Dysregulated Wnt Signalling in the Alzheimer’s Brain


Abstract
1. Introduction
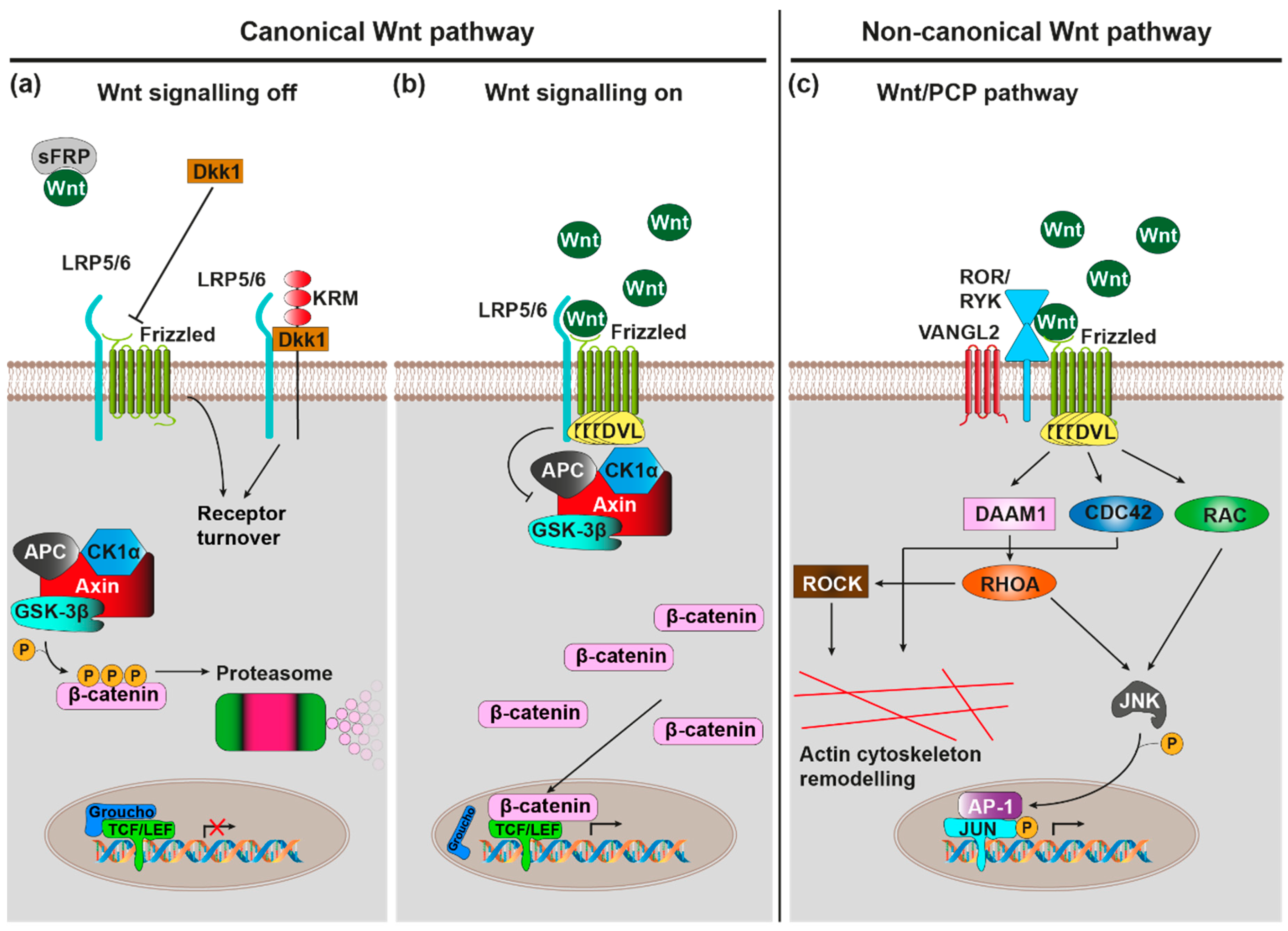
2. Wnt Signalling Pathways—An Overview
3. Wnt Signalling in the Brain
3.1. Wnt Signalling during Brain Development
3.2. Wnt Signalling in the Mature Brain
3.3. Wnt Signalling in the Ageing Brain
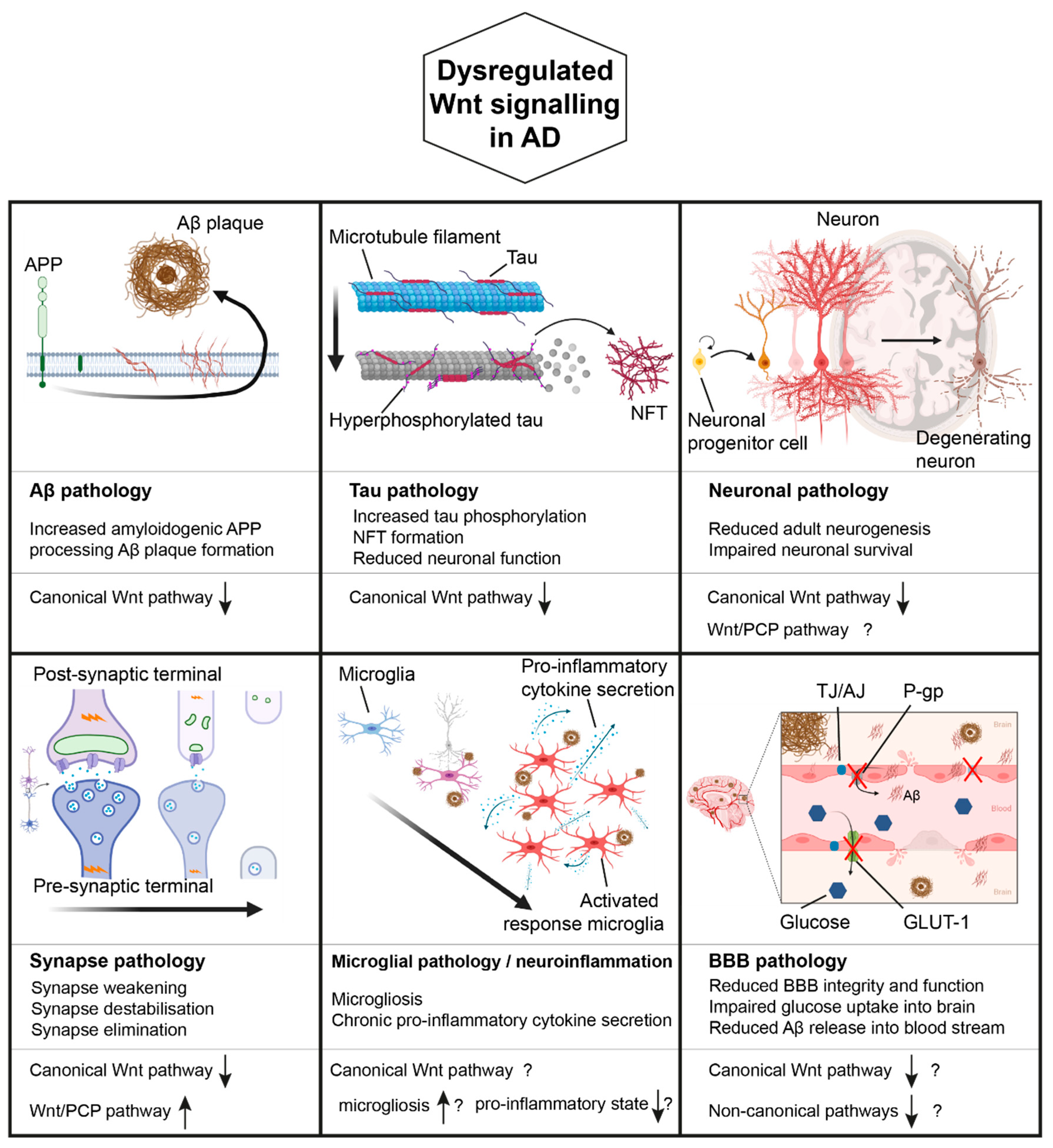
4. Wnt Signalling in AD
4.1. Wnt Signalling, APP Processing, and AD
4.2. Wnt Signalling, Tau Pathology, and AD
4.3. Wnt Signalling, Synapses, and AD
4.4. Wnt Signalling, Microglia/Neuroinflammation, and AD
4.5. Wnt Signalling, the Blood Brain Barrier, and AD
4.6. Are the Wnt Changes Seen in AD Simply an Exacerbated Consequence of Ageing?
5. Looking Forward: Does Wnt Signalling Offer Opportunities for New Therapies for AD?
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Fish, P.V.; Steadman, D.; Bayle, E.D.; Whiting, P. New approaches for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2019, 29, 125–133. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Hou, J.; Ping, J.; Cai, D. Advances in developing novel therapeutic strategies for Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 1–20. [Google Scholar] [CrossRef]
- Vermunt, L.; Sikkes, S.A.; Hout, A.V.D.; Handels, R.; Bos, I.; Van Der Flier, W.M.; Kern, S.; Ousset, P.-J.; Maruff, P.; Skoog, I.; et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimer’s Dement. 2019, 15, 888–898. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Sayas, C.L. Tau-based therapies for Alzheimer’s disease: Promising novel neuroprotective approaches. In Neuroprotection in Autism, Schizophrenia and Alzheimer’s Disease; Elsevier BV: Amsterdam, The Netherlands, 2020; pp. 245–272. [Google Scholar]
- Palomer, E.; Buechler, J.; Salinas, P.C. Wnt Signaling Deregulation in the Aging and Alzheimer’s Brain. Front. Cell. Neurosci. 2019, 13, 227. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Varela-Nallar, L. Wnt signaling in the nervous system and in Alzheimer’s disease. J. Mol. Cell Biol. 2014, 6, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Willert, K.; Brown, J.D.; Danenberg, E.; Duncan, A.W.; Weissman, I.L.; Reya, T.; Yates, J.R.; Nusse, R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nat. Cell Biol. 2003, 423, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012. [Google Scholar] [CrossRef] [PubMed]
- Schulte, G.; Bryja, V. The Frizzled family of unconventional G-protein-coupled receptors. Trends Pharmacol. Sci. 2007, 28, 518–525. [Google Scholar] [CrossRef]
- Macdonald, B.T.; He, X. Frizzled and LRP5/6 Receptors for Wnt/ -Catenin Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a007880. [Google Scholar] [CrossRef]
- Fradkin, L.G.; Dura, J.-M.; Noordermeer, J.N. Ryks: New partners for Wnts in the developing and regenerating nervous system. Trends Neurosci. 2010, 33, 84–92. [Google Scholar] [CrossRef]
- Green, J.L.; Kuntz, S.G.; Sternberg, P.W. Ror receptor tyrosine kinases: Orphans no more. Trends Cell Biol. 2008, 18, 536–544. [Google Scholar] [CrossRef]
- Bhanot, P.; Brink, M.; Samos, C.H.; Hsieh, J.-C.; Wang, Y.; Macke, J.P.; Andrew, D.; Nathans, J.; Nusse, R. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nat. Cell Biol. 1996, 382, 225–230. [Google Scholar] [CrossRef]
- Janda, C.Y.; Waghray, D.; Levin, A.M.; Thomas, C.; Garcia, K.C. Structural Basis of Wnt Recognition by Frizzled. Science 2012, 337, 59–64. [Google Scholar] [CrossRef]
- Dann, C.E.; Hsieh, J.-C.; Rattner, A.; Sharma, D.; Nathans, J.; Leahy, D.J. Insights into Wnt binding and signalling from the structures of two Frizzled cysteine-rich domains. Nat. Cell Biol. 2001, 412, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, A.; Yamamoto, H.; Sato, A.; Matsumoto, S. New Insights into the Mechanism of Wnt Signaling Pathway Activation. Int. Rev. Cell Mol. Biol. 2011, 291, 21–71. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, A.; Yamamoto, H.; Sato, A. Selective activation mechanisms of Wnt signaling pathways. Trends Cell Biol. 2009, 19, 119–129. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.-H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of β-Catenin Phosphorylation/Degradation by a Dual-Kinase Mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef]
- Huber, O.; Korn, R.; McLaughlin, J.; Ohsugi, M.; Herrmann, B.G.; Kemler, R. Nuclear localization of β-catenin by interaction with transcription factor LEF-1. Mech. Dev. 1996, 59, 3–10. [Google Scholar] [CrossRef]
- Molenaar, M.; Van De Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destrée, O.; Clevers, H. XTcf-3 Transcription Factor Mediates β-Catenin-Induced Axis Formation in Xenopus Embryos. Cell 1996, 86, 391–399. [Google Scholar] [CrossRef]
- Vlad, A.; Röhrs, S.; Klein-Hitpass, L.; Müller, O. The first five years of the Wnt targetome. Cell. Signal. 2008, 20, 795–802. [Google Scholar] [CrossRef]
- Macheda, M.L.; Sun, W.W.; Kugathasan, K.; Hogan, B.M.; Bower, N.I.; Halford, M.M.; Zhang, Y.F.; Jacques, B.E.; Lieschke, G.J.; Dabdoub, A.; et al. The Wnt Receptor Ryk Plays a Role in Mammalian Planar Cell Polarity Signaling. J. Biol. Chem. 2012, 287, 29312–29323. [Google Scholar] [CrossRef]
- Nishita, M.; Itsukushima, S.; Nomachi, A.; Endo, M.; Wang, Z.; Inaba, D.; Qiao, S.; Takada, S.; Kikuchi, A.; Minami, Y. Ror2/Frizzled Complex Mediates Wnt5a-Induced AP-1 Activation by Regulating Dishevelled Polymerization. Mol. Cell. Biol. 2010, 30, 3610–3619. [Google Scholar] [CrossRef]
- Green, J.; Nusse, R.; Van Amerongen, R. The Role of Ryk and Ror Receptor Tyrosine Kinases in Wnt Signal Transduction. Cold Spring Harb. Perspect. Biol. 2014, 6, a009175. [Google Scholar] [CrossRef] [PubMed]
- Strutt, D.; Weber, U.; Mlodzik, M. The role of RhoA in tissue polarity and Frizzled signalling. Nat. Cell Biol. 1997, 387, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Fanto, M.; Weber, U.; Strutt, D.; Mlodzik, M. Nuclear signaling by Rac and Rho GTPases is required in the establishment of epithelial planar polarity in the Drosophila eye. Curr. Biol. 2000, 10, 979–988. [Google Scholar] [CrossRef]
- Boutros, M.; Paricio, N.; Strutt, D.I.; Mlodzik, M. Dishevelled Activates JNK and Discriminates between JNK Pathways in Planar Polarity and wingless Signaling. Cell 1998, 94, 109–118. [Google Scholar] [CrossRef]
- Winter, C.G.; Wang, B.; Ballew, A.; Royou, A.; Karess, R.; Axelrod, J.D.; Luo, L. Drosophila Rho-Associated Kinase (Drok) Links Frizzled-Mediated Planar Cell Polarity Signaling to the Actin Cytoskeleton. Cell 2001, 105, 81–91. [Google Scholar] [CrossRef]
- Seifert, J.R.K.; Mlodzik, M. Frizzled/PCP signalling: A conserved mechanism regulating cell polarity and directed motility. Nat. Rev. Genet. 2007, 8, 126–138. [Google Scholar] [CrossRef]
- Slusarski, D.C.; Corces, V.G.; Moon, R.T. Interaction of Wnt and a Frizzled homologue triggers G-protein-linked phosphatidylinositol signalling. Nat. Cell Biol. 1997, 390, 410–413. [Google Scholar] [CrossRef]
- Kühl, M.; Sheldahl, L.C.; Park, M.; Miller, J.R.; Moon, R.T. The Wnt/Ca2+ pathway A new vertebrate Wnt signaling pathway takes shape. Trends Genet. 2000, 16, 279–283. [Google Scholar] [CrossRef]
- Slusarski, D.C.; Yang-Snyder, J.; Busa, W.B.; Moon, R.T. Modulation of Embryonic Intracellular Ca2+Signaling byWnt-5A. Dev. Biol. 1997, 182, 114–120. [Google Scholar] [CrossRef]
- Sheldahl, L.C.; Park, M.; Malbon, C.C.; Moon, R.T. Protein kinase C is differentially stimulated by Wnt and Frizzled homologs in aG-protein-dependent manner. Curr. Biol. 1999, 9, 695–698. [Google Scholar] [CrossRef]
- Kühl, M.; Sheldahl, L.C.; Malbon, C.C.; Moon, R.T. Ca2+/Calmodulin-dependent Protein Kinase II Is Stimulated by Wnt and Frizzled Homologs and Promotes Ventral Cell Fates inXenopus. J. Biol. Chem. 2000, 275, 12701–12711. [Google Scholar] [CrossRef] [PubMed]
- De, A. Wnt/Ca2+ signaling pathway: A brief overview. Acta Biochim. Biophys. Sin. 2011, 43, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Cruciat, C.-M.; Niehrs, C. Secreted and Transmembrane Wnt Inhibitors and Activators. Cold Spring Harb. Perspect. Biol. 2012, 5, a015081. [Google Scholar] [CrossRef] [PubMed]
- Semënov, M.V.; Tamai, K.; Brott, B.K.; Kühl, M.; Sokol, S.; He, X. Head inducer Dickkopf-1 is a ligand for Wnt coreceptor LRP6. Curr. Biol. 2001, 11, 951–961. [Google Scholar] [CrossRef]
- Mao, B.; Wu, W.; Davidson, G.; Marhold, J.; Li, M.; Mechler, B.M.; Delius, H.; Hoppe, D.; Stannek, P.; Walter, C.; et al. Kremen proteins are Dickkopf receptors that regulate Wnt/β-catenin signalling. Nat. Cell Biol. 2002, 417, 664–667. [Google Scholar] [CrossRef]
- Mao, B.; Niehrs, C. Kremen2 modulates Dickkopf2 activity during Wnt/lRP6 signaling. Gene 2003, 302, 179–183. [Google Scholar] [CrossRef]
- Hao, H.-X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nat. Cell Biol. 2012, 485, 195–200. [Google Scholar] [CrossRef]
- De Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The R-spondin/Lgr5/Rnf43 module: Regulator of Wnt signal strength. Genes Dev. 2014, 28, 305–316. [Google Scholar] [CrossRef]
- Ohkawara, B.; Glinka, A.; Niehrs, C. Rspo3 Binds Syndecan 4 and Induces Wnt/PCP Signaling via Clathrin-Mediated Endocytosis to Promote Morphogenesis. Dev. Cell 2011, 20, 303–314. [Google Scholar] [CrossRef]
- Xu, Q.; Wang, Y.; Dabdoub, A.; Smallwood, P.M.; Williams, J.; Woods, C.; Kelley, M.W.; Jiang, L.; Tasman, W.; Zhang, K.; et al. Vascular Development in the Retina and Inner Ear. Cell 2004, 116, 883–895. [Google Scholar] [CrossRef]
- Oliva, C.A.; Montecinos-Oliva, C.; Inestrosa, N.C. Wnt Signaling in the Central Nervous System: New Insights in Health and Disease. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2018; Volume 153, pp. 81–130. [Google Scholar]
- Brault, V.; Moore, R.; Kutsch, S.; Ishibashi, M.; Rowitch, D.H.; McMahon, A.P.; Sommer, L.; Boussadia, O.; Kemler, R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 2001, 128, 1253–1264. [Google Scholar] [PubMed]
- Thomas, K.R.; Capecchi, M.R. Targeted disruption of the murine int-1 proto-oncogene resulting in severe abnormalities in midbrain and cerebellar development. Nat. Cell Biol. 1990, 346, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Nordström, U.; Jessell, T.M.; Edlund, T. Progressive induction of caudal neural character by graded Wnt signaling. Nat. Neurosci. 2002, 5, 525–532. [Google Scholar] [CrossRef]
- Carron, C.; Shi, D.-L. Specification of anteroposterior axis by combinatorial signaling duringXenopusdevelopment. Wiley Interdiscip. Rev. Dev. Biol. 2015, 5, 150–168. [Google Scholar] [CrossRef] [PubMed]
- Glinka, A.; Wu, W.; Delius, H.; Monaghan, A.P.; Blumenstock, C.; Niehrs, C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nat. Cell Biol. 1998, 391, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Houart, C.; Caneparo, L.; Heisenberg, C.-P.; Barth, K.; Take-Uchi, M.; Wilson, S.W. Establishment of the Telencephalon during Gastrulation by Local Antagonism of Wnt Signaling. Neuron 2002, 35, 255–265. [Google Scholar] [CrossRef]
- Zhang, X.; Cheong, S.-M.; Amado, N.G.; Reis, A.H.; Macdonald, B.T.; Zebisch, M.; Jones, E.Y.; Abreu, J.G.; He, X. Notum Is Required for Neural and Head Induction via Wnt Deacylation, Oxidation, and Inactivation. Dev. Cell 2015, 32, 719–730. [Google Scholar] [CrossRef]
- Grove, E.A.; Tole, S.; Limon, J.; Yip, L.; Ragsdale, C.W. The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3-deficient mice. Development 1998, 125, 2315–2325. [Google Scholar]
- Lee, S.M.; Tole, S.; Grove, E.; McMahon, A.P. A local Wnt-3a signal is required for development of the mammalian hippocampus. Development 2000, 127, 457–467. [Google Scholar]
- Yoshinaga, Y.; Kagawa, T.; Shimizu, T.; Inoue, T.; Takada, S.; Kuratsu, J.-I.; Taga, T. Wnt3a Promotes Hippocampal Neurogenesis by Shortening Cell Cycle Duration of Neural Progenitor Cells. Cell. Mol. Neurobiol. 2010, 30, 1049–1058. [Google Scholar] [CrossRef]
- Galceran, J.; Miyashita-Lin, E.M.; Devaney, E.; Rubenstein, J.L.; Grosschedl, R. Hippocampus development and generation of dentate gyrus granule cells is regulated by LEF1. Development 2000, 127, 469–482. [Google Scholar] [PubMed]
- Hirabayashi, Y.; Gotoh, Y. Stage-dependent fate determination of neural precursor cells in mouse forebrain. Neurosci. Res. 2005, 51, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Zechner, D.; Fujita, Y.; Hülsken, J.; Müller, T.; Walther, I.; Taketo, M.M.; Crenshaw, E.B.; Birchmeier, W.; Birchmeier, C. β-Catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev. Biol. 2003, 258, 406–418. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, J.; Yang, G.-Y.; Wang, Q.-J.; Qian, L.; Chen, Y.-M.; Chen, F.; Tao, Y.; Hu, H.-S.; Wang, T.; et al. Dishevelled promotes axon differentiation by regulating atypical protein kinase C. Nat. Cell Biol. 2007, 9, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Slater, P.G.; Ramírez, V.T.; Gonzalez-Billault, C.; Varela-Nallar, L.; Inestrosa, N.C. Frizzled-5 Receptor Is Involved in Neuronal Polarity and Morphogenesis of Hippocampal Neurons. PLoS ONE 2013, 8, e78892. [Google Scholar] [CrossRef]
- Ohno, S. Extrinsic Wnt signalling controls the polarity component aPKC. Nat. Cell Biol. 2007, 9, 738–740. [Google Scholar] [CrossRef]
- Rosso, S.B.; Sussman, D.; Wynshaw-Boris, A.; Salinas, P.C. Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nat. Neurosci. 2005, 8, 34–42. [Google Scholar] [CrossRef]
- Viale, B.; Song, L.; Petrenko, V.; Combremont, A.-L.W.; Contestabile, A.; Bocchi, R.; Salmon, P.; Carleton, A.; An, L.; Vutskits, L.; et al. Transient Deregulation of Canonical Wnt Signaling in Developing Pyramidal Neurons Leads to Dendritic Defects and Impaired Behavior. Cell Rep. 2019, 27, 1487–1502.e6. [Google Scholar] [CrossRef]
- Ahmad-Annuar, A.; Ciani, L.; Simeonidis, I.; Herreros, J.; Ben Fredj, N.; Rosso, S.B.; Hall, A.; Brickley, S.; Salinas, P.C. Signaling across the synapse: A role for Wnt and Dishevelled in presynaptic assembly and neurotransmitter release. J. Cell Biol. 2006, 174, 127–139. [Google Scholar] [CrossRef]
- Cerpa, W.; Godoy, J.A.; Alfaro, I.; Farías, G.G.; Metcalfe, M.J.; Fuentealba, R.; Bonansco, C.; Inestrosa, N.C. Wnt-7aModulates the Synaptic Vesicle Cycle and Synaptic Transmission in Hippocampal Neurons. J. Biol. Chem. 2007, 283, 5918–5927. [Google Scholar] [CrossRef]
- Davis, E.K.; Zou, Y.; Ghosh, A. Wnts acting through canonical and noncanonical signaling pathways exert opposite effects on hippocampal synapse formation. Neural Dev. 2008, 3, 32. [Google Scholar] [CrossRef] [PubMed]
- Varela-Nallar, L.; Grabowski, C.P.; Alfaro, I.E.; Alvarez, A.; Inestrosa, N.C. Role of the Wnt receptor Frizzled-1 in presynaptic differentiation and function. Neural Dev. 2009, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Sahores, M.; Gibb, A.; Salinas, P.C. Frizzled-5, a receptor for the synaptic organizer Wnt7a, regulates activity-mediated synaptogenesis. Development 2010, 137, 2215–2225. [Google Scholar] [CrossRef]
- Varela-Nallar, L.; Alfaro, I.E.; Serrano, F.G.; Parodi, J.; Inestrosa, N.C. Wingless-type family member 5A (Wnt-5a) stimulates synaptic differentiation and function of glutamatergic synapses. Proc. Natl. Acad. Sci. USA 2010, 107, 21164–21169. [Google Scholar] [CrossRef] [PubMed]
- Farías, G.G.; Alfaro, I.E.; Cerpa, W.; Grabowski, C.P.; Godoy, J.A.; Bonansco, C.; Inestrosa, N.C. Wnt-5a/JNK Signaling Promotes the Clustering of PSD-95 in Hippocampal Neurons. J. Biol. Chem. 2009, 284, 15857–15866. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, V.T.; Ramos-Fernández, E.; Henríquez, J.P.; Lorenzo, A.; Inestrosa, N.C. Wnt-5a/Frizzled9 Receptor Signaling through the Gαo-Gβγ Complex Regulates Dendritic Spine Formation. J. Biol. Chem. 2016, 291, 19092–19107. [Google Scholar] [CrossRef] [PubMed]
- Ciani, L.; Boyle, K.A.; Dickins, E.; Sahores, M.; Anane, D.; Lopes, D.M.; Gibb, A.J.; Salinas, P.C. Wnt7a signaling promotes dendritic spine growth and synaptic strength through Ca2+/Calmodulin-dependent protein kinase II. Proc. Natl. Acad. Sci. USA 2011, 108, 10732–10737. [Google Scholar] [CrossRef] [PubMed]
- Oliva, C.A.; Inestrosa, N.C. A novel function for Wnt signaling modulating neuronal firing activity and the temporal structure of spontaneous oscillation in the entorhinal–hippocampal circuit. Exp. Neurol. 2015, 269, 43–55. [Google Scholar] [CrossRef]
- Cerpa, W.; Gambrill, A.; Inestrosa, N.C.; Barria, A. Regulation of NMDA-Receptor Synaptic Transmission by Wnt Signaling. J. Neurosci. 2011, 31, 9466–9471. [Google Scholar] [CrossRef]
- Gogolla, N.; Galimberti, I.; Deguchi, Y.; Caroni, P. Wnt Signaling Mediates Experience-Related Regulation of Synapse Numbers and Mossy Fiber Connectivities in the Adult Hippocampus. Neuron 2009, 62, 510–525. [Google Scholar] [CrossRef]
- McLeod, F.; Salinas, P.C. Wnt proteins as modulators of synaptic plasticity. Curr. Opin. Neurobiol. 2018, 53, 90–95. [Google Scholar] [CrossRef] [PubMed]
- McLeod, F.; Bossio, A.; Marzo, A.; Ciani, L.; Sibilla, S.; Hannan, S.; Wilson, G.A.; Palomer, E.; Smart, T.G.; Gibb, A.; et al. Wnt Signaling Mediates LTP-Dependent Spine Plasticity and AMPAR Localization through Frizzled-7 Receptors. Cell Rep. 2018, 23, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Ciani, L.; Marzo, A.; Boyle, K.; Stamatakou, E.; Lopes, D.M.; Anane, D.; McLeod, F.; Rosso, S.B.; Gibb, A.; Salinas, P.C. Wnt signalling tunes neurotransmitter release by directly targeting Synaptotagmin-1. Nat. Commun. 2015, 6, 8302. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Park, C.S.; Tang, S.-J. Activity-dependent Synaptic Wnt Release Regulates Hippocampal Long Term Potentiation. J. Biol. Chem. 2006, 281, 11910–11916. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.Y.; Fuenzalida, M.; Inestrosa, N.C. In vivo Activation of Wnt Signaling Pathway Enhances Cognitive Function of Adult Mice and Reverses Cognitive Deficits in an Alzheimer’s Disease Model. J. Neurosci. 2014, 34, 2191–2202. [Google Scholar] [CrossRef] [PubMed]
- Tabatadze, N.; Tomas, C.; McGonigal, R.; Lin, B.; Schook, A.; Routtenberg, A. Wnt transmembrane signaling and long-term spatial memory. Hippocampus 2011, 22, 1228–1241. [Google Scholar] [CrossRef] [PubMed]
- Fortress, A.M.; Schram, S.L.; Tuscher, J.J.; Frick, K.M. Canonical Wnt Signaling is Necessary for Object Recognition Memory Consolidation. J. Neurosci. 2013, 33, 12619–12626. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, J.T.; Schafer, S.T.; Gage, F.H. Adult Neurogenesis in the Hippocampus: From Stem Cells to Behavior. Cell 2016, 167, 897–914. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Pareek, V.; Faiq, M.A.; Ghosh, S.K.; Kumari, C. ADULT NEUROGENESIS IN HUMANS: A Review of Basic Concepts, History, Current Research, and Clinical Implications. Curr. Res. Clin. Implic. Innov. Clin. Neurosci. 2019, 16, 30–37. [Google Scholar]
- Lie, D.C.; Colamarino, S.A.; Song, H.-J.; Désiré, L.; Mira, H.; Consiglio, A.; Lein, E.S.; Jessberger, S.; Lansford, H.; Dearie, A.R.; et al. Wnt signalling regulates adult hippocampal neurogenesis. Nat. Cell Biol. 2005, 437, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.-H.; Bonaguidi, M.A.; Kitabatake, Y.; Sun, J.; Song, J.; Kang, E.; Jun, H.; Zhong, C.; Su, Y.; Guo, J.U.; et al. Secreted Frizzled-Related Protein 3 Regulates Activity-Dependent Adult Hippocampal Neurogenesis. Cell Stem Cell 2013, 12, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Jessberger, S.; Clark, R.E.; Broadbent, N.J.; Clemenson, J.G.D.; Consiglio, A.; Lie, D.C.; Squire, L.R.; Gage, F.H. Dentate gyrus-specific knockdown of adult neurogenesis impairs spatial and object recognition memory in adult rats. Learn. Mem. 2009, 16, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.H.; Bartholomae, A.; Heimrich, B.; Feldmeyer, D.; Druffel-Augustin, S.; Goebbels, S.; Naya, F.J.; Zhao, S.; Frotscher, M.; Tsai, M.-J.; et al. Neuronal Basic Helix-Loop-Helix Proteins (NEX and BETA2/Neuro D) Regulate Terminal Granule Cell Differentiation in the Hippocampus. J. Neurosci. 2000, 20, 3714–3724. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, T.; Hsieh, J.; Muotri, A.R.; Yeo, G.; Warashina, M.; Lie, D.C.; Moore, L.; Nakashima, K.; Asashima, M.; Gage, F.H. Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat. Neurosci. 2009, 12, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Sun, G.; Murai, K.; Ye, P.; Li, W.; Asuelime, G.; Cheung, Y.-T.; Shi, Y. Wnt7a Regulates Multiple Steps of Neurogenesis. Mol. Cell. Biol. 2013, 33, 2551–2559. [Google Scholar] [CrossRef]
- Mizrak, D.; Bayin, N.S.; Yuan, J.; Liu, Z.; Suciu, R.M.; Niphakis, M.J.; Ngo, N.; Lum, K.M.; Cravatt, B.F.; Joyner, A.L.; et al. Single-Cell Profiling and SCOPE-Seq Reveal Lineage Dynamics of Adult Ventricular-Subventricular Zone Neurogenesis and NOTUM as a Key Regulator. Cell Rep. 2020, 31, 107805. [Google Scholar] [CrossRef]
- García-Velázquez, L.; Arias, C. The emerging role of Wnt signaling dysregulation in the understanding and modification of age-associated diseases. Ageing Res. Rev. 2017, 37, 135–145. [Google Scholar] [CrossRef]
- Tobin, M.K.; Musaraca, K.; Disouky, A.; Shetti, A.; Bheri, A.; Honer, W.G.; Kim, N.; Dawe, R.J.; Bennett, D.A.; Arfanakis, K.; et al. Human Hippocampal Neurogenesis Persists in Aged Adults and Alzheimer’s Disease Patients. Cell Stem Cell 2019, 24, 974–982.e3. [Google Scholar] [CrossRef]
- Mathews, K.J.; Allen, K.M.; Boerrigter, D.; Ball, H.; Weickert, C.S.; Double, K.L. Evidence for reduced neurogenesis in the aging human hippocampus despite stable stem cell markers. Aging Cell 2017, 16, 1195–1199. [Google Scholar] [CrossRef]
- Petralia, R.S.; Mattson, M.P.; Yao, P.J. Communication breakdown: The impact of ageing on synapse structure. Ageing Res. Rev. 2014, 14, 31–42. [Google Scholar] [CrossRef]
- Morrison, J.H.; Baxter, M.G. The ageing cortical synapse: Hallmarks and implications for cognitive decline. Nat. Rev. Neurosci. 2012, 13, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.W.; McBryan, T.; Adams, P.D.; Sedivy, J.M. The effects of aging on the expression of Wnt pathway genes in mouse tissues. AGE 2014, 36, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Inoue, K.; Iwamura, H.; Terashima, K.; Soya, H.; Asashima, M.; Kuwabara, T. Reduction in paracrine Wnt3 factors during aging causes impaired adult neurogenesis. FASEB J. 2011, 25, 3570–3582. [Google Scholar] [CrossRef] [PubMed]
- Seib, D.R.; Corsini, N.S.; Ellwanger, K.; Plaas, C.; Mateos, A.; Pitzer, C.; Niehrs, C.; Celikel, T.; Martin-Villalba, A. Loss of Dickkopf-1 Restores Neurogenesis in Old Age and Counteracts Cognitive Decline. Cell Stem Cell 2013, 12, 204–214. [Google Scholar] [CrossRef]
- Orellana, A.M.M.; Vasconcelos, A.R.; Leite, J.A.; Lima, L.D.S.; Andreotti, D.Z.; Munhoz, C.D.; Kawamoto, E.M.; Scavone, C. Age-related neuroinflammation and changes in AKT-GSK-3β and WNT/ β-CATENIN signaling in rat hippocampus. Aging 2015, 7, 1094–1108. [Google Scholar] [CrossRef]
- Folke, J.; Pakkenberg, B.; Brudek, T. Impaired Wnt Signaling in the Prefrontal Cortex of Alzheimer’s Disease. Mol. Neurobiol. 2018, 56, 873–891. [Google Scholar] [CrossRef]
- Brann, D.W.; Dhandapani, K.; Wakade, C.; Mahesh, V.B.; Khan, M.M. Neurotrophic and neuroprotective actions of estrogen: Basic mechanisms and clinical implications. Steroids 2007, 72, 381–405. [Google Scholar] [CrossRef]
- Scott, E.L.; Zhang, Q.-G.; Han, N.; Desai, B.N.; Brann, D.W. Long-term estrogen deprivation leads to elevation of Dickkopf-1 and dysregulation of Wnt/β-Catenin signaling in hippocampal CA1 neurons. Steroids 2013, 78, 624–632. [Google Scholar] [CrossRef]
- Marzo, A.; Galli, S.; Lopes, D.; McLeod, F.; Podpolny, M.; Segovia-Roldan, M.; Ciani, L.; Purro, S.; Cacucci, F.; Gibb, A.; et al. Reversal of Synapse Degeneration by Restoring Wnt Signaling in the Adult Hippocampus. Curr. Biol. 2016, 26, 2551–2561. [Google Scholar] [CrossRef]
- Anderton, B.H.; Dayanandan, R.; Killick, R.; Lovestone, S. Does dysregulation of the Notch and wingless/Wnt pathways underlie the pathogenesis of Alzheimer’s disease? Mol. Med. Today 2000, 6, 54–59. [Google Scholar] [CrossRef]
- Van Der Flier, W.M. Epidemiology and risk factors of dementia. J. Neurol. Neurosurg. Psychiatry 2005, 76, v2–v7. [Google Scholar] [CrossRef] [PubMed]
- Mateo, I.; Infante, J.; Llorca, J.; Rodríguez, E.; Berciano, J.; Combarros, O. Association between Glycogen Synthase Kinase-3β Genetic Polymorphism and Late-Onset Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2006, 21, 228–232. [Google Scholar] [CrossRef] [PubMed]
- De Ferrari, G.V.; Papassotiropoulos, A.; Biechele, T.; De-Vrieze, F.W.; Avila, M.E.; Major, M.B.; Myers, A.; Sáez, K.; Henríquez, J.P.; Zhao, A.; et al. Common genetic variation within the Low-Density Lipoprotein Receptor-Related Protein 6 and late-onset Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 9434–9439. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, M.A.; Medina, M.A.; Hu, Q.; Avila, M.E.; Bustos, B.I.; Pérez-Palma, E.; Peralta, A.; Salazar, P.; Ugarte, G.D.; Reyes, A.E.; et al. A novel functional low-density lipoprotein receptor-related protein 6 gene alternative splice variant is associated with Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1709.e9–1709.e18. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Tsai, C.-W.; Deak, F.; Rogers, J.; Penuliar, M.; Sung, Y.M.; Maher, J.N.; Fu, Y.; Li, X.; Xu, H.; et al. Deficiency in LRP6-Mediated Wnt Signaling Contributes to Synaptic Abnormalities and Amyloid Pathology in Alzheimer’s Disease. Neuron 2014, 84, 63–77. [Google Scholar] [CrossRef]
- Buechler, J.; Salinas, P.C. Deficient Wnt Signaling and Synaptic Vulnerability in Alzheimer’s Disease: Emerging Roles for the LRP6 Receptor. Front. Synaptic Neurosci. 2018, 10, 38. [Google Scholar] [CrossRef]
- Caricasole, A.; Copani, A.; Caraci, F.; Aronica, E.; Rozemuller, A.J.; Caruso, A.; Storto, M.; Gaviraghi, G.; Terstappen, G.C.; Nicoletti, F. Induction of Dickkopf-1, a Negative Modulator of the Wnt Pathway, Is Associated with Neuronal Degeneration in Alzheimer’s Brain. J. Neurosci. 2004, 24, 6021–6027. [Google Scholar] [CrossRef]
- Rosi, M.C.; Luccarini, I.; Grossi, C.; Fiorentini, A.; Spillantini, M.G.; Prisco, A.; Scali, C.; Gianfriddo, M.; Caricasole, A.; Terstappen, G.C.; et al. Increased Dickkopf-1 expression in transgenic mouse models of neurodegenerative disease. J. Neurochem. 2010, 112, 1539–1551. [Google Scholar] [CrossRef]
- Purro, S.A.; Dickins, E.M.; Salinas, P.C. The Secreted Wnt Antagonist Dickkopf-1 Is Required for Amyloid -Mediated Synaptic Loss. J. Neurosci. 2012, 32, 3492–3498. [Google Scholar] [CrossRef]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.-H.; Haddick, P.C.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef]
- Lambert, J.-C.; The European Alzheimer’s Disease Initiative Investigators; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.O.; Combarros, O.; Zelenika, D.; Bullido, M.J.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.T.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; Van Duijn, C.M. Effects of Age, Sex, and Ethnicity on the Association Between Apolipoprotein E Genotype and Alzheimer Disease. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Schmechel, D.E.; Saunders, A.M.; Strittmatter, W.J.; Crain, B.J.; Hulette, C.M.; Joo, S.H.; Pericak-Vance, M.A.; Goldgaber, D.; Roses, A.D. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 9649–9653. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Holm, M.-L.; Liu, C.-C.; Shinohara, M.; Aikawa, T.; Oue, H.; Yamazaki, Y.; Martens, Y.A.; Murray, M.E.; Sullivan, P.M.; et al. APOE4-mediated amyloid-β pathology depends on its neuronal receptor LRP1. J. Clin. Investig. 2019, 129, 1272–1277. [Google Scholar] [CrossRef]
- Zilberberg, A.; Yaniv, A.; Gazit, A. The Low Density Lipoprotein Receptor-1, LRP1, Interacts with the Human Frizzled-1 (HFz1) and Down-regulates the Canonical Wnt Signaling Pathway. J. Biol. Chem. 2004, 279, 17535–17542. [Google Scholar] [CrossRef]
- Caruso, A.; Motolese, M.; Iacovelli, L.; Caraci, F.; Copani, A.; Nicoletti, F.; Terstappen, G.C.; Gaviraghi, G.; Caricasole, A. Inhibition of the canonical Wnt signaling pathway by apolipoprotein E4 in PC12 cells. J. Neurochem. 2006, 98, 364–371. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher-Moser, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.G.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Jia, L.; Liu, C.-C.; Rong, Z.; Zhong, L.; Yang, L.; Chen, X.-F.; Fryer, J.D.; Wang, X.; Zhang, Y.-W.; et al. TREM2 Promotes Microglial Survival by Activating Wnt/β-Catenin Pathway. J. Neurosci. 2017, 37, 1772–1784. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Wang, X.; Li, Y.; Chen, P.-C.; Yu, K.; Dey, K.K.; Yarbro, J.M.; Han, X.; Lutz, B.M.; Rao, S.; et al. Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 2020, 105, 975–991.e7. [Google Scholar] [CrossRef] [PubMed]
- Elliott, C.; Rojo, A.I.; Ribe, E.; Broadstock, M.; Xia, W.; Morin, P.; Semenov, M.; Baillie, G.; Cuadrado, A.; Al-Shawi, R.; et al. A role for APP in Wnt signalling links synapse loss with β-amyloid production. Transl. Psychiatry 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Xu, J.; Patassini, S.; Rustogi, N.; Riba-Garcia, I.; Hale, B.D.; Phillips, A.M.; Waldvogel, H.; Haines, R.; Bradbury, P.; Stevens, A.; et al. Regional protein expression in human Alzheimer’s brain correlates with disease severity. Commun. Biol. 2019, 2, 1–15. [Google Scholar] [CrossRef]
- Nakamura, R.E.; Hunter, D.D.; Yi, H.; Brunken, W.J.; Hackam, A.S. Identification of two novel activities of the Wnt signaling regulator Dickkopf 3 and characterization of its expression in the mouse retina. BMC Cell Biol. 2007, 8, 52. [Google Scholar] [CrossRef]
- Yue, W.; Sun, Q.; Dacic, S.; Landreneau, R.J.; Siegfried, J.M.; Yu, J.; Zhang, L. Downregulation of Dkk3 activates β-catenin/TCF-4 signaling in lung cancer. Carcinogenesis 2008, 29, 84–92. [Google Scholar] [CrossRef]
- Van Der Kant, R.; Goldstein, L.S.B. Cellular Functions of the Amyloid Precursor Protein from Development to Dementia. Dev. Cell 2015, 32, 502–515. [Google Scholar] [CrossRef]
- Obrien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Hooli, B.; Tanzi, R.E. The Genetic Basis of Alzheimer’s Disease: Findings From Genome-Wide Studies. In Genomics, Circuits, and Pathways in Clinical Neuropsychiatry; Elsevier Inc.: Amsterdam, The Netherlands, 2016; pp. 547–571. [Google Scholar]
- Sleegers, K.; Brouwers, N.; Gijselinck, I.; Theuns, J.; Goossens, D.; Wauters, J.; Del-Favero, J.; Cruts, M.; Van Duijn, C.M.; Van Broeckhoven, C. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain 2006, 129, 2977–2983. [Google Scholar] [CrossRef]
- Rovelet-Lecrux, A.; Hannequin, D.; Raux, G.; Le Meur, N.; Laquerrière, A.; Vital, A.; Dumanchin, C.; Feuillette, S.; Brice, A.; Vercelletto, M.; et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 2006, 38, 24–26. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Saftig, P.; Craessaerts, K.; Vanderstichele, H.; Guhde, G.; Annaert, W.; Von Figura, K.; Van Leuven, F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nat. Cell Biol. 1998, 391, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Parr, C.J.; Birch, A.M.; Goldfinger, M.H.; Sastre, M. The amyloid precursor protein binds to β-catenin and modulates its cellular distribution. Neurosci. Lett. 2018, 685, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hartmann, H.; Do, V.M.; Abramowski, D.; Sturchler-Pierrat, C.; Staufenbiel, M.; Sommer, B.; Van De Wetering, M.; Clevers, H.; Saftig, P.; et al. Destabilization of β-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nat. Cell Biol. 1998, 395, 698–702. [Google Scholar] [CrossRef]
- Nishimura, M.; Yu, G.; Levesque, G.; Zhang, D.; Ruel, L.; Chen, F.; Milman, P.; Holmes, E.; Liang, Y.; Kawarai, T.; et al. Presenilin mutations associated with Alzheimer disease cause defective intracellular trafficking of β-catenin,a component of the presenilin protein complex. Nat. Med. 1999, 5, 164–169. [Google Scholar] [CrossRef]
- Kang, D.E.; Soriano, S.; Frosch, M.P.; Collins, T.; Naruse, S.; Sisodia, S.S.; Leibowitz, G.; Levine, F.; Koo, E.H. Presenilin 1 Facilitates the Constitutive Turnover of β-Catenin: Differential Activity of Alzheimer’s Disease–Linked PS1 Mutants in the β-Catenin–Signaling Pathway. J. Neurosci. 1999, 19, 4229–4237. [Google Scholar] [CrossRef]
- Teo, J.-L.; Ma, H.; Nguyen, C.; Lam, C.; Kahn, M. Specific inhibition of CBP/-catenin interaction rescues defects in neuronal differentiation caused by a presenilin-1 mutation. Proc. Natl. Acad. Sci. USA 2005, 102, 12171–12176. [Google Scholar] [CrossRef]
- Boonen, R.A.; Van Tijn, P.; Zivkovic, D. Wnt signaling in Alzheimer’s disease: Up or down, that is the question. Ageing Res. Rev. 2009, 8, 71–82. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Burgos, P.V.; Inestrosa, N.C. Inhibition of Wnt signaling induces amyloidogenic processing of amyloid precursor protein and the production and aggregation of Amyloid-β (Aβ)42peptides. J. Neurochem. 2016, 139, 1175–1191. [Google Scholar] [CrossRef]
- Parr, C.; Mirzaei, N.; Christian, M.; Sastre, M. Activation of the Wnt/β-catenin pathway represses the transcription of the β-amyloid precursor protein cleaving enzyme (BACE1) via binding of T-cell factor-4 to BACE1 promoter. FASEB J. 2014, 29, 623–635. [Google Scholar] [CrossRef]
- Vargas, J.Y.; Ahumada, J.; Arrázola, M.S.; Fuenzalida, M.; Inestrosa, N.C. WASP-1, a canonical Wnt signaling potentiator, rescues hippocampal synaptic impairments induced by Aβ oligomers. Exp. Neurol. 2015, 264, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Rojas, C.; Inestrosa, N.C. Wnt signaling loss accelerates the appearance of neuropathological hallmarks of Alzheimer’s disease in J20-APP transgenic and wild-type mice. J. Neurochem. 2018, 144, 443–465. [Google Scholar] [CrossRef] [PubMed]
- De Ferrari, G.V.; Chacon, M.A.; Barría, M.I.; Garrido, J.L.; Godoy, J.A.; Olivares, G.H.; Reyes, A.E.; Alvarez, A.R.; Bronfman, M.; Inestrosa, N.C. Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by β-amyloid fibrils. Mol. Psychiatry 2003, 8, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.R.; Godoy, J.A.; Mullendorff, K.; Olivares, G.H.; Bronfman, M.; Inestrosa, N.C. Wnt-3a overcomes β-amyloid toxicity in rat hippocampal neurons. Exp. Cell Res. 2004, 297, 186–196. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Muñoz, F.J.; Metcalfe, M.J.; Hitschfeld, M.; Olivares, G.; Godoy, J.A.; Inestrosa, N.C. Trolox and 17 -Estradiol Protect against Amyloid -Peptide Neurotoxicity by a Mechanism That Involves Modulation of the Wnt Signaling Pathway. J. Biol. Chem. 2005, 280, 11615–11625. [Google Scholar] [CrossRef]
- Magdesian, M.H.; Carvalho, M.M.V.F.; Mendes, F.A.; Saraiva, L.M.; Juliano, M.A.; Juliano, L.; Garcia-Abreu, J.; Ferreira, S.T. Amyloid-β Binds to the Extracellular Cysteine-rich Domain of Frizzled and Inhibits Wnt/β-Catenin Signaling. J. Biol. Chem. 2008, 283, 9359–9368. [Google Scholar] [CrossRef] [PubMed]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nat. Cell Biol. 1998, 393, 702–705. [Google Scholar] [CrossRef]
- Small, S.A.; Duff, K. Linking Aβ and Tau in Late-Onset Alzheimer’s Disease: A Dual Pathway Hypothesis. Neuron 2008, 60, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; Van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic Function of Tau Mediates Amyloid-β Toxicity in Alzheimer’s Disease Mouse Models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.-Q.; et al. Amyloid- /Fyn-Induced Synaptic, Network, and Cognitive Impairments Depend on Tau Levels in Multiple Mouse Models of Alzheimer’s Disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.-Q.; Mucke, L. Reducing Endogenous Tau Ameliorates Amyloid -Induced Deficits in an Alzheimer’s Disease Mouse Model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Pickett, E.K.; Herrmann, A.G.; McQueen, J.; Abt, K.; Dando, O.; Tulloch, J.; Jain, P.; Dunnett, S.; Sohrabi, S.; Fjeldstad, M.P.; et al. Amyloid Beta and Tau Cooperate to Cause Reversible Behavioral and Transcriptional Deficits in a Model of Alzheimer’s Disease. Cell Rep. 2019, 29, 3592–3604.e5. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Lee, V.M.-Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Weigand, A.J.; Bangen, K.J.; Thomas, K.R.; Delano-Wood, L.; Gilbert, P.E.; Brickman, A.M.; Bondi, M.W.; Initiative, A.D.N. Is tau in the absence of amyloid on the Alzheimer’s continuum?: A study of discordant PET positivity. Brain Commun. 2020, 2, fcz046. [Google Scholar] [CrossRef]
- Van Der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 2020, 21, 21–35. [Google Scholar] [CrossRef]
- Cavallini, A.; Brewerton, S.; Bell, A.; Sargent, S.; Glover, S.; Hardy, C.; Moore, R.; Calley, J.; Ramachandran, D.; Poidinger, M.; et al. An Unbiased Approach to Identifying Tau Kinases That Phosphorylate Tau at Sites Associated with Alzheimer Disease. J. Biol. Chem. 2013, 288, 23331–23347. [Google Scholar] [CrossRef]
- Asuni, A.A.; Hooper, C.; Reynolds, C.H.; Lovestone, S.; Anderton, B.H.; Killick, R. GSK3α exhibits β-catenin and tau directed kinase activities that are modulated by Wnt. Eur. J. Neurosci. 2006, 24, 3387–3392. [Google Scholar] [CrossRef] [PubMed]
- Salcedo-Tello, P.; Hernández-Ortega, K.; Arias, C. Susceptibility to GSK3β-Induced Tau Phosphorylation Differs Between the Young and Aged Hippocampus after Wnt Signaling Inhibition. J. Alzheimer’s Dis. 2014, 39, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Scali, C.; Caraci, F.; Gianfriddo, M.; Diodato, E.; Roncarati, R.; Pollio, G.; Gaviraghi, G.; Copani, A.; Nicoletti, F.; Terstappen, G.C.; et al. Inhibition of Wnt signaling, modulation of Tau phosphorylation and induction of neuronal cell death by DKK1. Neurobiol. Dis. 2006, 24, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Garrido, J.L.; Godoy, J.A.; Alvarez, A.; Bronfman, M.; Inestrosa, N.C. Protein kinase C inhibits amyloid β-peptide neurotoxicity by acting on members of the Wnt pathway. FASEB J. 2002, 16, 1982–1984. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Martín, M.; Jurado, J.; Hernández, F.; Ávila, J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A.; Murayama, M.; Murayama, O.; Kohno, T.; Honda, T.; Yasutake, K.; Nihonmatsu, N.; Mercken, M.; Yamaguchi, H.; Sugihara, S.; et al. Presenilin 1 associates with glycogen synthase kinase-3β and its substrate tau. Proc. Natl. Acad. Sci. USA 1998, 95, 9637–9641. [Google Scholar] [CrossRef]
- Cohen, T.J.; Friedmann, D.; Hwang, A.W.; Marmorstein, R.; Lee, V.M.Y. The microtubule-associated tau protein has intrinsic acetyltransferase activity. Nat. Struct. Mol. Biol. 2013, 20, 756–762. [Google Scholar] [CrossRef]
- Liu, E.; Zhou, Q.; Xie, A.; Li, X.; Li, M.; Ye, J.; Li, S.; Ke, D.; Wang, Q.; Xu, Z.; et al. Tau acetylates and stabilizes β-catenin thereby promoting cell survival. EMBO Rep. 2020, 21, e48328. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef]
- Rodriguez, J.L.; Karikari, T.K.; Suárez-Calvet, M.; Troakes, C.; King, A.; Emersic, A.; Aarsland, D.; Hye, A.; Zetterberg, H.; Blennow, K.; et al. Plasma p-tau181 accurately predicts Alzheimer’s disease pathology at least 8 years prior to post-mortem and improves the clinical characterisation of cognitive decline. Acta Neuropathol. 2020, 140, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.; Sabatini, B.L. Natural Oligomers of the Alzheimer Amyloid-Protein Induce Reversible Synapse Loss by Modulating an NMDA-Type Glutamate Receptor-Dependent Signaling Pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, A.S.; Velasco, P.T.; Wood, M.; Viola, K.L.; Klein, W.L. A Oligomer-Induced Aberrations in Synapse Composition, Shape, and Density Provide a Molecular Basis for Loss of Connectivity in Alzheimer’s Disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s Disease Is a Synaptic Failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Hsia, A.Y.; Masliah, E.; McConlogue, L.; Yu, G.-Q.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Malenka, R.C.; Nicoll, R.A.; Mucke, L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 1999, 96, 3228–3233. [Google Scholar] [CrossRef] [PubMed]
- Hermann, D.; Both, M.; Ebert, U.; Gross, G.; Schoemaker, H.; Draguhn, A.; Wicke, K.; Nimmrich, V. Synaptic transmission is impaired prior to plaque formation in amyloid precursor protein–overexpressing mice without altering behaviorally-correlated sharp wave–ripple complexes. Neuroscience 2009, 162, 1081–1090. [Google Scholar] [CrossRef]
- Cerpa, W.; Farías, G.G.; Godoy, J.A.; Fuenzalida, M.; Bonansco, C.; Inestrosa, N.C. Wnt-5a occludes Aβ oligomer-induced depression of glutamatergic transmission in hippocampal neurons. Mol. Neurodegener. 2010, 5, 3. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Mufson, E.J. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; Bs, R.D.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- Forner, S.; Baglietto-Vargas, D.; Martini, A.C.; Trujillo-Estrada, L.; LaFerla, F.M. Synaptic Impairment in Alzheimer’s Disease: A Dysregulated Symphony. Trends Neurosci. 2017, 40, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Killick, R.; Ribe, E.M.; Alshawi, R.; Malik, B.; Hooper, C.; Fernandes, C.P.D.; Dobson, R.; Nolan, P.M.; Lourdusamy, A.; Furney, S.J.; et al. Clusterin regulates β-amyloid toxicity via Dickkopf-1-driven induction of the wnt–PCP–JNK pathway. Mol. Psychiatry 2014, 19, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Rose, J.; Tulloch, J.; Henstridge, C.; Smith, C.; Spires-Jones, T.L. Clusterin accumulates in synapses in Alzheimer’s disease and is increased in apolipoprotein E4 carriers. Brain Commun. 2019, 1, fcz003. [Google Scholar] [CrossRef]
- Galli, S.; Lopes, D.M.; Ammari, R.; Kopra, J.; Millar, S.E.; Gibb, A.; Salinas, P.C. Deficient Wnt signalling triggers striatal synaptic degeneration and impaired motor behaviour in adult mice. Nat. Commun. 2014, 5, 4992. [Google Scholar] [CrossRef]
- Mao, B.; Wu, W.; Li, Y.; Hoppe, D.; Stannek, P.; Glinka, A.; Niehrs, C. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nat. Cell Biol. 2001, 411, 321–325. [Google Scholar] [CrossRef]
- Bafico, A.; Liu, G.; Yaniv, A.; Gazit, A.; Aaronson, S.A. Novel mechanism of Wnt signalling inhibition mediated by Dickkopf-1 interaction with LRP6/Arrow. Nat. Cell Biol. 2001, 3, 683–686. [Google Scholar] [CrossRef]
- Ross, S.P.; Baker, K.E.; Fisher, A.; Hoff, L.; Pak, E.S.; Murashov, A.K. miRNA-431 Prevents Amyloid-β-Induced Synapse Loss in Neuronal Cell Culture Model of Alzheimer’s Disease by Silencing Kremen1. Front. Cell. Neurosci. 2018, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Sellers, K.J.; Elliott, C.; Jackson, J.; Ghosh, A.; Ribe, E.; Rojo, A.I.; Jarosz-Griffiths, H.H.; Watson, I.A.; Xia, W.; Semenov, M.; et al. Amyloid β synaptotoxicity is Wnt-PCP dependent and blocked by fasudil. Alzheimer’s Dement. 2018, 14, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Siskova, Z.; Tremblay, M.-È. Microglia and Synapse: Interactions in Health and Neurodegeneration. Neural Plast. 2013, 2013, 425845. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, E.M.; Initiative, T.A.D.N.; Chibnik, L.B.; Keenan, B.T.; Ottoboni, L.; Raj, T.; Tang, A.; Rosenkrantz, L.L.; Imboywa, S.; Lee, M.; et al. CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat. Neurosci. 2013, 16, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Hyman, B.T.; Spires-Jones, T.L. Beyond the neuron–cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2019, 20, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Frigerio, C.S.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.-T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019, 27, 1293–1306.e6. [Google Scholar] [CrossRef]
- Meilandt, W.J.; Ngu, H.; Gogineni, A.; Lalehzadeh, G.; Lee, S.-H.; Srinivasan, K.; Imperio, J.; Wu, T.; Weber, M.; Kruse, A.J.; et al. Trem2 Deletion Reduces Late-Stage Amyloid Plaque Accumulation, Elevates the Aβ42:Aβ40 Ratio, and Exacerbates Axonal Dystrophy and Dendritic Spine Loss in the PS2APP Alzheimer’s Mouse Model. J. Neurosci. 2020, 40, 1956–1974. [Google Scholar] [CrossRef]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef]
- Paresce, D.M.; Ghosh, R.N.; Maxfield, F.R. Microglial Cells Internalize Aggregates of the Alzheimer’s Disease Amyloid β-Protein Via a Scavenger Receptor. Neuron 1996, 17, 553–565. [Google Scholar] [CrossRef]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A Cell Surface Receptor Complex for Fibrillar β-Amyloid Mediates Microglial Activation. J. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.-L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e7. [Google Scholar] [CrossRef]
- Okun, E.; Griffioen, K.J.; Lathia, J.D.; Tang, S.-C.; Mattson, M.P.; Arumugam, T.V. Toll-like receptors in neurodegeneration. Brain Res. Rev. 2009, 59, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Yuan, P.; Schain, A.; Grutzendler, J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 2015, 6, 7176. [Google Scholar] [CrossRef]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.P.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain 2016, 139, 1265–1281. [Google Scholar] [CrossRef] [PubMed]
- Grathwohl, S.A.; Kälin, R.E.; Bolmont, T.; Prokop, S.; Winkelmann, G.; Kaeser, S.A.; Odenthal, J.; Radde, R.; Eldh, T.; Gandy, S.; et al. Formation and maintenance of Alzheimer’s disease β-amyloid plaques in the absence of microglia. Nat. Neurosci. 2009, 12, 1361–1363. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Hu, W.; Tsai, J.; Li, W.; Gan, W.-B. Microglia limit the expansion of β-amyloid plaques in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 1–11. [Google Scholar] [CrossRef]
- Czirr, E.; Castello, N.A.; Mosher, K.I.; Castellano, J.M.; Hinkson, I.V.; Lucin, K.M.; Baeza-Raja, B.; Ryu, J.K.; Li, L.; Farina, S.N.; et al. Microglial complement receptor 3 regulates brain Aβ levels through secreted proteolytic activity. J. Exp. Med. 2017, 214, 1081–1092. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Rajendran, L.; Paolicelli, R.C. Microglia-Mediated Synapse Loss in Alzheimer’s Disease. J. Neurosci. 2018, 38, 2911–2919. [Google Scholar] [CrossRef]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/β-Catenin and NF-κB Signaling Pathway during Inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef]
- Zolezzi, J.M.; Inestrosa, N.C. Wnt/TLR Dialog in Neuroinflammation, Relevance in Alzheimer’s Disease. Front. Immunol. 2017, 8, 187. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Y.; Hao, W.; Wolf, L.; Kiliaan, A.J.; Penke, B.; Rübe, C.E.; Walter, J.; Heneka, M.T.; Hartmann, T.; et al. TLR2 Is a Primary Receptor for Alzheimer’s Amyloid β Peptide To Trigger Neuroinflammatory Activation. J. Immunol. 2011, 188, 1098–1107. [Google Scholar] [CrossRef]
- Reed-Geaghan, E.G.; Savage, J.C.; Hise, A.G.; Landreth, G.E. CD14 and Toll-Like Receptors 2 and 4 Are Required for Fibrillar A -Stimulated Microglial Activation. J. Neurosci. 2009, 29, 11982–11992. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Fey, M.; Hottiger, M.O. WNT/β-catenin signaling inhibits CBP-mediated RelA acetylation and expression of proinflammatory NF-κB target genes. J. Cell Sci. 2015, 128, 2430–2436. [Google Scholar] [CrossRef] [PubMed]
- Noubissi, F.K.; Elcheva, I.; Bhatia, N.; Shakoori, A.; Ougolkov, A.; Liu, J.; Minamoto, T.; Ross, J.; Fuchs, S.Y.; Spiegelman, V.S. CRD-BP mediates stabilization of βTrCP1 and c-myc mRNA in response to β-catenin signalling. Nat. Cell Biol. 2006, 441, 898–901. [Google Scholar] [CrossRef] [PubMed]
- Van Steenwinckel, J.; Schang, A.-L.; Krishnan, M.L.; Degos, V.; Delahaye-Duriez, A.; Bokobza, C.; Csaba, Z.; Verdonk, F.; Montané, A.; Sigaut, S.; et al. Decreased microglial Wnt/β-catenin signalling drives microglial pro-inflammatory activation in the developing brain. Brain 2019, 142, 3806–3833. [Google Scholar] [CrossRef]
- Zhang, D.; Lu, Z.; Man, J.; Cui, K.; Fu, X.; Yu, L.; Gao, Y.; Liao, L.; Xiao, Q.; Guo, R.; et al. Wnt-3a alleviates neuroinflammation after ischemic stroke by modulating the responses of microglia/macrophages and astrocytes. Int. Immunopharmacol. 2019, 75, 105760. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Johnson, A.M.; Keep, R.F.; Andjelkovic, A.V. Junctional proteins of the blood-brain barrier: New insights into function and dysfunction. Tissue Barriers 2016, 4, e1154641. [Google Scholar] [CrossRef]
- Liu, L.; Wan, W.; Xia, S.; Kalionis, B.; Li, Y. Dysfunctional Wnt/β-catenin signaling contributes to blood–brain barrier breakdown in Alzheimer’s disease. Neurochem. Int. 2014, 75, 19–25. [Google Scholar] [CrossRef]
- Schinkel, A.H. P-Glycoprotein, a gatekeeper in the blood–brain barrier. Adv. Drug Deliv. Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef]
- Galochkina, T.; Chong, M.N.F.; Challali, L.; Abbar, S.; Etchebest, C. New insights into GluT1 mechanics during glucose transfer. Sci. Rep. 2019, 9, 998. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood–brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 2018, 21, 1318–1331. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Agalliu, D.; Zhou, L.; Kuhnert, F.; Kuo, C.J.; Barres, B.A. Wnt/-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 641–646. [Google Scholar] [CrossRef]
- Stenman, J.M.; Rajagopal, J.; Carroll, T.J.; Ishibashi, M.; McMahon, J.; McMahon, A.P. Canonical Wnt Signaling Regulates Organ-Specific Assembly and Differentiation of CNS Vasculature. Science 2008, 322, 1247–1250. [Google Scholar] [CrossRef]
- Liebner, S.; Corada, M.; Bangsow, T.; Babbage, J.; Taddei, A.; Czupalla, C.J.; Reis, M.; Felici, A.; Wolburg, H.; Fruttiger, M.; et al. Wnt/β-catenin signaling controls development of the blood–brain barrier. J. Cell Biol. 2008, 183, 409–417. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Y.; Tischfield, M.; Williams, J.; Smallwood, P.M.; Rattner, A.; Taketo, M.M.; Nathans, J. Canonical WNT signaling components in vascular development and barrier formation. J. Clin. Investig. 2014, 124, 3825–3846. [Google Scholar] [CrossRef]
- Zhou, Y.; Nathans, J. Gpr124 Controls CNS Angiogenesis and Blood-Brain Barrier Integrity by Promoting Ligand-Specific Canonical Wnt Signaling. Dev. Cell 2014, 31, 248–256. [Google Scholar] [CrossRef]
- Cho, C.; Wang, Y.; Smallwood, P.M.; Williams, J.; Nathans, J. Molecular determinants in Frizzled, Reck, and Wnt7a for ligand-specific signaling in neurovascular development. eLife 2019, 8, e47300. [Google Scholar] [CrossRef]
- Cho, C.; Smallwood, P.M.; Nathans, J. Reck and Gpr124 Are Essential Receptor Cofactors for Wnt7a/Wnt7b-Specific Signaling in Mammalian CNS Angiogenesis and Blood-Brain Barrier Regulation. Neuron 2017, 95, 1056–1073.e5. [Google Scholar] [CrossRef]
- Paolinelli, R.; Corada, M.; Ferrarini, L.; Devraj, K.; Artus, C.; Czupalla, C.J.; Rudini, N.; Maddaluno, L.; Papa, E.; Engelhardt, B.; et al. Wnt Activation of Immortalized Brain Endothelial Cells as a Tool for Generating a Standardized Model of the Blood Brain Barrier In Vitro. PLoS ONE 2013, 8, e70233. [Google Scholar] [CrossRef] [PubMed]
- Hübner, K.; Cabochette, P.; Diéguez-Hurtado, R.; Wiesner, C.; Wakayama, Y.; Grassme, K.S.; Hubert, M.; Guenther, S.; Belting, H.-G.; Affolter, M.; et al. Wnt/β-catenin signaling regulates VE-cadherin-mediated anastomosis of brain capillaries by counteracting S1pr1 signaling. Nat. Commun. 2018, 9, 4860. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.C.; Kania, K.D.; Wijesuriya, H.; Chawla, S.; Sethi, J.K.; Pulaski, L.; Romero, I.A.; Couraud, P.O.; Weksler, B.B.; Hladky, S.B.; et al. Activation of -catenin signalling by GSK-3 inhibition increases P-glycoprotein expression in brain endothelial cells. J. Neurochem. 2008, 106, 1855–1865. [Google Scholar] [CrossRef] [PubMed]
- Laksitorini, M.D.; Yathindranath, V.; Xiong, W.; Hombach-Klonisch, S.; Miller, D.W. Modulation of Wnt/β-catenin signaling promotes blood-brain barrier phenotype in cultured brain endothelial cells. Sci. Rep. 2019, 9, 4860. [Google Scholar] [CrossRef] [PubMed]
- Pinzón-Daza, M.L.; Salaroglio, I.C.; Kopecka, J.; Garzòn, R.; Couraud, P.-O.; Ghigo, D.; Riganti, C. The Cross-Talk between Canonical and Non-Canonical Wnt-Dependent Pathways Regulates P-Glycoprotein Expression in Human Blood–Brain Barrier Cells. Br. J. Pharmacol. 2014, 34, 1258–1269. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.A.; Zhang, X.; Predescu, D.; Huang, X.; Machado, R.F.; Göthert, J.R.; Malik, A.B.; Valyi-Nagy, T.; Zhao, Y.-Y. Endothelial β-Catenin Signaling Is Required for Maintaining Adult Blood–Brain Barrier Integrity and Central Nervous System Homeostasis. Circulation 2016, 133, 177–186. [Google Scholar] [CrossRef]
- Wang, Y.; Cho, C.; Williams, J.; Smallwood, P.M.; Zhang, C.; Junge, H.J.; Nathans, J. Interplay of the Norrin and Wnt7a/Wnt7b signaling systems in blood–brain barrier and blood–retina barrier development and maintenance. Proc. Natl. Acad. Sci. USA 2018, 115, E11827–E11836. [Google Scholar] [CrossRef]
- Kania, K.D.; Wijesuriya, H.C.; Hladky, S.B.; Barrand, M.A. Beta amyloid effects on expression of multidrug efflux transporters in brain endothelial cells. Brain Res. 2011, 1418, 1–11. [Google Scholar] [CrossRef]
- Wijesuriya, H.C.; Bullock, J.Y.; Faull, R.L.; Hladky, S.B.; Barrand, M.A. ABC efflux transporters in brain vasculature of Alzheimer’s subjects. Brain Res. 2010, 1358, 228–238. [Google Scholar] [CrossRef]
- Lim, J.C.; Mickute, Z.; Zaman, M.; Hopkins, S.; Wijesuriya, H.; Steckler, T.; Moechars, D.; Van Leuven, F.; Sarnyai, Z.; Hladky, S.B.; et al. Decreased expression of multidrug efflux transporters in the brains of GSK-3β transgenic mice. Brain Res. 2009, 1276, 1–10. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid- deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2005, 115, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.C.; Tavares, R.; Johanson, C.E.; Hovanesian, V.; Donahue, J.E.; Gonzalez, L.; Silverberg, G.D.; Stopa, E.G. Hippocampal RAGE immunoreactivity in early and advanced Alzheimer’s disease. Brain Res. 2008, 1230, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.S.; Miller, D.S.; Bauer, B. Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer’s disease. Mol. Pharmacol. 2010, 77, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.; Chung, H.; Shah, G. GLUT-1 Expression in the Cerebra of Patients with Alzheimer’s Disease. Neurobiol. Aging 1997, 18, 469–474. [Google Scholar] [CrossRef]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Mu, Q.; Chen, Y.; Wang, J. Deciphering Brain Complexity Using Single-cell Sequencing. Genom. Proteom. Bioinform. 2019, 17, 344–366. [Google Scholar] [CrossRef]
- Tay, L.; Leung, B.; Yeo, A.; Chan, M.; Lim, W.S. Elevations in Serum Dickkopf-1 and Disease Progression in Community-Dwelling Older Adults With Mild Cognitive Impairment and Mild-to-Moderate Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 278. [Google Scholar] [CrossRef]
- Zhu, Z.; Guo, D.; Zhong, C.; Wang, A.; Xie, X.; Xu, T.; Chen, C.-S.; Peng, Y.; Peng, H.; Li, Q.; et al. Serum Dkk-1 (Dickkopf-1) Is a Potential Biomarker in the Prediction of Clinical Outcomes Among Patients With Acute Ischemic Stroke. Arter. Thromb. Vasc. Biol. 2019, 39, 285–293. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Fratila, O.; Brata, R.; Bungau, S. Exploring the Potential of Therapeutic Agents Targeted towards Mitigating the Events Associated with Amyloid-β Cascade in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7443. [Google Scholar] [CrossRef] [PubMed]
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef] [PubMed]
- Hosseinpour, S.; Rad, M.R.; Khojasteh, A.; Zadeh, H. Antibody Administration for Bone Tissue Engineering: A Systematic Review. Curr. Stem Cell Res. Ther. 2018, 13, 292–315. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Rong, D.; Wang, Y. Targeting Protein-Protein Interaction with Covalent Small-Molecule Inhibitors. Curr. Top. Med. Chem. 2019, 19, 1872–1876. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.C.; Lobo, D.D.; Martins, I.M.; Lopes, S.M.; Henriques, C.; Duarte, S.P.; Dodart, J.-C.; Nobre, R.; De Almeida, L.P. Antisense oligonucleotide therapeutics in neurodegenerative diseases: The case of polyglutamine disorders. Brain 2019, 143, 407–429. [Google Scholar] [CrossRef]
- Lewcock, J.W.; Schlepckow, K.; Di Paolo, G.; Tahirovic, S.; Monroe, K.M.; Haass, C. Emerging Microglia Biology Defines Novel Therapeutic Approaches for Alzheimer’s Disease. Neuron 2020, 6273, 30753–30754. [Google Scholar] [CrossRef]
- Lengfeld, J.E.; Lutz, S.E.; Smith, J.R.; Diaconu, C.; Scott, C.; Kofman, S.B.; Choi, C.; Walsh, C.M.; Raine, C.S.; Agalliu, I.; et al. Endothelial Wnt/β-catenin signaling reduces immune cell infiltration in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1168–E1177. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, N.J.; Menet, R.; Picard, K.; Parent, G.; Tremblay, M.-È.; ElAli, A. Canonical Wnt Pathway Maintains Blood-Brain Barrier Integrity upon Ischemic Stroke and Its Activation Ameliorates Tissue Plasminogen Activator Therapy. Mol. Neurobiol. 2019, 56, 6521–6538. [Google Scholar] [CrossRef]
- Salehi, A.; Jullienne, A.; Baghchechi, M.; Hamer, M.; Walsworth, M.; Donovan, V.; Tang, J.; Zhang, J.H.; Pearce, W.J.; Obenaus, A. Up-regulation of Wnt/β-catenin expression is accompanied with vascular repair after traumatic brain injury. Br. J. Pharmacol. 2017, 38, 274–289. [Google Scholar] [CrossRef]
- Wold, E.A.; Chen, J.; Cunningham, K.A.; Zhou, J. Allosteric Modulation of Class A GPCRs: Targets, Agents, and Emerging Concepts. J. Med. Chem. 2019, 62, 88–127. [Google Scholar] [CrossRef]
- Molinuevo, J.L.; Ayton, S.; Batrla, R.; Bednar, M.M.; Bittner, T.; Cummings, J.; Fagan, A.M.; Hampel, H.; Mielke, M.M.; Mikulskis, A.; et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018, 136, 821–853. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Han, S.M.; Kim, C.E. New fluid biomarkers tracking non-amyloid-β and non-tau pathology in Alzheimer’s disease. Exp. Mol. Med. 2020, 52, 556–568. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Reference | Type of Manipulation | Experimental Model | Experimental Outcome |
|---|---|---|---|
| Parr et al., 2014 [153] | Wnt3a, β-catenin over-expression | N2Asw murine neuroblastoma cell line | ↓ Aβ levels, ↓BACE1 levels |
| Alvarez et al., 2004 [157] | Wnt3a treatment | Rat primary hippocampal neurons (treated with Aβ) | ↑ neuronal survival, ↓ GSK-3β phosphorylation, ↓ tau phosphorylation, restored cytosolic β-catenin levels, ↑ increased expression of Wnt target gene engrailed-1 |
| Zhang et al., 2019 [230] | Wnt3a | Ischemic stroke (tMCAO) mouse model (in vivo) | ↓ infarct volume, ↓ neurologic deficits, ↓ peri-infarct apoptosis, ↓ microglial activation, ↓ pro-inflammatory cytokine secretion (iNos, TNFα), ↓ astrogliosis |
| Zheng et al., 2017 [134] | Wnt3a, LiCl, TDZD-8 | Trem2-/- mouse primary microglia | ↑ β-catenin levels, ↑ microglial survival, ↑ microglial proliferation |
| Cerpa et al., 2010 [190] | Wnt5a | Rat primary hippocampal neurons and hippocampal slices (treated with Aβ) | ↑ fEPSP & (AMPAR, NMDAR) EPSC amplitudes, ↑ post-synaptic PSD-95 clusters |
| Marzo et al., 2016 [111] | 6-BIO (GSK-3β inhibitor) | Rat primary hippocampal neurons (treated with Dkk1) | ↑ excitatory synapse numbers |
| Salcedo-Tello et al., 2014 [175] | 6-BIO | Rat hippocampal slices at 3 months (young) and 18–20 months (aged) | ↓ GSK-3β activity, ↑ β-catenin levels |
| Quintanilla et al., 2005 [158] | 17β-estradiol, Trolox (antioxidant) | Rat primary hippocampal neurons (treated with Aβ) | ↑ neuronal survival, ↓ endoperoxide production, ↓ GSK-3β activity, ↑ cytoplasmic β-catenin, ↑ Wnt5a & Wnt7a levels (17β-estradiol), ↓ Ca2+ mediated toxicity |
| Wnt7a | protection against Ca2+ mediated toxicity | ||
| Magdesian et al., 2008 [159] | cSP5 (Aβ-binding peptide) | N2Asmurine neuroblastoma cell line (differentiated) | ↑ total & nuclear β-catenin |
| Elliott et al., 2018 [136] | Fasudil (Rock inhibitor) | Rat primary cortical neurons (treated with Dkk1) | Protection against Dkk1-mediated dendritic spine loss, and Aβ production |
| 3xTg AD mice (in vivo) | ↓ soluble and insoluble Aβ | ||
| Vargas et al., 2014 [187] | FOXY-5 (peptide) | APP/PS1 mouse model (in vivo & hippocampal slices) | Rescued memory retention, ↑ fEPSP, rescued LTP deficit |
| Van Steenwinckel et al., 2019 [229] | L803mts (Wnt/β-catenin activator; targeted to microglia by 3DNA) | Mouse developmental encephalopathy model (IL-1β-mediated; in vivo) | ↓ microglial expression of pro-inflammatory cytokines (iNos, Cox2), restored myelination, restored short- & long-term memory retention |
| De Ferrari et al., 2003 [156] | LiCl (GSK-3β inhibitor), Wnt3a, PMA (PKC activator) | Rat primary hippocampal neurons (treated with Aβ) | ↑ neuronal survival, restored cytosolic β-catenin levels |
| LiCl | Intrahippocampal injection of Aβ fibrils in rats (in vivo) | ↑ neuronal survival, improved deficit in spatial learning (Morris water maze) induced by Aβ | |
| Scali et al., 2006 [176] | LiCl | Rats (3 months) injected with recombinant human DKK1 (in vivo) | ↓ DKK1-stimulated GSK-3β activity, protects against DKK1-mediated neurodegeneration, tau hyperphosphorylation, and astrogliosis |
| Ross et al., 2018 [200] | miRNA-431 (silences Dkk1 receptor Krm1) | Mouse primary cortico-hippocampal neurons from 3xTg AD mouse model (in vivo) | ↓ Aβ- and Dkk1-mediated synapse loss, ↓ neurite degeneration |
| Vargas et al., 2015 [154] | WASP-1 small molecule | Rat primary hippocampal neurons | ↑ synaptic transmission, rescued hippocampal LTP impairments due to Aβ |
| APP/PS1 mouse model (in vivo) | ↓ synaptic protein loss, ↓ tau phosphorylation | ||
| Y27632 (Rock inhibitor) | Rat primary hippocampal neurons (treated with Dkk1) | ↑ excitatory synapse numbers | |
| Sellers el al., 2018 [201] | Y27632, Fasudil | Rat primary cortical neurons (treated with Aβ or Dkk1) | Protection against Aβ- or Dkk1- mediated spine density reduction, ↑ PSD-95 puncta, ↑ GluA1 puncta |
| Fasudil | Rats (treated with Aβ; in vivo) | Protection against Aβ-driven cognitive impairment, ↑ performance in NOR task | |
| LiCl | Trem2-/- mice (in vivo) | ↑ microglia cell # in hippocampus and cortex, ↑ β-catenin, ↓ GSK-3β activation, ↑ cell cycle markers (c-Myc, Cyclin D1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aghaizu, N.D.; Jin, H.; Whiting, P.J. Dysregulated Wnt Signalling in the Alzheimer’s Brain. Brain Sci. 2020, 10, 902. https://doi.org/10.3390/brainsci10120902
Aghaizu ND, Jin H, Whiting PJ. Dysregulated Wnt Signalling in the Alzheimer’s Brain. Brain Sciences. 2020; 10(12):902. https://doi.org/10.3390/brainsci10120902
Chicago/Turabian StyleAghaizu, Nozie D., Hanqing Jin, and Paul J. Whiting. 2020. "Dysregulated Wnt Signalling in the Alzheimer’s Brain" Brain Sciences 10, no. 12: 902. https://doi.org/10.3390/brainsci10120902
APA StyleAghaizu, N. D., Jin, H., & Whiting, P. J. (2020). Dysregulated Wnt Signalling in the Alzheimer’s Brain. Brain Sciences, 10(12), 902. https://doi.org/10.3390/brainsci10120902