Fatal Status Epilepticus in Dravet Syndrome

 ,
,
Abstract
1. Introduction
2. Materials and Methods
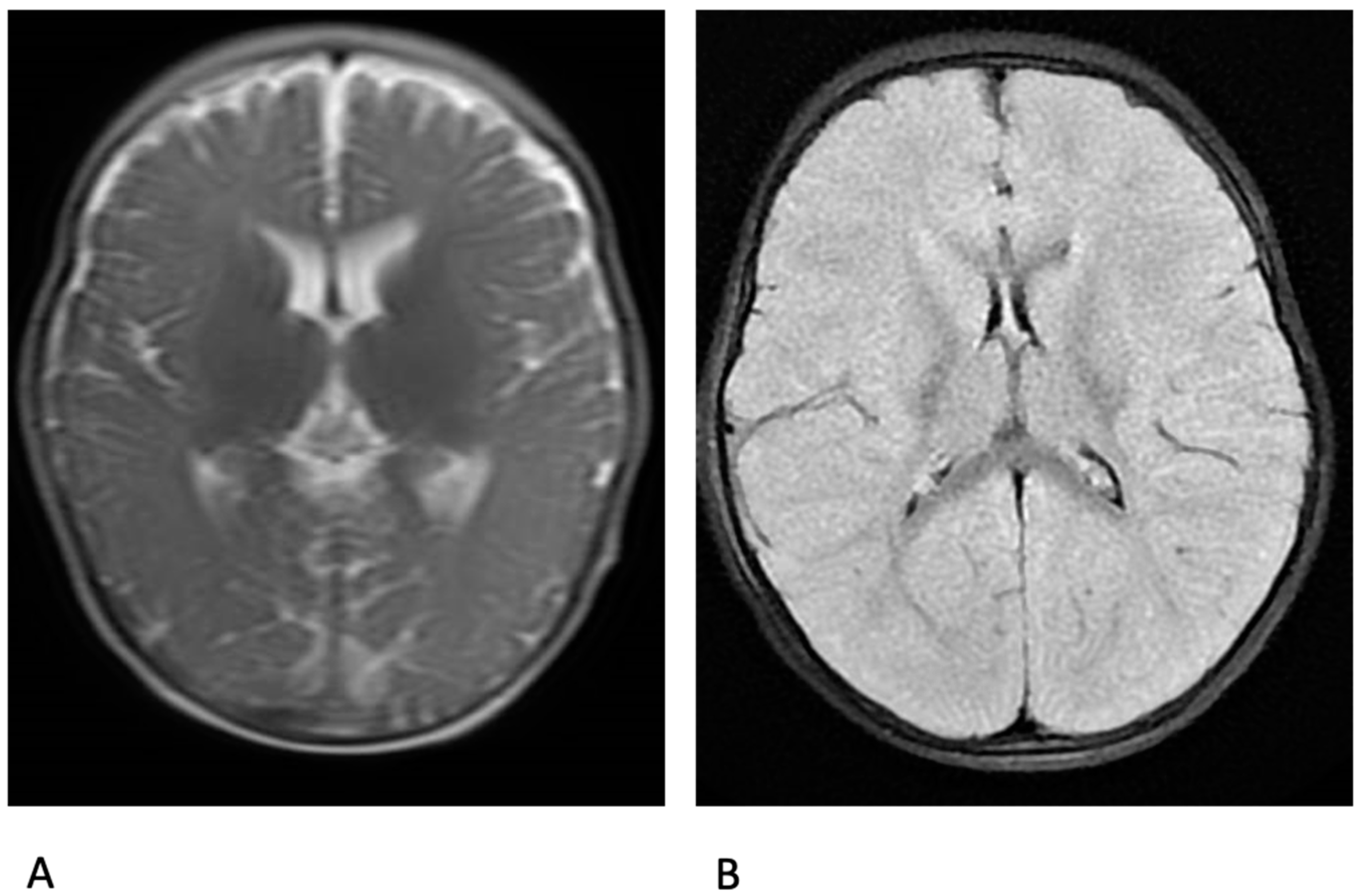
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AE | acute encephalopathy |
| ASMs | anti-seizure medications |
| DEE | developmental and epileptic encephalopathy |
| DS | Dravet syndrome |
| EpiCARE | European Reference Network on Rare and Complex Epilepsies |
| GEFS+ | generalized epilepsy with febrile seizures plus |
| GCSE | generalized convulsive status epileptic |
| MRI | magnetic resonance imaging |
| SE | status epilepticus |
| SUDEP | sudden unexpected death in epilepsy |
References
- Dravet, C.; Oguni, H.; Cokar, O.; Guerrini, R. Dravet Syndrome (Previously Severe Myoclonic Epilepsy in Infancy). In Epileptic Syndromes in Infancy, Childhood and Adolescence; John Libbey Eurotext Ltd.: Montrouge, France, 2019; pp. 139–172. [Google Scholar]
- Sakauchi, M.; Oguni, H.; Kato, I.; Osawa, M.; Hirose, S.; Kaneko, S.; Takahashi, Y.; Takayama, R.; Fujiwara, T. Mortality in Dravet syndrome: Search for risk factors in Japanese patients. Epilepsia 2011, 52 (Suppl. 2), 50–54. [Google Scholar] [CrossRef]
- Shmuely, S.; Sisodiya, S.M.; Gunning, W.B.; Sander, J.W.; Thijs, R.D. Mortality in Dravet syndrome: A review. Epilepsy Behav. 2016, 64, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Okumura, A.; Uematsu, M.; Imataka, G.; Tanaka, M.; Okanishi, T.; Kubota, T.; Sudo, A.; Tohyama, J.; Tsuji, M.; Ohmori, I.; et al. Acute encephalopathy in children with Dravet syndrome. Epilepsia 2012, 53, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Chipaux, M.; Villeneuve, N.; Sabouraud, P.; Desguerre, I.; Boddaert, N.; Depienne, C.; Chiron, C.; Dulac, O.; Nabbout, R. Unusual consequences of status epilepticus in Dravet syndrome. Seizure 2010, 19, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.A.; McMahon, J.M.; Mandelstam, S.A.; Mackay, M.T.; Kalnins, R.M.; Leventer, R.J.; Scheffer, I.E. Fatal Cerebral Edema With Status Epilepticus in Children With Dravet Syndrome: Report of 5 Cases. Pediatrics 2017, 139, e20161933. [Google Scholar] [CrossRef]
- Tian, X.; Ye, J.; Zeng, Q.; Zhang, J.; Yang, X.; Liu, A.; Yang, Z.; Liu, X.; Wu, X.; Zhang, Y. The clinical outcome and neuroimaging of acute encephalopathy after status epilepticus in Dravet syndrome. Dev. Med. Child. Neurol. 2018, 60, 566–573. [Google Scholar] [CrossRef]
- Do, H.; Huynh, T.T.K.; Le, T.K. Acute encephalopathy in Dravet syndrome: Case reports and literature review. Neurol. Asia 2016, 21, 181–185. [Google Scholar]
- Büren, C.; Kamp, M.A.; Munoz-Bendix, C.; Steiger, H.-J.; Windolf, J.; Dibué-Adjei, M. Can the combination of hyperthermia, seizures and ion channel dysfunction cause fatal post-ictal cerebral edema in patients with SCN1A mutations? Epilepsy Behav. Case Rep. 2018, 9, 29–32. [Google Scholar] [CrossRef]
- Catarino, C.B.; Liu, J.Y.W.; Liagkouras, I.; Gibbons, V.S.; Labrum, R.W.; Ellis, R.; Woodward, C.; Davis, M.B.; Smith, S.J.; Cross, J.H.; et al. Dravet syndrome as epileptic encephalopathy: Evidence from long-term course and neuropathology. Brain 2011, 134, 2982–3010. [Google Scholar] [CrossRef]
- Genton, P.; Velizarova, R.; Dravet, C. Dravet syndrome: The long-term outcome. Epilepsia 2011, 52 (Suppl. 2), 44–49. [Google Scholar] [CrossRef]
- Skluzacek, J.V.; Watts, K.P.; Parsy, O.; Wical, B.; Camfield, P. Dravet syndrome and parent associations: The IDEA League experience with comorbid conditions, mortality, management, adaptation, and grief. Epilepsia 2011, 52 (Suppl. 2), 95–101. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.S.; Mcintosh, A.; Crompton, D.E.; McMahon, J.M.; Schneider, A.; Farrell, K.; Ganesan, V.; Gill, D.; Kivity, S.; Lerman-Sagie, T.; et al. Mortality in Dravet syndrome. Epilepsy Res. 2016, 128, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Oguni, H.; Hayashi, K.; Awaya, Y.; Fukuyama, Y.; Osawa, M. Severe myoclonic epilepsy in infants--a review based on the Tokyo Women’s Medical University series of 84 cases. Brain Dev. 2001, 23, 736–748. [Google Scholar] [CrossRef]
- Akiyama, M.; Kobayashi, K.; Yoshinaga, H.; Ohtsuka, Y. A long-term follow-up study of Dravet syndrome up to adulthood. Epilepsia 2010, 51, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Dravet, C.; Bureau, M.; Guerrini, R.; Giraud, N.; Roger, J. Severe Myoclonic Epilepsy in Infancy. In Epileptic Syndromes in Infancy, Childhood and Adolescence; John Libbey Eurotext Ltd.: Montrouge, France, 1992; pp. 75–88. [Google Scholar]
- Lee, J.H.; Cui, H.S.; Shin, S.K.; Kim, J.M.; Kim, S.Y.; Lee, J.E.; Koo, B.-N. Effect of propofol post-treatment on blood-brain barrier integrity and cerebral edema after transient cerebral ischemia in rats. Neurochem. Res. 2013, 38, 2276–2286. [Google Scholar] [CrossRef]
- De Liso, P.; Chemaly, N.; Laschet, J.; Barnerias, C.; Hully, M.; Leunen, D.; Desguerre, I.; Chiron, C.; Dulac, O.; Nabbout, R. Patients with dravet syndrome in the era of stiripentol: A French cohort cross-sectional study. Epilepsy Res. 2016, 125, 42–46. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Yamanouchi, H.; Ichiyama, T.; Shiomi, M. Acute encephalopathy associated with influenza and other viral infections. Acta Neurol. Scand. Suppl. 2007, 186, 45–56. [Google Scholar] [CrossRef]
- Berkovic, S.F.; Harkin, L.; McMahon, J.M.; Pelekanos, J.T.; Zuberi, S.M.; Wirrell, E.C.; Gill, D.S.; Iona, X.; Mulley, J.C.; Scheffer, I.E. De-novo mutations of the sodium channel gene SCN1A in alleged vaccine encephalopathy: A retrospective study. Lancet Neurol. 2006, 5, 488–492. [Google Scholar] [CrossRef]
- McIntosh, A.M.; McMahon, J.; Dibbens, L.M.; Iona, X.; Mulley, J.C.; Scheffer, I.E.; Berkovic, S.F. Effects of vaccination on onset and outcome of Dravet syndrome: A retrospective study. Lancet Neurol. 2010, 9, 592–598. [Google Scholar] [CrossRef]
- Tsuji, M.; Mazaki, E.; Ogiwara, I.; Wada, T.; Iai, M.; Okumura, A.; Yamashita, S.; Yamakawa, K.; Osaka, H. Acute encephalopathy in a patient with Dravet syndrome. Neuropediatrics 2011, 42, 78–81. [Google Scholar] [CrossRef]
- Deng, L.; Ma, A.; Wood, N.; Ardern-Holmes, S. Vaccination management in an asymptomatic child with a novel SCN1A variant and family history of status epilepticus following vaccination: A case report on a potential new direction in personalised medicine. Seizure 2020, 78, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Trivisano, M.; Specchio, N. The role of PCDH19 in refractory status epilepticus. Epilepsy Behav. 2019, 101, 106539. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.A.; Shevell, M.I.; Sébire, G. Sudden unexpected death in GEFS+ families with sodium channel pathogenic variants. Epilepsy Res. 2019, 150, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Riuró, H.; Campuzano, O.; Arbelo, E.; Iglesias, A.; Batlle, M.; Pérez-Villa, F.; Brugada, J.; Pérez, G.J.; Scornik, F.S.; Brugada, R. A missense mutation in the sodium channel β1b subunit reveals SCN1B as a susceptibility gene underlying long QT syndrome. Heart Rhythm 2014, 11, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.A.; Bello-Espinosa, L.E.; Symonds, J.D.; Zuberi, S.M.; Clegg, R.; Sadleir, L.G.; Buchhalter, J.; Scheffer, I.E. Heart rate variability in epilepsy: A potential biomarker of sudden unexpected death in epilepsy risk. Epilepsia 2018, 59, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- Wirrell, E.C.; Laux, L.; Donner, E.; Jette, N.; Knupp, K.; Meskis, M.A.; Miller, I.; Sullivan, J.; Welborn, M.; Berg, A.T. Optimizing the Diagnosis and Management of Dravet Syndrome: Recommendations From a North American Consensus Panel. Pediatr. Neurol. 2017, 68, 18–34. [Google Scholar] [CrossRef]


{kind=link}
| Pt | Sex | Age at Epilepsy Onset (m) | Age at Death (y) | SCN1A Variant | Seizures during the 6 Months before Death | Ongoing ASMs | Previous SE | Treatment of Previous SE | Trigger of SE | SE Duration (min) | Body Temperature (°C) | Neuroimaging after SE Onset | SE Treatment |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | NA | 23.4 | c.5414-5415delTT (p.Phe1805Stop) | No seizures | VPA, CLB, STP, TPM, PER | Yes | DZP | Flu | 120 | 42 | np | Rectal DZP (at home), IV MDZ |
| 2 | M | 4 | 2.5 | c.17_18dupTT (p.Val7Leufsx86) | focal, 1/m, <10 min | VPA, TPM | Yes | DZP | NSFI | 80 | 38 | np | VPA, TPM |
| 3 | M | NA | 4.8 | c.664T>C (p.Arg222*) | GTC, 1-3/m, 8–30 min; focal, 2–3/m, 10–20 min | VPA, CLB, STP | Yes | DZP, MDZ | NSFI | NA | NA | np | PHT, DZP, MDZ, propofol |
| 4 | F | NA | 11.9 | c.2503T>A (p.Leu835Met) | GTC, rare, <10 min | VPA, CLB, STP | Yes | DZP, MDZ, PHT | Fever, respiratory distress, ab ingestis pneumoniae | NA | 40 | np | Rectal DZP (at home), IV MDZ, IV LEV, IV PHT |
| 5 | F | 4 | 2.5 | c.4934G>A (p.Arg1645Gln) | no seizures | VPA, CLB, STP | Yes | MDZ, Propofol | Fever, vaccination (DTPa-HBV-IPV-Hib) | 150 | 38 | np | Rectal DZP, IV MDZ, propofol |
| 6 | F | 5 | 2.3 | c.1837C>T (p.Arg613*) | GTC, monthly, long lasting; focal, rare, brief | VPA, CLB, STP, CLZ | Yes | MDZ, DZP, CLZ PHT | RSV infection | (25 days) | 42 | MRI: significant cytotoxic oedema in the grey matter and cortex | Oromucosal MDZ, CZP; PHT; MDZ; propofol; LEV; KD |
| 7 | F | 7 | 1.3 | c.4427A>G (p.Asn1476Ser) | GTC rare brief | VPA, CLB, STP, LEV | Yes | DZP, MDZ, CLZ | NSFI | 540 | 40 | CT scan: diffuse cerebral oedema | MDZ |
| Author | Sex | Age at Epilepsy Onset (m) | Age at Death (y) | SCN1A Variant | History of SE | Seizures Frequency | Ongoing ASMs at Death | Fatal SE Trigger | Duration of SE (h) | Temp (°C) | Fatal SE Treatment |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Okumura, 2012 [4] | M | 3 | 4.4 | R568X | 3 | monthly * | VPA, CZP, KBr | Flu | 4 | NA | DZP, PHT, TL MDZ |
| Okumura, 2012 [4] | F | 3 | 1.1 | R701X | 0 | none * | VPA, CZP, PB | NSFI | 3 | NA | DZP, MDZ, PHT, PTB, TP |
| Okumura, 2012 [4] | M | 5 | 15.3 | np | 1 | monthly * | VPA, ZNS, NZP | URI | 1 | NA | DZP, MDZ |
| Okumura, 2012 [4] | M | 4 | 3.6 | np | 7 | monthly * | VPA, ZNS, CLB | NSFI | 5 | NA | DZP, MDZ, PB, TP |
| Myers, 2017 [6] | NA | NA | 5 | c.5347G>A, p. Ala1783thr | several | seizure free ** | VPA, TPM | URI, Flu (A+) | 1.25 | 40 | MDZ, PHT, TP |
| Myers, 2017 [6] | NA | NA | 8 | c.5741_5742delAA, p.Gln1914fs*1943 | 3 | 1–2/year | VPA, TPM | abdominal pain, diarrhoea | 1.5 | 43.7 | MDZ, PHT |
| Myers, 2017 [6] | NA | NA | 11 | c. 4633A>G | several | seizure free ^ | VPA, TPM, STP | sore throat | 2 | 41 | MDZ, PB |
| Myers, 2017 [6] | NA | NA | 5 | c.4970G>A, p. Arg1657His | 12 | 2 seizures ^^ | LTG, VPA | viral URI | 4 | 40 | MDZ, DZP, PHT, TP, PB |
| Myers, 2017 [6] | NA | NA | 0.8 | c.3136delG, p.Asp1046Metfs*1055 | 0 | 1–2/m, long lasting | TPM, LEV | viral URI | 1.5 | 40 | CZP, MDZ, PHT |
| Tian, 2018 [7] | F | 5 | 3 | R101W | 2–3/year | monthly | VPA, LEV | NA | 2 | 39.3 | NA |
| Tian, 2018 [7] | M | 2.5 | 4 | V422A | 3–4/year | monthly | VPA, LEV | NA | 2 | 40.5 | NA |
| Tian, 2018 [7] | M | 5.5 | 5 | L556fsX | 4–5/year | monthly | VPA, TPM | NA | 4 | 40.4 | NA |
| Tian, 2018 [7] | F | 5 | 10 | R612X | 1–2/year | monthly | VPA, LEV, CZP | NA | 2 | 39.5 | NA |
| Tian, 2018 [7] | F | 4 | 3 | W1286X | 4–5/year | monthly | VPA, LEV | NA | 2 | 39.3 | NA |
| Tian, 2018 [7] | F | 5 | 5 | A1783T | 2–3/year | yearly | VPA, TPM | NA | 3 | 40.6 | NA |
| Tian, 2018 [7] | M | 2 | 4.2 | c659-1G>A | 1–2/year | monthly | VPA, TPM, CZP | NA | 3 | 39.5 | NA |
| Tian, 2018 [7] | F | 4 | 5.2 | R1892X | 2–3/year | monthly | VPA, TPM | NA | 2 | normal | NA |
| Tian, 2018 [7] | M | 5 | 3.4 | F1699S | 2–3/year | monthly | VPA, LEV | NA | 5 | 39.5 | NA |
| Tian, 2018 [7] | M | 5 | 1.3 | Q1904X | 2–3/year | yearly | VPA, TPM | NA | 5 | 40.2 | NA |
| Tian, 2018 [7] | F | 3 | 2.7 | S243Y | 1–2/year | monthly | CZP, LEV, ZNS | NA | 12 | 40.3 | NA |
| Tian, 2018 [7] | F | 4.5 | 3.3 | I1810N | 1–2/year | monthly | VPA, LEV | NA | 2 | 39.5 | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Liso, P.; Pironi, V.; Mastrangelo, M.; Battaglia, D.; Craiu, D.; Trivisano, M.; Specchio, N.; Nabbout, R.; Vigevano, F. Fatal Status Epilepticus in Dravet Syndrome. Brain Sci. 2020, 10, 889. https://doi.org/10.3390/brainsci10110889
De Liso P, Pironi V, Mastrangelo M, Battaglia D, Craiu D, Trivisano M, Specchio N, Nabbout R, Vigevano F. Fatal Status Epilepticus in Dravet Syndrome. Brain Sciences. 2020; 10(11):889. https://doi.org/10.3390/brainsci10110889
Chicago/Turabian StyleDe Liso, Paola, Virginia Pironi, Massimo Mastrangelo, Domenica Battaglia, Dana Craiu, Marina Trivisano, Nicola Specchio, Rima Nabbout, and Federico Vigevano. 2020. "Fatal Status Epilepticus in Dravet Syndrome" Brain Sciences 10, no. 11: 889. https://doi.org/10.3390/brainsci10110889
APA StyleDe Liso, P., Pironi, V., Mastrangelo, M., Battaglia, D., Craiu, D., Trivisano, M., Specchio, N., Nabbout, R., & Vigevano, F. (2020). Fatal Status Epilepticus in Dravet Syndrome. Brain Sciences, 10(11), 889. https://doi.org/10.3390/brainsci10110889