Do the Bugs in Your Gut Eat Your Memories? Relationship between Gut Microbiota and Alzheimer’s Disease


Abstract
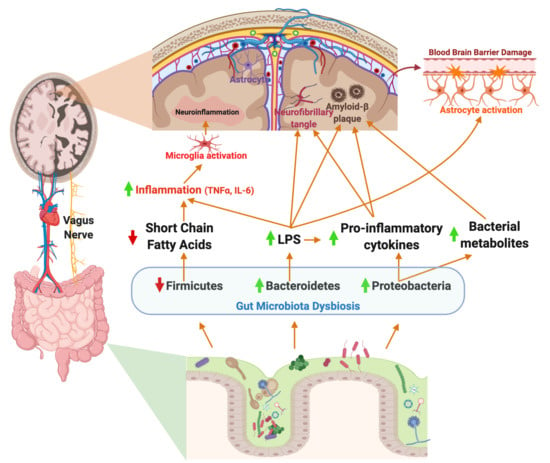
1. Introduction
Methodology
2. Characterization of Alzheimer’s Disease
3. Techniques for Microbiome Analysis
4. Gut Microbiota–Brain Axis in Alzheimer’s Disease
Enteric Nervous System: Vagus Nerve
5. Gut Microbiota Composition and Diversity in Individuals with AD
6. Gut Microbiota Composition and Diversity in Murine Models of Key AD Pathologies
7. Potential Role of the Gut Microbiome in Neuroinflammation in AD
7.1. Neuroinflammation: Microglial Activation and the Gut Microbiome
7.2. Neuroinflammation: Astrocyte Activation and Gut Microbiome
8. Microbial Etiology Hypothesis in AD
9. Potential for Microbiome-Based Therapeutics
10. Limitations of Current Research
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, S.M. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787. [Google Scholar] [CrossRef] [PubMed]
- Barcel’o-Coblijn, G.; Amedei, A. The Interplay of Microbiome and Immune Response in Health and Diseases; MDPI: Basel, Switzerland, 2019; ISBN 9783039216468. [Google Scholar]
- Dinan, T.G.; Cryan, J.F. The Microbiome-Gut-Brain Axis in Health and Disease. Gastroenterol. Clin. N. Am. 2017, 46, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Ubeda, C.; Djukovic, A.; Isaac, S. Roles of the intestinal microbiota in pathogen protection. Clin. Transl. Immunol. 2017, 6, e128. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, K.; Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: Major fermentation by-products and their impact on host health. Microbiome 2019, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Kho, Z.Y.; Lal, S.K. The Human Gut Microbiome—A Potential Controller of Wellness and Disease. Front. Microbiol. 2018, 9, 1835. [Google Scholar] [CrossRef]
- Tap, J.; Mondot, S.; Levenez, F.; Pelletier, E.; Caron, C.; Furet, J.-P.; Ugarte, E.; Muñoz-Tamayo, R.; Paslier, D.L.E.; Nalin, R.; et al. Towards the human intestinal microbiota phylogenetic core. Environ. Microbiol. 2009, 11, 2574–2584. [Google Scholar] [CrossRef]
- Soto-Martin, E.C.; Warnke, I.; Farquharson, F.M.; Christodoulou, M.; Horgan, G.; Derrien, M.; Faurie, J.-M.; Flint, H.J.; Duncan, S.H.; Louis, P. Vitamin Biosynthesis by Human Gut Butyrate-Producing Bacteria and Cross-Feeding in Synthetic Microbial Communities. MBio 2020, 11. [Google Scholar] [CrossRef]
- Yoshii, K.; Hosomi, K.; Sawane, K.; Kunisawa, J. Metabolism of Dietary and Microbial Vitamin B Family in the Regulation of Host Immunity. Front. Nutr. 2019, 6, 48. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of Colonic Regulatory T Cells by Indigenous Clostridium Species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Makki, K.; Deehan, E.C.; Walter, J.; Bäckhed, F. The Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell Host Microbe 2018, 23, 705–715. [Google Scholar] [CrossRef]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef]
- Trompette, A.; Gollwitzer, E.S.; Pattaroni, C.; Lopez-Mejia, I.C.; Riva, E.; Pernot, J.; Ubags, N.; Fajas, L.; Nicod, L.P.; Marsland, B.J. Dietary Fiber Confers Protection against Flu by Shaping Ly6c− Patrolling Monocyte Hematopoiesis and CD8 T Cell Metabolism. Immunity 2018, 48, 992–1005.e8. [Google Scholar] [CrossRef]
- Yu, L.C.-H. Microbiota dysbiosis and barrier dysfunction in inflammatory bowel disease and colorectal cancers: Exploring a common ground hypothesis. J. Biomed. Sci. 2018, 25, 79. [Google Scholar] [CrossRef]
- Henke, M.T.; Kenny, D.J.; Cassilly, C.D.; Vlamakis, H.; Xavier, R.J.; Clardy, J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc. Natl. Acad. Sci. USA 2019, 116, 12672–12677. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, K.; Mulak, A. Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef]
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen–gut microbiome axis: Physiological and clinical implications. Maturitas 2017, 103, 45–53. [Google Scholar] [CrossRef]
- Flores, R.; Shi, J.; Fuhrman, B.; Xu, X.; Veenstra, T.D.; Gail, M.H.; Gajer, P.; Ravel, J.; Goedert, J.J. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: A cross-sectional study. J. Transl. Med. 2012, 10, 253. [Google Scholar] [CrossRef]
- Fülling, C.; Dinan, T.G.; Cryan, J.F. Gut Microbe to Brain Signaling: What Happens in Vagus…. Neuron 2019, 101, 998–1002. [Google Scholar]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, E.; Kloosterhuis, N.J.; Koster, M.; Dekker, D.C.; Gijbels, M.J.J.; van der Velden, S.; Ríos-Morales, M.; van Faassen, M.J.R.; Loreti, M.G.; de Bruin, A.; et al. A Proinflammatory Gut Microbiota Increases Systemic Inflammation and Accelerates Atherosclerosis. Circ. Res. 2019, 124, 94–100. [Google Scholar] [CrossRef]
- Ma, Q.; Xing, C.; Long, W.; Wang, H.Y.; Liu, Q.; Wang, R.-F. Impact of microbiota on central nervous system and neurological diseases: The gut-brain axis. J. Neuroinflamm. 2019, 16, 53. [Google Scholar] [CrossRef]
- Alkasir, R.; Li, J.; Li, X.; Jin, M.; Zhu, B. Human gut microbiota: The links with dementia development. Protein Cell 2017, 8, 90–102. [Google Scholar] [CrossRef]
- Saji, N.; Murotani, K.; Hisada, T.; Kunihiro, T.; Tsuduki, T.; Sugimoto, T.; Kimura, A.; Niida, S.; Toba, K.; Sakurai, T. Relationship between dementia and gut microbiome-associated metabolites: A cross-sectional study in Japan. Sci. Rep. 2020, 10, 8088. [Google Scholar] [CrossRef]
- Li, S.; Shao, Y.; Li, K.; HuangFu, C.; Wang, W.; Liu, Z.; Cai, Z.; Zhao, B. Vascular Cognitive Impairment and the Gut Microbiota. J. Alzheimers Dis. 2018, 63, 1209–1222. [Google Scholar] [CrossRef] [PubMed]
- Daulatzai, M.A. Non-celiac gluten sensitivity triggers gut dysbiosis, neuroinflammation, gut-brain axis dysfunction, and vulnerability for dementia. CNS Neurol. Disord. Drug Targets 2015, 14, 110–131. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.; Okeoma, C.M.; Sestak, K. Dietary Gluten and Neurodegeneration: A Case for Preclinical Studies. Int. J. Mol. Sci. 2020, 21, 5407. [Google Scholar] [CrossRef]
- Pennisi, M.; Bramanti, A.; Cantone, M.; Pennisi, G.; Bella, R.; Lanza, G. Neurophysiology of the “Celiac Brain”: Disentangling Gut-Brain Connections. Front. Neurosci. 2017, 11, 498. [Google Scholar] [CrossRef]
- Lanza, G.; Bella, R.; Cantone, M.; Pennisi, G.; Ferri, R.; Pennisi, M. Cognitive Impairment and Celiac Disease: Is Transcranial Magnetic Stimulation a Trait d’Union between Gut and Brain? Int. J. Mol. Sci. 2018, 19, 2243. [Google Scholar] [CrossRef]
- Lyon, L. “All disease begins in the gut”: Was Hippocrates right? Brain 2018, 141, e20. [Google Scholar] [CrossRef]
- Hippius, H.; Neundörfer, G. The discovery of Alzheimer’s disease. Dialogues Clin. Neurosci. 2003, 5, 101–108. [Google Scholar]
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020, 5, 234–270. [Google Scholar] [CrossRef]
- Lewis, T.J.; Trempe, C.L. The Amyloid Cascade Hypothesis. The End of Alzheimer’s: The Brain and Beyond; Academic Press: Cambridge, MA, USA, 2017; pp. 26–50. [Google Scholar]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef] [PubMed]
- Bitan, G.; Kirkitadze, M.D.; Lomakin, A.; Vollers, S.S.; Benedek, G.B.; Teplow, D.B. Amyloid beta -protein (Abeta) assembly: Abeta 40 and Abeta 42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Sochocka, M.; Donskow-Łysoniewska, K.; Diniz, B.S.; Kurpas, D.; Brzozowska, E.; Leszek, J. The Gut Microbiome Alterations and Inflammation-Driven Pathogenesis of Alzheimer’s Disease—A Critical Review. Mol. Neurobiol. 2019, 56, 1841–1851. [Google Scholar] [CrossRef]
- Hashemiaghdam, A.; Mroczek, M. Microglia heterogeneity and neurodegeneration: The emerging paradigm of the role of immunity in Alzheimer’s disease. J. Neuroimmunol. 2020, 341, 577185. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers. Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Jill, E.; Clarridge, I.I.I. Impact of 16S rRNA Gene Sequence Analysis for Identification of Bacteria on Clinical Microbiology and Infectious Diseases. Clin. Microbiol. Rev. 2004, 17, 840–862. [Google Scholar]
- Ihrmark, K.; Bödeker, I.T.M.; Cruz-Martinez, K.; Friberg, H.; Kubartova, A.; Schenck, J.; Strid, Y.; Stenlid, J.; Brandström-Durling, M.; Clemmensen, K.E.; et al. New primers to amplify the fungal ITS2 region–evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 2012, 82, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Hillmann, B.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Zhu, Q.; Gohl, D.M.; Beckman, K.B.; Knight, R.; Knights, D. Evaluating the Information Content of Shallow Shotgun Metagenomics. MSystems 2018, 3. [Google Scholar] [CrossRef]
- Aguiar-Pulido, V.; Huang, W.; Suarez-Ulloa, V.; Cickovski, T.; Mathee, K.; Narasimhan, G. Metagenomics, Metatranscriptomics, and Metabolomics Approaches for Microbiome Analysis. Evol. Bioinform. Online 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. Author Correction: History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef]
- Sun, M.; Ma, K.; Wen, J.; Wang, G.; Zhang, C.; Li, Q.; Bao, X.; Wang, H. A Review of the Brain-Gut-Microbiome Axis and the Potential Role of Microbiota in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 73, 849–865. [Google Scholar] [CrossRef]
- Giau, V.; Wu, S.; Jamerlan, A.; An, S.; Kim, S.; Hulme, J. Gut Microbiota and Their Neuroinflammatory Implications in Alzheimer’s Disease. Nutrients 2018, 10, 1765. [Google Scholar] [CrossRef] [PubMed]
- Domingues, C.; da Cruz e Silva, O.A.B.; Henriques, A.G. Impact of Cytokines and Chemokines on Alzheimer’s Disease Neuropathological Hallmarks. Curr. Alzheimer Res. 2017, 14, 870–882. [Google Scholar] [CrossRef]
- Sheng, J.G.; Ito, K.; Skinner, R.D.; Mrak, R.E.; Rovnaghi, C.R.; Van Eldik, L.J.; Griffin, W.S. In vivo and in vitro evidence supporting a role for the inflammatory cytokine interleukin-1 as a driving force in Alzheimer pathogenesis. Neurobiol. Aging 1996, 17, 761–766. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Orellana, D.I.; González-Billault, C.; Maccioni, R.B. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 2004, 295, 245–257. [Google Scholar] [CrossRef]
- Steardo, L.; Bronzuoli, M.R.; Iacomino, A.; Esposito, G.; Steardo, L.; Scuderi, C. Does neuroinflammation turn on the flame in Alzheimer’s disease? Focus on astrocytes. Front. Neurosci. 2015, 9, 259. [Google Scholar] [CrossRef]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef]
- Vogt, N.M.; Romano, K.A.; Darst, B.F.; Engelman, C.D.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Blennow, K.; Zetterberg, H.; Bendlin, B.B.; et al. The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 124. [Google Scholar] [CrossRef]
- Ho, L.; Ono, K.; Tsuji, M.; Mazzola, P.; Singh, R.; Pasinetti, G.M. Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type beta-amyloid neuropathological mechanisms. Expert Rev. Neurother. 2018, 18, 83–90. [Google Scholar] [CrossRef]
- Rao, M.; Gershon, M.D. The bowel and beyond: The enteric nervous system in neurological disorders. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Brookes, S.J.; Hennig, G.W. Anatomy and physiology of the enteric nervous system. Gut 2000, 47 (Suppl. 4), iv15–iv19. [Google Scholar] [CrossRef]
- Abdel-Haq, R.; Schlachetzki, J.C.M.; Glass, C.K.; Mazmanian, S.K. Microbiome–microglia connections via the gut–brain axis. J. Exp. Med. 2019, 216, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B.; Sinniger, V.; Pellissier, S. The Vagus Nerve in the Neuro-Immune Axis: Implications in the Pathology of the Gastrointestinal Tract. Front. Immunol. 2017, 8, 1452. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B.; Bazin, T.; Pellissier, S. The Vagus Nerve at the Interface of the Microbiota-Gut-Brain Axis. Front. Neurosci. 2018, 12, 49. [Google Scholar] [CrossRef]
- Browning, K.N. Role of central vagal 5-HT3 receptors in gastrointestinal physiology and pathophysiology. Front. Neurosci. 2015, 9, 413. [Google Scholar] [CrossRef]
- Merrill, C.A.; Jonsson, M.A.G.; Minthon, L.; Ejnell, H.; C-son Silander, H.; Blennow, K.; Karlsson, M.; Nordlund, A.; Rolstad, S.; Warkentin, S.; et al. Vagus nerve stimulation in patients with Alzheimer’s disease: Additional follow-up results of a pilot study through 1 year. J. Clin. Psychiatry 2006, 67, 1171–1178. [Google Scholar] [CrossRef]
- Sjögren, M.J.C.; Hellström, P.T.O.; Jonsson, M.A.G.; Runnerstam, M.; Silander, H.C.-S.; Ben-Menachem, E. Cognition-enhancing effect of vagus nerve stimulation in patients with Alzheimer’s disease: A pilot study. J. Clin. Psychiatry 2002, 63, 972–980. [Google Scholar] [CrossRef]
- Desbeaumes Jodoin, V.; Richer, F.; Miron, J.-P.; Fournier-Gosselin, M.-P.; Lespérance, P. Long-term Sustained Cognitive Benefits of Vagus Nerve Stimulation in Refractory Depression. J. ECT 2018, 34, 283–290. [Google Scholar] [CrossRef]
- Kaczmarczyk, R.; Tejera, D.; Simon, B.J.; Heneka, M.T. Microglia modulation through external vagus nerve stimulation in a murine model of Alzheimer’s disease. J. Neurochem. 2018, 146, 76–85. [Google Scholar] [CrossRef]
- Tran, T.T.T.; Corsini, S.; Kellingray, L.; Hegarty, C.; Le Gall, G.; Narbad, A.; Müller, M.; Tejera, N.; O’Toole, P.W.; Minihane, A.-M.; et al. APOE genotype influences the gut microbiome structure and function in humans and mice: Relevance for Alzheimer’s disease pathophysiology. FASEB J. 2019, 33, 8221–8231. [Google Scholar] [CrossRef]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef]
- Liu, P.; Wu, L.; Peng, G.; Han, Y.; Tang, R.; Ge, J.; Zhang, L.; Jia, L.; Yue, S.; Zhou, K.; et al. Altered microbiomes distinguish Alzheimer’s disease from amnestic mild cognitive impairment and health in a Chinese cohort. Brain Behav. Immun. 2019, 80, 633–643. [Google Scholar] [CrossRef]
- Zhuang, Z.-Q.; Shen, L.-L.; Li, W.-W.; Fu, X.; Zeng, F.; Gui, L.; Lü, Y.; Cai, M.; Zhu, C.; Tan, Y.-L.; et al. Gut Microbiota is Altered in Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2018, 63, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Haran, J.P.; Bhattarai, S.K.; Foley, S.E.; Dutta, P.; Ward, D.V.; Bucci, V.; McCormick, B.A. Alzheimer’s Disease Microbiome Is Associated with Dysregulation of the Anti-Inflammatory P-Glycoprotein Pathway. MBio 2019, 10. [Google Scholar] [CrossRef]
- Emery, D.C.; Shoemark, D.K.; Batstone, T.E.; Waterfall, C.M.; Coghill, J.A.; Cerajewska, T.L.; Davies, M.; West, N.X.; Allen, S.J. 16S rRNA Next Generation Sequencing Analysis Shows Bacteria in Alzheimer’s Post-Mortem Brain. Front. Aging Neurosci. 2017, 9, 195. [Google Scholar] [CrossRef] [PubMed]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef]
- Chen, Y.; Fang, L.; Chen, S.; Zhou, H.; Fan, Y.; Lin, L.; Li, J.; Xu, J.; Chen, Y.; Ma, Y.; et al. Gut Microbiome Alterations Precede Cerebral Amyloidosis and Microglial Pathology in a Mouse Model of Alzheimer’s Disease. BioMed. Res. Int. 2020, 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Brandscheid, C.; Schuck, F.; Reinhardt, S.; Schäfer, K.-H.; Pietrzik, C.U.; Grimm, M.; Hartmann, T.; Schwiertz, A.; Endres, K. Altered Gut Microbiome Composition and Tryptic Activity of the 5xFAD Alzheimer’s Mouse Model. J. Alzheimer’s Dis. 2017, 56, 775–788. [Google Scholar] [CrossRef]
- Parikh, I.J.; Estus, J.L.; Zajac, D.J.; Malik, M.; Maldonado Weng, J.; Tai, L.M.; Chlipala, G.E.; LaDu, M.J.; Green, S.J.; Estus, S. Murine Gut Microbiome Association with APOE Alleles. Front. Immunol. 2020, 11, 200. [Google Scholar] [CrossRef]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Erratum: Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 41802. [Google Scholar] [CrossRef]
- Bonfili, L.; Cecarini, V.; Berardi, S.; Scarpona, S.; Suchodolski, J.S.; Nasuti, C.; Fiorini, D.; Boarelli, M.C.; Rossi, G.; Eleuteri, A.M. Microbiota modulation counteracts Alzheimer’s disease progression influencing neuronal proteolysis and gut hormones plasma levels. Sci. Rep. 2017, 7, 2426. [Google Scholar] [CrossRef]
- Sun, J.; Liu, S.; Ling, Z.; Wang, F.; Ling, Y.; Gong, T.; Fang, N.; Ye, S.; Si, J.; Liu, J. Fructooligosaccharides Ameliorating Cognitive Deficits and Neurodegeneration in APP/PS1 Transgenic Mice through Modulating Gut Microbiota. J. Agric. Food Chem. 2019, 67, 3006–3017. [Google Scholar] [CrossRef]
- Sanguinetti, E.; Collado, M.C.; Marrachelli, V.G.; Monleon, D.; Selma-Royo, M.; Pardo-Tendero, M.M.; Burchielli, S.; Iozzo, P. Microbiome-metabolome signatures in mice genetically prone to develop dementia, fed a normal or fatty diet. Sci. Rep. 2018, 8, 4907. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xu, J.; Ling, Y.; Wang, F.; Gong, T.; Yang, C.; Ye, S.; Ye, K.; Wei, D.; Song, Z.; et al. Fecal microbiota transplantation alleviated Alzheimer’s disease-like pathogenesis in APP/PS1 transgenic mice. Transl. Psychiatry 2019, 9, 189. [Google Scholar] [CrossRef]
- Chen, W.-W.; Zhang, X.; Huang, W.-J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef]
- Guo, W.; Wang, H.; Watanabe, M.; Shimizu, K.; Zou, S.; LaGraize, S.C.; Wei, F.; Dubner, R.; Ren, K. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J. Neurosci. 2007, 27, 6006–6018. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Ma, N.; Yu, B.; Zhang, W.; Wan, J. Transcriptomic profiling of microglia and astrocytes throughout aging. J. Neuroinflamm. 2020, 17, 97. [Google Scholar] [CrossRef]
- Salas, I.H.; Burgado, J.; Allen, N.J. Glia: Victims or villains of the aging brain? Neurobiol. Dis. 2020, 143, 105008. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Larry Sparks, D. Dystrophic microglia in the aging human brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Fransen, F.; van Beek, A.A.; Borghuis, T.; Aidy, S.E.; Hugenholtz, F.; van der Gaast-de Jongh, C.; Savelkoul, H.F.J.; De Jonge, M.I.; Boekschoten, M.V.; Smidt, H.; et al. Aged Gut Microbiota Contributes to Systemical Inflammaging after Transfer to Germ-Free Mice. Front. Immunol. 2017, 8, 1385. [Google Scholar] [CrossRef]
- Cerovic, M.; Forloni, G.; Balducci, C. Neuroinflammation and the Gut Microbiota: Possible Alternative Therapeutic Targets to Counteract Alzheimer’s Disease? Front. Aging Neurosci. 2019, 11, 284. [Google Scholar] [CrossRef]
- Arcuri, C.; Mecca, C.; Bianchi, R.; Giambanco, I.; Donato, R. The Pathophysiological Role of Microglia in Dynamic Surveillance, Phagocytosis and Structural Remodeling of the Developing CNS. Front. Mol. Neurosci. 2017, 10, 191. [Google Scholar] [CrossRef] [PubMed]
- Leyns, C.E.G.; Holtzman, D.M. Glial contributions to neurodegeneration in tauopathies. Mol. Neurodegener. 2017, 12, 50. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Wang, Y.; Li, F.; Jia, J.; Song, X.; Qin, S.; Wang, R.; Jin, F.; Kitazato, K.; et al. The Gut-Microglia Connection: Implications for Central Nervous System Diseases. Front. Immunol. 2018, 9, 2325. [Google Scholar] [CrossRef]
- Hemonnot, A.-L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities. Front. Aging Neurosci. 2019, 11, 233. [Google Scholar] [CrossRef]
- Lai, A.Y.; McLaurin, J. Clearance of amyloid-β peptides by microglia and macrophages: The issue of what, when and where. Future Neurol. 2012, 7, 165–176. [Google Scholar] [CrossRef]
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and Inflammation in Alzheimers Disease. CNS Neurol. Disord.-Drug Targets 2010, 9, 156–167. [Google Scholar] [CrossRef]
- Hristovska, I.; Pascual, O. Deciphering Resting Microglial Morphology and Process Motility from a Synaptic Prospect. Front. Integr. Neurosci. 2015, 9, 73. [Google Scholar] [CrossRef]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S.T. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflammation 2004, 1, 14. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Meneses, G.; Bautista, M.; Florentino, A.; Díaz, G.; Acero, G.; Besedovsky, H.; Meneses, D.; Fleury, A.; Del Rey, A.; Gevorkian, G.; et al. Electric stimulation of the vagus nerve reduced mouse neuroinflammation induced by lipopolysaccharide. J. Inflamm. 2016, 13, 33. [Google Scholar] [CrossRef]
- Huffman, W.J.; Subramaniyan, S.; Rodriguiz, R.M.; Wetsel, W.C.; Grill, W.M.; Terrando, N. Modulation of neuroinflammation and memory dysfunction using percutaneous vagus nerve stimulation in mice. Brain Stimul. 2019, 12, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Zu, H.-B. Microglial polarization: Novel therapeutic mechanism against Alzheimer’s disease. Inflammopharmacology 2020, 28, 95–110. [Google Scholar] [CrossRef]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Oré, M.-V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Sävman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Xiayu, X.; Shi, C.; Chen, W.; Song, N.; Fu, X.; Zhou, R.; Xu, Y.-F.; Huang, L.; et al. Altered Gut Microbiota in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 60, 1241–1257. [Google Scholar] [CrossRef]
- Amici, S.A.; Dong, J.; Guerau-de-Arellano, M. Molecular Mechanisms Modulating the Phenotype of Macrophages and Microglia. Front. Immunol. 2017, 8, 1520. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef]
- Sharma, R.K.; Yang, T.; Oliveira, A.C.; Lobaton, G.O.; Aquino, V.; Kim, S.; Richards, E.M.; Pepine, C.J.; Sumners, C.; Raizada, M.K. Microglial Cells Impact Gut Microbiota and Gut Pathology in Angiotensin II-Induced Hypertension. Circ. Res. 2019, 124, 727–736. [Google Scholar] [CrossRef]
- Siracusa, R.; Fusco, R.; Cuzzocrea, S. Astrocytes: Role and Functions in Brain Pathologies. Front. Pharmacol. 2019, 10, 1114. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef]
- Michinaga, S.; Koyama, Y. Dual Roles of Astrocyte-Derived Factors in Regulation of Blood-Brain Barrier Function after Brain Damage. Int. J. Mol. Sci. 2019, 20, 571. [Google Scholar] [CrossRef] [PubMed]
- Csipo, T.; Lipecz, A.; Ashpole, N.M.; Balasubramanian, P.; Tarantini, S. Astrocyte senescence contributes to cognitive decline. GeroScience 2020, 42, 51–55. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef]
- Van de Haar, H.J.; Burgmans, S.; Jansen, J.F.A.; van Osch, M.J.P.; van Buchem, M.A.; Muller, M.; Hofman, P.A.M.; Verhey, F.R.J.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2017, 282, 615. [Google Scholar] [CrossRef] [PubMed]
- Yarlagadda, A.; Alfson, E.; Clayton, A.H. The blood brain barrier and the role of cytokines in neuropsychiatry. Psychiatry 2009, 6, 18–22. [Google Scholar] [PubMed]
- Pan, W.; Stone, K.P.; Hsuchou, H.; Manda, V.K.; Zhang, Y.; Kastin, A.J. Cytokine Signaling Modulates Blood-Brain Barrier Function. Curr. Pharm. Des. 2011, 17, 3729–3740. [Google Scholar] [CrossRef]
- Hyvärinen, T.; Hagman, S.; Ristola, M.; Sukki, L.; Veijula, K.; Kreutzer, J.; Kallio, P.; Narkilahti, S. Co-stimulation with IL-1β and TNF-α induces an inflammatory reactive astrocyte phenotype with neurosupportive characteristics in a human pluripotent stem cell model system. Sci. Rep. 2019, 9, 16944. [Google Scholar] [CrossRef]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef]
- Consonni, A.; Morara, S.; Codazzi, F.; Grohovaz, F.; Zacchetti, D. Inhibition of lipopolysaccharide-induced microglia activation by calcitonin gene related peptide and adrenomedullin. Mol. Cell. Neurosci. 2011, 48, 151–160. [Google Scholar] [CrossRef]
- You, L.-H.; Yan, C.-Z.; Zheng, B.-J.; Ci, Y.-Z.; Chang, S.-Y.; Yu, P.; Gao, G.-F.; Li, H.-Y.; Dong, T.-Y.; Chang, Y.-Z. Astrocyte hepcidin is a key factor in LPS-induced neuronal apoptosis. Cell Death Dis. 2017, 8, e2676. [Google Scholar] [CrossRef]
- Ryu, K.-Y.; Lee, H.-J.; Woo, H.; Kang, R.-J.; Han, K.-M.; Park, H.; Lee, S.M.; Lee, J.-Y.; Jeong, Y.J.; Nam, H.-W.; et al. Dasatinib regulates LPS-induced microglial and astrocytic neuroinflammatory responses by inhibiting AKT/STAT3 signaling. J. Neuroinflamm. 2019, 16, 190. [Google Scholar] [CrossRef] [PubMed]
- Tomova, A.; Soltys, K.; Repiská, G.; Palkova, L.; Filcikova, D.; Minarik, G.; Turna, J.; Prochotska, K.; Babinska, K.; Ostatnikova, D. Specificity of gut microbiota in children with autism spectrum disorder in Slovakia and its correlation with astrocytes activity marker and specific behavioural patterns. Physiol. Behav. 2020, 214, 112745. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, J.; Liu, Y.; Xiao, N.; Suo, H.; Xie, K.; Yang, C.; Wu, C. Short-Chain Fatty Acids Suppress Lipopolysaccharide-Induced Production of Nitric Oxide and Proinflammatory Cytokines Through Inhibition of NF-κB Pathway in RAW264.7 Cells. Inflammation 2012, 35, 1676–1684. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, R.; Cheng, M.; Wang, L.; Chao, J.; Li, J.; Zheng, P.; Xie, P.; Zhang, Z.; Yao, H. Gut microbiota from NLRP3-deficient mice ameliorates depressive-like behaviors by regulating astrocyte dysfunction via circHIPK2. Microbiome 2019, 7, 116. [Google Scholar] [CrossRef]
- Clark, R.I.; Salazar, A.; Yamada, R.; Fitz-Gibbon, S.; Morselli, M.; Alcaraz, J.; Rana, A.; Rera, M.; Pellegrini, M.; Ja, W.W.; et al. Distinct Shifts in Microbiota Composition during Drosophila Aging Impair Intestinal Function and Drive Mortality. Cell Rep. 2015, 12, 1656–1667. [Google Scholar] [CrossRef]
- Conley, M.N.; Wong, C.P.; Duyck, K.M.; Hord, N.; Ho, E.; Sharpton, T.J. Aging and serum MCP-1 are associated with gut microbiome composition in a murine model. PeerJ 2016, 4, e1854. [Google Scholar] [CrossRef] [PubMed]
- Thevaranjan, N.; Puchta, A.; Schulz, C.; Naidoo, A.; Szamosi, J.C.; Verschoor, C.P.; Loukov, D.; Schenck, L.P.; Jury, J.; Foley, K.P.; et al. Age-Associated Microbial Dysbiosis Promotes Intestinal Permeability, Systemic Inflammation, and Macrophage Dysfunction. Cell Host Microbe 2017, 21, 455–466.e4. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.-S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef]
- Friedland, R.P.; Chapman, M.R. The role of microbial amyloid in neurodegeneration. PLoS Pathog. 2017, 13, e1006654. [Google Scholar] [CrossRef]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.; Ren, J.-J.; Hammer, N.D.; Chapman, M.R. Gatekeeper residues in the major curlin subunit modulate bacterial amyloid fiber biogenesis. Proc. Natl. Acad. Sci. USA. 2010, 107, 163–168. [Google Scholar] [CrossRef]
- Chapman, M. P2-024: Protein misfolding done right: The biogenesis of bacterial amyloid fibers. Alzheimer’s Dement. 2006, 2, S239. [Google Scholar] [CrossRef]
- Andreasen, M.; Meisl, G.; Taylor, J.D.; Michaels, T.C.T.; Levin, A.; Otzen, D.E.; Chapman, M.R.; Dobson, C.M.; Matthews, S.J.; Knowles, T.P.J. Physical Determinants of Amyloid Assembly in Biofilm Formation. MBio 2019, 10. [Google Scholar] [CrossRef]
- Biesecker, S.G.; Nicastro, L.K.; Wilson, R.P.; Tükel, Ç. The Functional Amyloid Curli Protects Escherichia coli against Complement-Mediated Bactericidal Activity. Biomolecules 2018, 8, 5. [Google Scholar] [CrossRef]
- Lundmark, K.; Westermark, G.T.; Olsén, A.; Westermark, P. Protein fibrils in nature can enhance amyloid protein a amyloidosis in mice: Cross-seeding as a disease mechanism. Proc. Natl. Acad. Sci. USA 2005, 102, 6098–6102. [Google Scholar] [CrossRef]
- Zhou, Y.; Smith, D.; Leong, B.J.; Brännström, K.; Almqvist, F.; Chapman, M.R. Promiscuous cross-seeding between bacterial amyloids promotes interspecies biofilms. J. Biol. Chem. 2012, 287, 35092–35103. [Google Scholar] [CrossRef]
- Nishimori, J.H.; Newman, T.N.; Oppong, G.O.; Rapsinski, G.J.; Yen, J.-H.; Biesecker, S.G.; Wilson, R.P.; Butler, B.P.; Winter, M.G.; Tsolis, R.M.; et al. Microbial amyloids induce interleukin 17A (IL-17A) and IL-22 responses via Toll-like receptor 2 activation in the intestinal mucosa. Infect. Immun. 2012, 80, 4398–4408. [Google Scholar] [CrossRef]
- Oppong, G.O.; Rapsinski, G.J.; Tursi, S.A.; Biesecker, S.G.; Klein-Szanto, A.J.; Goulian, M.; McCauley, C.; Healy, C.; Wilson, R.P.; Tükel, C. Biofilm-associated bacterial amyloids dampen inflammation in the gut: Oral treatment with curli fibres reduces the severity of hapten-induced colitis in mice. NPJ Biofilms Microbiomes 2015, 1, 15019. [Google Scholar] [CrossRef]
- Brown, G.C. The endotoxin hypothesis of neurodegeneration. J. Neuroinflamm. 2019, 16, 180. [Google Scholar] [CrossRef]
- Banks, W.A.; Gray, A.M.; Erickson, M.A.; Salameh, T.S.; Damodarasamy, M.; Sheibani, N.; Meabon, J.S.; Wing, E.E.; Morofuji, Y.; Cook, D.G.; et al. Lipopolysaccharide-induced blood-brain barrier disruption: Roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J. Neuroinflamm. 2015, 12, 223. [Google Scholar] [CrossRef]
- Salguero, M.V.; Al-Obaide, M.A.I.; Singh, R.; Siepmann, T.; Vasylyeva, T.L. Dysbiosis of Gram-negative gut microbiota and the associated serum lipopolysaccharide exacerbates inflammation in type 2 diabetic patients with chronic kidney disease. Exp. Ther. Med. 2019, 18, 3461–3469. [Google Scholar] [CrossRef]
- Lukiw, W.J. Bacteroides fragilis Lipopolysaccharide and Inflammatory Signaling in Alzheimer’s Disease. Front. Microbiol. 2016, 7, 1544. [Google Scholar] [CrossRef]
- Daulatzai, M.A. Chronic functional bowel syndrome enhances gut-brain axis dysfunction, neuroinflammation, cognitive impairment, and vulnerability to dementia. Neurochem. Res. 2014, 39, 624–644. [Google Scholar] [CrossRef]
- Coquenlorge, S.; Duchalais, E.; Chevalier, J.; Cossais, F.; Rolli-Derkinderen, M.; Neunlist, M. Modulation of lipopolysaccharide-induced neuronal response by activation of the enteric nervous system. J. Neuroinflamm. 2014, 11, 202. [Google Scholar] [CrossRef]
- Maguire, M.; Maguire, G. Gut dysbiosis, leaky gut, and intestinal epithelial proliferation in neurological disorders: Towards the development of a new therapeutic using amino acids, prebiotics, probiotics, and postbiotics. Rev. Neurosci. 2019, 30, 179–201. [Google Scholar] [CrossRef]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; de Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell. Neurosci. 2018, 12, 329. [Google Scholar] [CrossRef]
- Wang, L.-M.; Wu, Q.; Kirk, R.A.; Horn, K.P.; Ebada Salem, A.H.; Hoffman, J.M.; Yap, J.T.; Sonnen, J.A.; Towner, R.A.; Bozza, F.A.; et al. Lipopolysaccharide endotoxemia induces amyloid-β and p-tau formation in the rat brain. Am. J. Nucl. Med. Mol. Imaging 2018, 8, 86–99. [Google Scholar]
- Zhan, X.; Stamova, B.; Sharp, F.R. Lipopolysaccharide Associates with Amyloid Plaques, Neurons and Oligodendrocytes in Alzheimer’s Disease Brain: A Review. Front. Aging Neurosci. 2018, 10, 42. [Google Scholar] [CrossRef]
- Sochocka, M.; Zwolińska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef]
- Wang, X.-L.; Zeng, J.; Feng, J.; Tian, Y.-T.; Liu, Y.-J.; Qiu, M.; Yan, X.; Yang, Y.; Xiong, Y.; Zhang, Z.-H.; et al. Helicobacter pylori filtrate impairs spatial learning and memory in rats and increases β-amyloid by enhancing expression of presenilin-2. Front. Aging Neurosci. 2014, 6, 66. [Google Scholar] [CrossRef]
- Pisa, D.; Alonso, R.; Rábano, A.; Rodal, I.; Carrasco, L. Different Brain Regions are Infected with Fungi in Alzheimer’s Disease. Sci. Rep. 2015, 5, 15015. [Google Scholar] [CrossRef] [PubMed]
- Pisa, D.; Alonso, R.; Fernández-Fernández, A.M.; Rábano, A.; Carrasco, L. Polymicrobial Infections In Brain Tissue From Alzheimer’s Disease Patients. Sci. Rep. 2017, 7, 5559. [Google Scholar] [CrossRef]
- Bittar, A.; Sengupta, U.; Kayed, R. Prospects for strain-specific immunotherapy in Alzheimer’s disease and tauopathies. NPJ Vaccines 2018, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D. Immunotherapy for Alzheimer’s Disease. J. Intern. Med. 2011, 269, 54–63. [Google Scholar] [CrossRef]
- Cytokine Inhibition for Treatment of Alzheimer’s Disease. Available online: http://www.medscape.com/viewarticle/530141 (accessed on 28 August 2020).
- Mengel, D.; Röskam, S.; Neff, F.; Balakrishnan, K.; Deuster, O.; Gold, M.; Oertel, W.H.; Bacher, M.; Bach, J.-P.; Dodel, R. Naturally occurring autoantibodies interfere with β-amyloid metabolism and improve cognition in a transgenic mouse model of Alzheimer’s disease 24 h after single treatment. Transl. Psychiatry 2013, 3, e236. [Google Scholar] [CrossRef]
- Rosenberg, R.N.; Fu, M.; Lambracht-Washington, D. Active full-length DNA Aβ42 immunization in 3xTg-AD mice reduces not only amyloid deposition but also tau pathology. Alzheimer’s Res. Ther. 2018, 10, 115. [Google Scholar] [CrossRef]
- McAlpine, F.E.; Lee, J.-K.; Harms, A.S.; Ruhn, K.A.; Blurton-Jones, M.; Hong, J.; Das, P.; Golde, T.E.; LaFerla, F.M.; Oddo, S.; et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 2009, 34, 163–177. [Google Scholar] [CrossRef]
- Overk, C.; Masliah, E. Could changing the course of Alzheimer’s disease pathology with immunotherapy prevent dementia? Brain 2019, 142, 1853–1855. [Google Scholar] [CrossRef]
- Bostanciklioğlu, M. The role of gut microbiota in pathogenesis of Alzheimer’s disease. J. Appl. Microbiol. 2019, 127, 954–967. [Google Scholar] [CrossRef]
- Bonfili, L.; Cecarini, V.; Gogoi, O.; Berardi, S.; Scarpona, S.; Angeletti, M.; Rossi, G.; Eleuteri, A.M. Gut microbiota manipulation through probiotics oral administration restores glucose homeostasis in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2020, 87, 35–43. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Yanckello, L.M.; Chlipala, G.; Hammond, T.C.; McCulloch, S.D.; Parikh, I.; Sun, S.; Morganti, J.M.; Green, S.J.; Lin, A.-L. Dietary inulin alters the gut microbiome, enhances systemic metabolism and reduces neuroinflammation in an APOE4 mouse model. PLoS ONE 2019, 14, e0221828. [Google Scholar] [CrossRef]
- Tamtaji, O.R.; Heidari-Soureshjani, R.; Mirhosseini, N.; Kouchaki, E.; Bahmani, F.; Aghadavod, E.; Tajabadi-Ebrahimi, M.; Asemi, Z. Probiotic and selenium co-supplementation, and the effects on clinical, metabolic and genetic status in Alzheimer’s disease: A randomized, double-blind, controlled trial. Clin. Nutr. 2019, 38, 2569–2575. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kinoshita, T.; Matsumoto, A.; Yoshino, K.; Saito, I.; Xiao, J.-Z. Bifidobacterium Breve A1 Supplementation Improved Cognitive Decline in Older Adults with Mild Cognitive Impairment: An Open-Label, Single-Arm Study. J. Prev. Alzheimers Dis. 2019, 6, 70–75. [Google Scholar] [PubMed]
- Hazan, S. Rapid improvement in Alzheimer’s disease symptoms following fecal microbiota transplantation: A case report. J. Int. Med Res. 2020, 48, 030006052092593. [Google Scholar] [CrossRef]
- Pistollato, F.; Battino, M. Role of plant-based diets in the prevention and regression of metabolic syndrome and neurodegenerative diseases. Trends Food Sci. Technol. 2014, 40, 62–81. [Google Scholar] [CrossRef]
- Mariotti, F. Vegetarian and Plant-Based Diets in Health and Disease Prevention; Academic Press: Cambridge, MA, USA, 2017; ISBN 9780128039694. [Google Scholar]
- Anastasiou, C.A.; Yannakoulia, M.; Kosmidis, M.H.; Dardiotis, E.; Hadjigeorgiou, G.M.; Sakka, P.; Arampatzi, X.; Bougea, A.; Labropoulos, I.; Scarmeas, N. Mediterranean diet and cognitive health: Initial results from the Hellenic Longitudinal Investigation of Ageing and Diet. PLoS ONE 2017, 12, e0182048. [Google Scholar] [CrossRef]
- Pistollato, F.; Iglesias, R.C.; Ruiz, R.; Aparicio, S.; Crespo, J.; Lopez, L.D.; Manna, P.P.; Giampieri, F.; Battino, M. Nutritional patterns associated with the maintenance of neurocognitive functions and the risk of dementia and Alzheimer’s disease: A focus on human studies. Pharmacol. Res. 2018, 131, 32–43. [Google Scholar] [CrossRef]
- Scarmeas, N.; Stern, Y.; Mayeux, R.; Manly, J.J.; Schupf, N.; Luchsinger, J.A. Mediterranean diet and mild cognitive impairment. Arch. Neurol. 2009, 66, 216–225. [Google Scholar] [CrossRef]
- Farooqui, T.; Farooqui, A.A. Role of the Mediterranean Diet in the Brain and Neurodegenerative Diseases; Academic Press: Cambridge, MA, USA, 2017; ISBN 9780128119600. [Google Scholar]
- Tangney, C.C.; Li, H.; Wang, Y.; Barnes, L.; Schneider, J.A.; Bennett, D.A.; Morris, M.C. Relation of DASH- and Mediterranean-like dietary patterns to cognitive decline in older persons. Neurology 2014, 83, 1410–1416. [Google Scholar] [CrossRef]
- McEvoy, C.T.; Guyer, H.; Langa, K.M.; Yaffe, K. Neuroprotective Diets Are Associated with Better Cognitive Function: The Health and Retirement Study. J. Am. Geriatr. Soc. 2017, 65, 1857–1862. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Courchesne-Loyer, A.; St-Pierre, V.; Vandenberghe, C.; Pierotti, T.; Fortier, M.; Croteau, E.; Castellano, C.-A. Can ketones compensate for deteriorating brain glucose uptake during aging? Implications for the risk and treatment of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2016, 1367, 12–20. [Google Scholar] [CrossRef]
- Owen, O.E.; Morgan, A.P.; Kemp, H.G.; Sullivan, J.M.; Herrera, M.G.; Cahill, G.F., Jr. Brain metabolism during fasting. J. Clin. Investig. 1967, 46, 1589–1595. [Google Scholar] [CrossRef]
- Castellano, C.-A.; Nugent, S.; Paquet, N.; Tremblay, S.; Bocti, C.; Lacombe, G.; Imbeault, H.; Turcotte, É.; Fulop, T.; Cunnane, S.C. Lower brain 18F-fluorodeoxyglucose uptake but normal 11C-acetoacetate metabolism in mild Alzheimer’s disease dementia. J. Alzheimers. Dis. 2015, 43, 1343–1353. [Google Scholar] [CrossRef]
- Ogawa, M.; Fukuyama, H.; Ouchi, Y.; Yamauchi, H.; Kimura, J. Altered energy metabolism in Alzheimer’s disease. J. Neurol. Sci. 1996, 139, 78–82. [Google Scholar] [CrossRef]
- Henderson, S.T. Ketone bodies as a therapeutic for Alzheimer’s disease. Neurotherapeutics 2008, 5, 470–480. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Bergman, C.; Lee, J.-H.; Wan, R.; King, M.T.; Mughal, M.R.; Okun, E.; Clarke, K.; Mattson, M.P.; Veech, R.L. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1530–1539. [Google Scholar] [CrossRef]
- Włodarek, D. Role of Ketogenic Diets in Neurodegenerative Diseases (Alzheimer’s Disease and Parkinson’s Disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef]
- Murray, A.J.; Knight, N.S.; Cole, M.A.; Cochlin, L.E.; Carter, E.; Tchabanenko, K.; Pichulik, T.; Gulston, M.K.; Atherton, H.J.; Schroeder, M.A.; et al. Novel ketone diet enhances physical and cognitive performance. FASEB J. 2016, 30, 4021–4032. [Google Scholar] [CrossRef]
- Grammatikopoulou, M.G.; Goulis, D.G.; Gkiouras, K.; Theodoridis, X.; Gkouskou, K.K.; Evangeliou, A.; Dardiotis, E.; Bogdanos, D.P. To Keto or Not to Keto? A Systematic Review of Randomized Controlled Trials Assessing the Effects of Ketogenic Therapy on Alzheimer Disease. Adv. Nutr. 2020, nmaa073. [Google Scholar] [CrossRef]
- Ma, D.; Wang, A.C.; Parikh, I.; Green, S.J.; Hoffman, J.D.; Chlipala, G.; Murphy, M.P.; Sokola, B.S.; Bauer, B.; Hartz, A.M.S.; et al. Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Sci. Rep. 2018, 8, 6670. [Google Scholar] [CrossRef]
- Nagpal, R.; Neth, B.J.; Wang, S.; Craft, S.; Yadav, H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 2019, 47, 529–542. [Google Scholar] [CrossRef]
- Neth, B.J.; Mintz, A.; Whitlow, C.; Jung, Y.; Solingapuram Sai, K.; Register, T.C.; Kellar, D.; Lockhart, S.N.; Hoscheidt, S.; Maldjian, J.; et al. Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer’s disease: A pilot study. Neurobiol. Aging 2020, 86, 54–63. [Google Scholar] [CrossRef]
- Li, X.; Leonardi, I.; Semon, A.; Doron, I.; Gao, I.H.; Putzel, G.G.; Kim, Y.; Kabata, H.; Artis, D.; Fiers, W.D.; et al. Response to Fungal Dysbiosis by Gut-Resident CX3CR1 Mononuclear Phagocytes Aggravates Allergic Airway Disease. Cell Host Microbe 2018, 24, 847–856.e4. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Rutayisire, E.; Huang, K.; Liu, Y.; Tao, F. The mode of delivery affects the diversity and colonization pattern of the gut microbiota during the first year of infants’ life: A systematic review. BMC Gastroenterol. 2016, 16, 86. [Google Scholar] [CrossRef]
- Moise, A.M.R. The Gut Microbiome: Exploring the Connection between Microbes, Diet, and Health; ABC-CLIO: Santa Barbara, CA, USA, 2017; ISBN 9781440842658. [Google Scholar]
- Lach, G.; Schellekens, H.; Dinan, T.G.; Cryan, J.F. Anxiety, Depression, and the Microbiome: A Role for Gut Peptides. Neurotherapeutics 2018, 15, 36–59. [Google Scholar] [CrossRef]
- Hasan, N.; Yang, H. Factors affecting the composition of the gut microbiota, and its modulation. PeerJ 2019, 7, e7502. [Google Scholar] [CrossRef]
- Kim, C.H.; Park, J.; Kim, M. Gut Microbiota-Derived Short-Chain Fatty Acids, T Cells, and Inflammation. Immune Netw. 2014, 14, 277. [Google Scholar] [CrossRef]


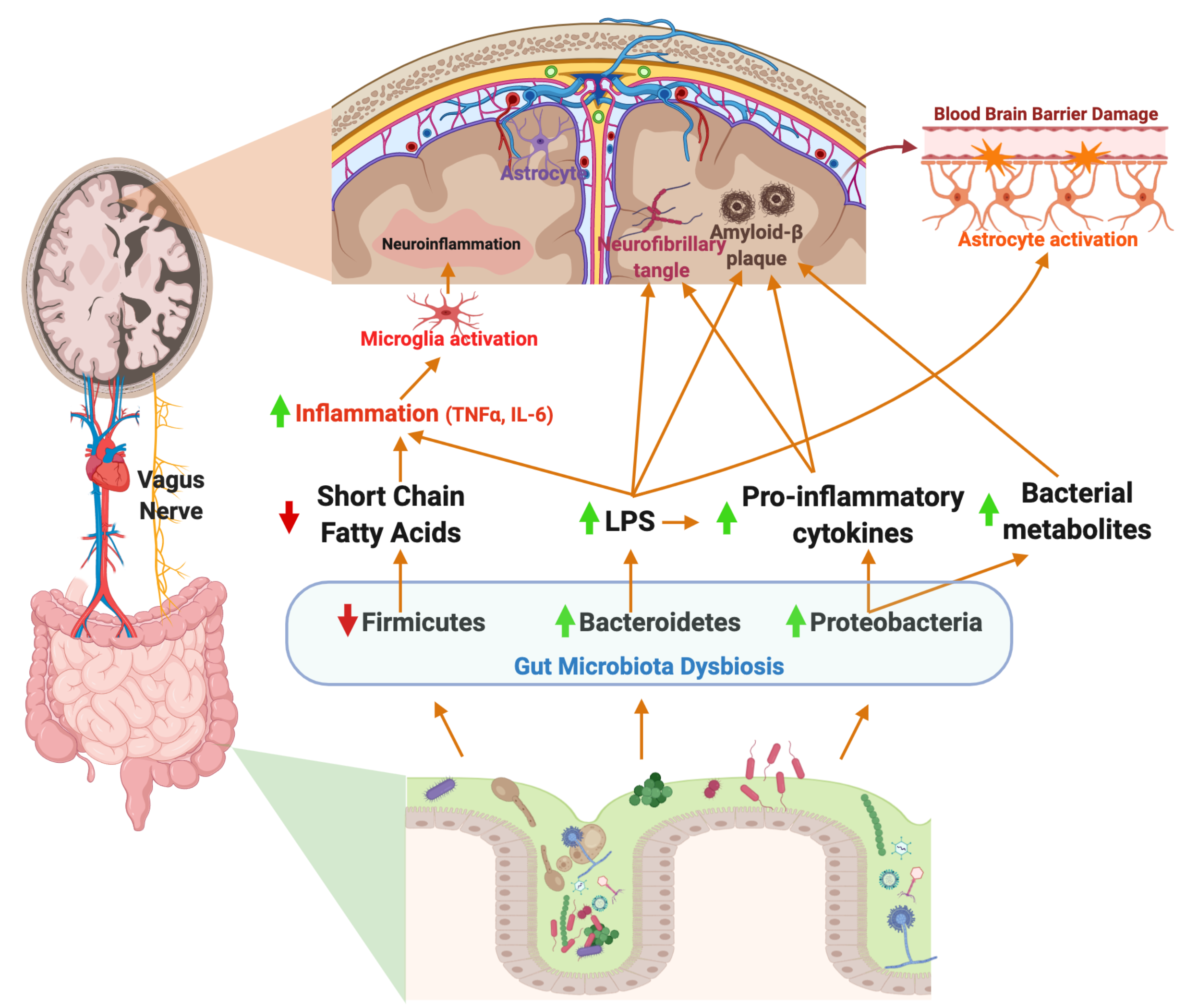{kind=link}
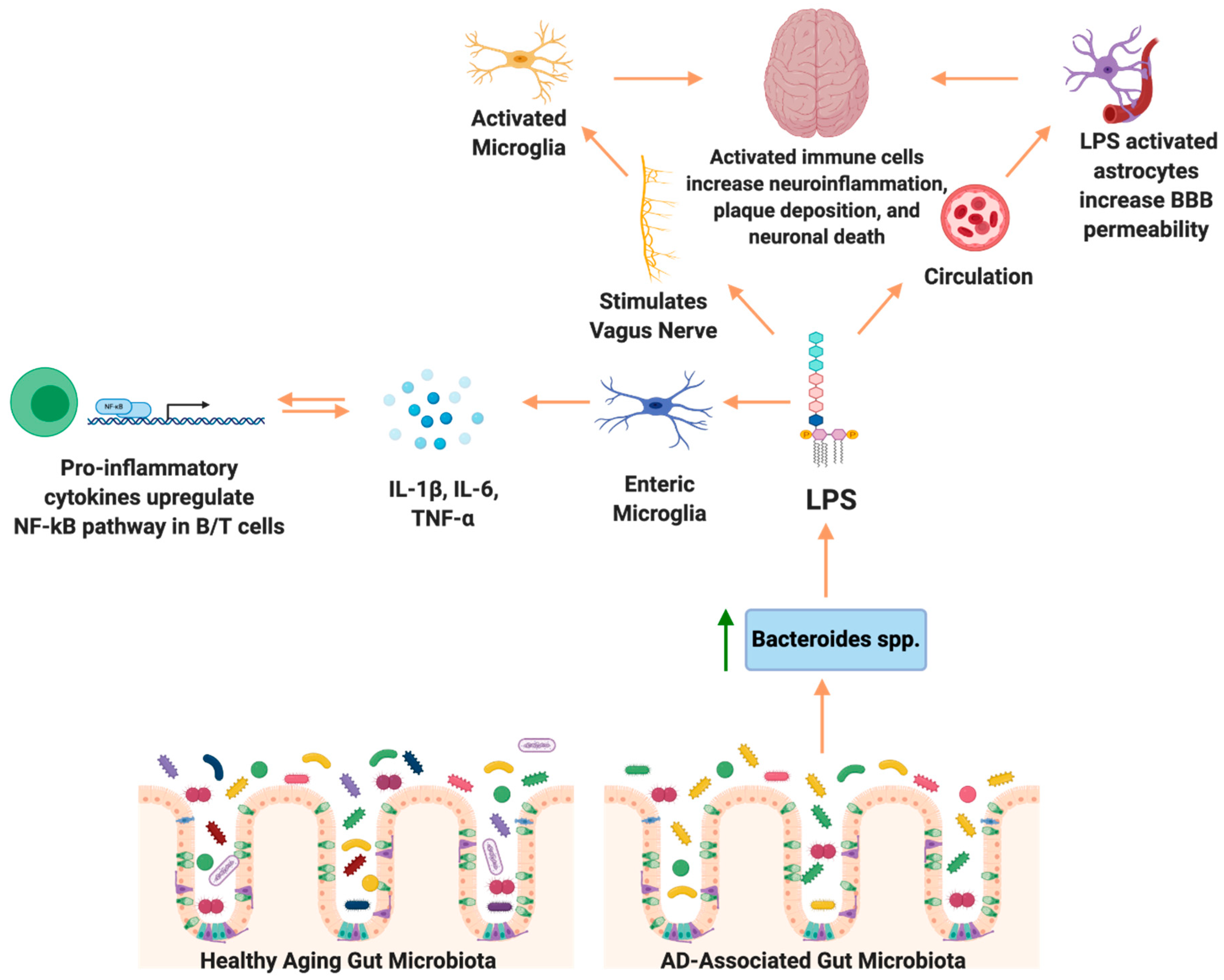{kind=link}
| Species | Increased Abundance | Decreased Abundance | Citation |
|---|---|---|---|
| Human | g.Blautia, g.Alistipes, g.Bacteroides | g.Turicibacter, g.Clostridium, g.Dialister | Vogt et al. [66] |
| Human | g.Gammaproteobacteria, g.Enterobacteriales and f.Enterobacteriaceae | g.Ruminococcus f.Clostridiaceae, f.Lachnospiraceae | Liu et al. [67] |
| Human | g.Ruminococcus, g.Subdoligranulum | f.Lachnospiraceae, g.Lachnoclostridium, g.Bacteroides | Zhuang [68] |
| Human | g.Bacteroides, g.Alistipes, g.Odoribacter | g.Butyrivibrio, g.Eubacterium, g.Lachnoclostridium, | Haran et al. [69] |
| APP/PS1 | g.Desulfovibrio, g.Akkermansia, f.Lachnospiraceae, g.Ruminococcus | g.Alistiples | Chen et al. [72] |
| Species | Increased Abundance | Decreased Abundance | Intervention | Citation |
|---|---|---|---|---|
| 3xTg-AD | g.Bifidobacterium | o.Campylobacterales | SLAB51 probiotic | Bonfili et al. [76] |
| APP/PS1 | g.Lactobacillus | g.Helicobacter | FOS supplement | Sun et al. [77] |
| 3xTg-AD | f. Rikenellaceae, f.Lachnospiraceae, f.Enterococcaceae and f.S24.7 | g.Bifidobacterium, g.Lactobacillus | Fatty diet | Sanguinetti et al. [78] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borsom, E.M.; Lee, K.; Cope, E.K. Do the Bugs in Your Gut Eat Your Memories? Relationship between Gut Microbiota and Alzheimer’s Disease. Brain Sci. 2020, 10, 814. https://doi.org/10.3390/brainsci10110814
Borsom EM, Lee K, Cope EK. Do the Bugs in Your Gut Eat Your Memories? Relationship between Gut Microbiota and Alzheimer’s Disease. Brain Sciences. 2020; 10(11):814. https://doi.org/10.3390/brainsci10110814
Chicago/Turabian StyleBorsom, Emily M., Keehoon Lee, and Emily K. Cope. 2020. "Do the Bugs in Your Gut Eat Your Memories? Relationship between Gut Microbiota and Alzheimer’s Disease" Brain Sciences 10, no. 11: 814. https://doi.org/10.3390/brainsci10110814
APA StyleBorsom, E. M., Lee, K., & Cope, E. K. (2020). Do the Bugs in Your Gut Eat Your Memories? Relationship between Gut Microbiota and Alzheimer’s Disease. Brain Sciences, 10(11), 814. https://doi.org/10.3390/brainsci10110814