conditional Knock-out of Kir4.1 Leads to Glial Membrane Depolarization, Inhibition of Potassium and Glutamate Uptake, and Enhanced Short-Term Synaptic Potentiation. J. Neurosci. 2007, 27, 11354–11365. [Google Scholar] [CrossRef] [PubMed]
- Inyushin, M.; Kucheryavykh, L.Y.; Kucheryavykh, Y.V.; Nichols, C.G.; Buono, R.J.; Ferraro, T.N.; Skatchkov, S.N.; Eaton, M.J. Potassium Channel Activity and Glutamate Uptake Are Impaired in Astrocytes of Seizure-Susceptible DBA/2 Mice. Epilepsia 2010, 51, 1707–1713. [Google Scholar] [CrossRef]
- Seifert, G.; Carmignoto, G.; Steinhauser, C. Astrocyte Dysfunction in Epilepsy. Brain Res. Rev. 2010, 63, 212–221. [Google Scholar] [CrossRef]
- Olsen, M.L.; Khakh, B.S.; Skatchkov, S.N.; Zhou, M.; Lee, C.J.; Rouach, N. New Insights on Astrocyte Ion Channels: Critical for Homeostasis and Neuron-Glia Signaling. J. Neurosci. 2015, 35, 13827–13835. [Google Scholar] [CrossRef]
- Seifert, G.; Huttmann, K.; Binder, D.K.; Hartmann, C.; Wyczynski, A.; Neusch, C.; Steinhauser, C. Analysis of Astroglial K+ Channel Expression in the Developing Hippocampus Reveals a Predominant Role of the Kir4.1 Subunit. J. Neurosci. 2009, 29, 7474–7488. [Google Scholar] [CrossRef]
- Chen, K.C.; Nicholson, C. Spatial Buffering of Potassium Ions in Brain Extracellular Space. Biophys. J. 2000, 78, 2776–2797. [Google Scholar] [CrossRef]
- Skatchkov, S.N.; Krusek, J.; Reichenbach, A.; Orkand, R.K. Potassium Buffering by Muller Cells Isolated from the Center and Periphery of the Frog Retina. Glia 1999, 27, 171–180. [Google Scholar] [CrossRef]
- Scholl, U.I.; Choi, M.; Liu, T.; Ramaekers, V.T.; Hausler, M.G.; Grimmer, J.; Tobe, S.W.; Farhi, A.; Nelson-Williams, C.; Lifton, R.P. Seizures, Sensorineural Deafness, Ataxia, Mental Retardation, and Electrolyte Imbalance (SeSAME Syndrome) Caused by Mutations in KCNJ10. Proc. Natl. Acad. Sci. USA 2009, 106, 5842–5847. [Google Scholar] [CrossRef]
- Bockenhauer, D.; Feather, S.; Stanescu, H.C.; Bandulik, S.; Zdebik, A.A.; Reichold, M.; Tobin, J.; Lieberer, E.; Sterner, C.; Landoure, G.; et al. Epilepsy, Ataxia, Sensorineural Deafness, Tubulopathy, and KCNJ10 Mutations. N. Engl. J. Med. 2009, 360, 1960–1970. [Google Scholar] [CrossRef] [PubMed]
- Sala-Rabanal, M.; Kucheryavykh, L.Y.; Skatchkov, S.N.; Eaton, M.J.; Nichols, C.G. Molecular Mechanisms of EAST/SeSAME Syndrome Mutations in Kir4.1 (KCNJ10). J. Biol. Chem. 2010, 285, 36040–36048. [Google Scholar] [CrossRef]
- Mir, A.; Chaudhary, M.; Alkhaldi, H.; Alhazmi, R.; Albaradie, R.; Housawi, Y. Epilepsy in Patients with EAST Syndrome Caused by Mutation in the KCNJ10. Brain Dev. 2019, 41, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.M.; Strobos, R.J. Epilepsia Partialis Continua Associated with Nonketotic Hyperglycemia: Clinical and Biochemical Profile of 21 Patients. Ann. Neurol. 1980, 8, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Kamha, A. Non Ketotic Hyperosmolar Hyperglycemia Presenting as Epilepsia Partialis Continua: An Unusual Presentation of a Common Disorder. Libyan J. Med. 2008, 3, 111–112. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Aponte, D.E.; Mendez-Gonzalez, M.P.; Rivera-Pagan, A.F.; Kucheryavykh, Y.V.; Kucheryavykh, L.Y.; Skatchkov, S.N.; Eaton, M.J. Hyperglycemia Reduces Functional Expression of Astrocytic Kir4.1 Channels and Glial Glutamate Uptake. Neuroscience 2015, 310, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Hummel, K.P.; Dickie, M.M.; Coleman, D.L. Diabetes, a New Mutation in the Mouse. Science 1966, 153, 1127–1128. [Google Scholar] [CrossRef] [PubMed]
- Hibino, H.; Fujita, A.; Iwai, K.; Yamada, M.; Kurachi, Y. Differential Assembly of Inwardly Rectifying K+ Channel Subunits, Kir4.1 and Kir5.1, in Brain Astrocytes. J. Biol. Chem. 2004, 279, 44065–44073. [Google Scholar] [CrossRef]
- Fan, H.; Huang, A.; Villegas, C.; Wright, J.A. The R1 Component of Mammalian Ribonucleotide Reductase Has Malignancy-Suppressing Activity as Demonstrated by Gene Transfer Experiments. Proc. Natl. Acad. Sci. USA 1997, 94, 13181–13186. [Google Scholar] [CrossRef]
- Zhou, M.; Schools, G.P.; Kimelberg, H.K. Development of GLAST(+) Astrocytes and NG2(+) Glia in Rat Hippocampus CA1: Mature Astrocytes Are Electrophysiologically Passive. J. Neurophysiol. 2006, 95, 134–143. [Google Scholar] [CrossRef]
- Battefeld, A.; Klooster, J.; Kole, M.H.P. Myelinating Satellite Oligodendrocytes Are Integrated in a Glial Syncytium Constraining Neuronal High-Frequency Activity. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, P.P.; Velez-Fort, M.; Levavasseur, F.; Angulo, M.C. Oligodendrocyte Precursor Cells Are Accurate Sensors of Local K+ in Mature Gray Matter. J. Neurosci. 2013, 33, 2432–2442. [Google Scholar] [CrossRef] [PubMed]
- Feliciano, P.; Andrade, R.; Bykhovskaia, M. Synapsin II and Rab3a Cooperate in the Regulation of Epileptic and Synaptic Activity in the CA1 Region of the Hippocampus. J. Neurosci. 2013, 33, 18319–18330. [Google Scholar] [CrossRef] [PubMed]
- Verrotti, A.; Scaparrotta, A.; Olivieri, C.; Chiarelli, F. Seizures and Type 1 Diabetes Mellitus: Current State of Knowledge. Eur. J. Endocrinol. 2012, 167, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Moien-Afshari, F.; Tellez-Zenteno, J.F. Occipital Seizures Induced by Hyperglycemia: A Case Report and Review of Literature. Seizure 2009, 18, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Margineanu, D.G.; Niespodziany, I.; Wulfert, E. Hippocampal Slices from Long-Term Streptozotocin-Injected Rats Are Prone to Epileptiform Responses. Neurosci. Lett. 1998, 252, 183–186. [Google Scholar] [CrossRef]
- Huang, C.W.; Cheng, J.T.; Tsai, J.J.; Wu, S.N.; Huang, C.C. Diabetic Hyperglycemia Aggravates Seizures and Status Epilepticus-Induced Hippocampal Damage. Neurotox. Res. 2009, 15, 71–81. [Google Scholar] [CrossRef]
- Schwechter, E.M.; Veliskova, J.; Velisek, L. Correlation between Extracellular Glucose and Seizure Susceptibility in Adult Rats. Ann. Neurol. 2003, 53, 91–101. [Google Scholar] [CrossRef]
- Lin, C.W.; Sim, S.; Ainsworth, A.; Okada, M.; Kelsch, W.; Lois, C. Genetically Increased Cell-Intrinsic Excitability Enhances Neuronal Integration into Adult Brain Circuits. Neuron 2010, 65, 32–39. [Google Scholar] [CrossRef]
- Narla, C.; Scidmore, T.; Jeong, J.; Everest, M.; Chidiac, P.; Poulter, M.O. A Switch in G Protein Coupling for Type 1 Corticotropin-Releasing Factor Receptors Promotes Excitability in Epileptic Brains. Sci. Signal. 2016, 9, ra60. [Google Scholar] [CrossRef]
- Howlett, E.; Lin, C.C.; Lavery, W.; Stern, M. A PI3-Kinase-Mediated Negative Feedback Regulates Neuronal Excitability. PLoS Genet. 2008, 4, e1000277. [Google Scholar] [CrossRef] [PubMed]
- Arakaki, X.; Foster, H.; Su, L.; Do, H.; Wain, A.J.; Fonteh, A.N.; Zhou, F.; Harrington, M.G. Extracellular Sodium Modulates the Excitability of Cultured Hippocampal Pyramidal Cells. Brain Res. 2011, 1401, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Toni, N.; Buchs, P.A.; Nikonenko, I.; Bron, C.R.; Muller, D. LTP Promotes Formation of Multiple Spine Synapses between a Single Axon Terminal and a Dendrite. Nature 1999, 402, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Bacci, A.; Verderio, C.; Pravettoni, E.; Matteoli, M. The Role of Glial Cells in Synaptic Function. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 403–409. [Google Scholar] [CrossRef][Green Version]
- Jayanarayanan, S.; Smijin, S.; Peeyush, K.T.; Anju, T.R.; Paulose, C.S. NMDA and AMPA Receptor Mediated Excitotoxicity in Cerebral Cortex of Streptozotocin Induced Diabetic Rat: Ameliorating Effects of Curcumin. Chem. Biol. Interact. 2013, 201, 39–48. [Google Scholar] [CrossRef]
- Bach, E.C.; Halmos, K.C.; Smith, B.N. Enhanced NMDA Receptor-Mediated Modulation of Excitatory Neurotransmission in the Dorsal Vagal Complex of Streptozotocin-Treated, Chronically Hyperglycemic Mice. PLoS ONE 2015, 10, e0121022. [Google Scholar] [CrossRef]
- Wanrooy, B.J.; Kumar, K.P.; Wen, S.W.; Qin, C.X.; Ritchie, R.H.; Wong, C.H.Y. Distinct Contributions of Hyperglycemia and High-Fat Feeding in Metabolic Syndrome-Induced Neuroinflammation. J. Neuroinflamm. 2018, 15, 293. [Google Scholar] [CrossRef]
- Bogdanov, P.; Corraliza, L.; Villena, J.A.; Carvalho, A.R.; Garcia-Arumí, J.; Ramos, D.; Ruberte, J.; Simó, R.; Hernández, C. The Db/Db Mouse: A Useful Model for the Study of Diabetic Retinal Neurodegeneration. PLoS ONE 2014, 9, e97302. [Google Scholar] [CrossRef]
- Pannicke, T.; Iandiev, I.; Wurm, A.; Uckermann, O.; vom Hagen, F.; Reichenbach, A.; Wiedemann, P.; Hammes, H.P.; Bringmann, A. Diabetes Alters Osmotic Swelling Characteristics and Membrane Conductance of Glial Cells in Rat Retina. Diabetes 2006, 55, 633–639. [Google Scholar] [CrossRef]
- Kucheryavykh, Y.V.; Pearson, W.L.; Kurata, H.T.; Eaton, M.J.; Skatchkov, S.N.; Nichols, C.G. Polyamine Permeation and Rectification of Kir4.1 Channels. Channels 2007, 1, 172–178. [Google Scholar] [CrossRef]
- Thomzig, A.; Wenzel, M.; Karschin, C.; Eaton, M.J.; Skatchkov, S.N.; Karschin, A.; Veh, R.W. Kir6.1 is the principal pore-forming subunit of astrocyte but not neuronal plasma membrane K-ATP channels. Mol. Cell. Neurosci. 2001, 18, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Kucheryavykh, L.Y.; Kucheryavykh, Y.V.; Inyushin, M.; Shuba, Y.M.; Sanabria, P.; Cubano, L.A.; Skatchkov, S.N.; Eaton, M.J. Ischemia Increases TREK-2 Channel Expression in Astrocytes: Relevance to Glutamate Clearance. Open Neurosci. J. 2009, 3, 40–47. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, M.; Xu, G.; Xie, M.; Zhang, X.; Schools, G.P.; Ma, L.; Kimelberg, H.K.; Chen, H. TWIK-1 and TREK-1 Are Potassium Channels Contributing Significantly to Astrocyte Passive Conductance in Rat Hippocampal Slices. J. Neurosci. 2009, 29, 8551–8564. [Google Scholar] [CrossRef] [PubMed]
- Sibille, J.; Pannasch, U.; Rouach, N. Astroglial Potassium Clearance Contributes to Short-Term Plasticity of Synaptically Evoked Currents at the Tripartite Synapse. J. Physiol. 2014, 592 Pt 1, 87–102. [Google Scholar] [CrossRef]
- Witthoft, A.; Filosa, J.A.; Karniadakis, G.E. Potassium Buffering in the Neurovascular Unit: Models and Sensitivity Analysis. Biophys. J. 2013, 105, 2046–2054. [Google Scholar] [CrossRef]
- Du, M.; Li, J.; Chen, L.; Yu, Y.; Wu, Y. Astrocytic Kir4.1 Channels and Gap Junctions Account for Spontaneous Epileptic Seizure. PLoS Comput. Biol. 2018, 14, e1005877. [Google Scholar] [CrossRef]
- Mennerick, S.; Shen, W.; Xu, W.; Benz, A.; Tanaka, K.; Shimamoto, K.; Isenberg, K.E.; Krause, J.E.; Zorumski, C.F. Substrate Turnover by Transporters Curtails Synaptic Glutamate Transients. J. Neurosci. 1999, 19, 9242–9251. [Google Scholar] [CrossRef]
- Bordey, A.; Sontheimer, H. Modulation of Glutamatergic Transmission by Bergmann Glial Cells in Rat Cerebellum in Situ. J. Neurophysiol. 2003, 89, 979–988. [Google Scholar] [CrossRef]
- Ferraro, T.N.; Golden, G.T.; Smith, G.G.; Martin, J.F.; Lohoff, F.W.; Gieringer, T.A.; Zamboni, D.; Schwebel, C.L.; Press, D.M.; Kratzer, S.O.; et al. Fine Mapping of a Seizure Susceptibility Locus on Mouse Chromosome 1: Nomination of Kcnj10 as a Causative Gene. Mamm. Genome 2004, 15, 239–251. [Google Scholar] [CrossRef]
- Buono, R.J.; Lohoff, F.W.; Sander, T.; Sperling, M.R.; O’Connor, M.J.; Dlugos, D.J.; Ryan, S.G.; Golden, G.T.; Zhao, H.; Scattergood, T.M.; et al. Association between Variation in the Human KCNJ10 Potassium Ion Channel Gene and Seizure Susceptibility. Epilepsy Res. 2004, 58, 175–183. [Google Scholar] [CrossRef]
- Heinemann, U.; Lux, H.D. Ceiling of Stimulus Induced Rises in Extracellular Potassium Concentration in the Cerebral Cortex of Cat. Brain Res. 1977, 120, 231–249. [Google Scholar] [CrossRef]




 ) indicates significant difference after application of 4-AP from the same non-diabetic or diabetic ACSF group (p < 0.05; Jonckheere–Terpstra k independent groups test).
) indicates significant difference after application of 4-AP from the same non-diabetic or diabetic ACSF group (p < 0.05; Jonckheere–Terpstra k independent groups test).
) indicates significant difference after application of 4-AP from the same non-diabetic or diabetic ACSF group (p < 0.05; Jonckheere–Terpstra k independent groups test).
) indicates significant difference after application of 4-AP from the same non-diabetic or diabetic ACSF group (p < 0.05; Jonckheere–Terpstra k independent groups test).
 ) indicates significant difference after application of 4-AP from the same non-diabetic or diabetic ACSF group (p < 0.05; Jonckheere–Terpstra k independent groups test). (B) Large EPSC frequency (Hz) recorded in CA1 pyramidal neurons from control (open bars) or diabetic (filled blue bars) mice. Data were obtained from 11 cells from 8 db/+ control and 12 cells from 5 db/db diabetic mice. The large EPSCs recorded from CA1 pyramidal neurons were significantly greater after 4-AP in both control and diabetic mice although there was no difference in the frequency between control and diabetic mice at any time point examined either before or after 4-AP.
) indicates significant difference after application of 4-AP from the same non-diabetic or diabetic ACSF group (p < 0.05; Jonckheere–Terpstra k independent groups test). (B) Large EPSC frequency (Hz) recorded in CA1 pyramidal neurons from control (open bars) or diabetic (filled blue bars) mice. Data were obtained from 11 cells from 8 db/+ control and 12 cells from 5 db/db diabetic mice. The large EPSCs recorded from CA1 pyramidal neurons were significantly greater after 4-AP in both control and diabetic mice although there was no difference in the frequency between control and diabetic mice at any time point examined either before or after 4-AP.
) indicates significant difference after application of 4-AP from the same non-diabetic or diabetic ACSF group (p < 0.05; Jonckheere–Terpstra k independent groups test). (B) Large EPSC frequency (Hz) recorded in CA1 pyramidal neurons from control (open bars) or diabetic (filled blue bars) mice. Data were obtained from 11 cells from 8 db/+ control and 12 cells from 5 db/db diabetic mice. The large EPSCs recorded from CA1 pyramidal neurons were significantly greater after 4-AP in both control and diabetic mice although there was no difference in the frequency between control and diabetic mice at any time point examined either before or after 4-AP.
) indicates significant difference after application of 4-AP from the same non-diabetic or diabetic ACSF group (p < 0.05; Jonckheere–Terpstra k independent groups test). (B) Large EPSC frequency (Hz) recorded in CA1 pyramidal neurons from control (open bars) or diabetic (filled blue bars) mice. Data were obtained from 11 cells from 8 db/+ control and 12 cells from 5 db/db diabetic mice. The large EPSCs recorded from CA1 pyramidal neurons were significantly greater after 4-AP in both control and diabetic mice although there was no difference in the frequency between control and diabetic mice at any time point examined either before or after 4-AP.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Méndez-González, M.P.; Rivera-Aponte, D.E.; Benedikt, J.; Maldonado-Martínez, G.; Tejeda-Bayron, F.; Skatchkov, S.N.; Eaton, M.J. Downregulation of Astrocytic Kir4.1 Potassium Channels Is Associated with Hippocampal Neuronal Hyperexcitability in Type 2 Diabetic Mice. Brain Sci. 2020, 10, 72. https://doi.org/10.3390/brainsci10020072
Méndez-González MP, Rivera-Aponte DE, Benedikt J, Maldonado-Martínez G, Tejeda-Bayron F, Skatchkov SN, Eaton MJ. Downregulation of Astrocytic Kir4.1 Potassium Channels Is Associated with Hippocampal Neuronal Hyperexcitability in Type 2 Diabetic Mice. Brain Sciences. 2020; 10(2):72. https://doi.org/10.3390/brainsci10020072
Chicago/Turabian StyleMéndez-González, Miguel P., David E. Rivera-Aponte, Jan Benedikt, Geronimo Maldonado-Martínez, Flavia Tejeda-Bayron, Serguei N. Skatchkov, and Misty J. Eaton. 2020. "Downregulation of Astrocytic Kir4.1 Potassium Channels Is Associated with Hippocampal Neuronal Hyperexcitability in Type 2 Diabetic Mice" Brain Sciences 10, no. 2: 72. https://doi.org/10.3390/brainsci10020072
APA StyleMéndez-González, M. P., Rivera-Aponte, D. E., Benedikt, J., Maldonado-Martínez, G., Tejeda-Bayron, F., Skatchkov, S. N., & Eaton, M. J. (2020). Downregulation of Astrocytic Kir4.1 Potassium Channels Is Associated with Hippocampal Neuronal Hyperexcitability in Type 2 Diabetic Mice. Brain Sciences, 10(2), 72. https://doi.org/10.3390/brainsci10020072