First Principles Study on the Effect of Pressure on the Structure, Elasticity, and Magnetic Properties of Cubic GaFe(CN)6 Prussian Blue Analogue


Abstract
1. Introduction
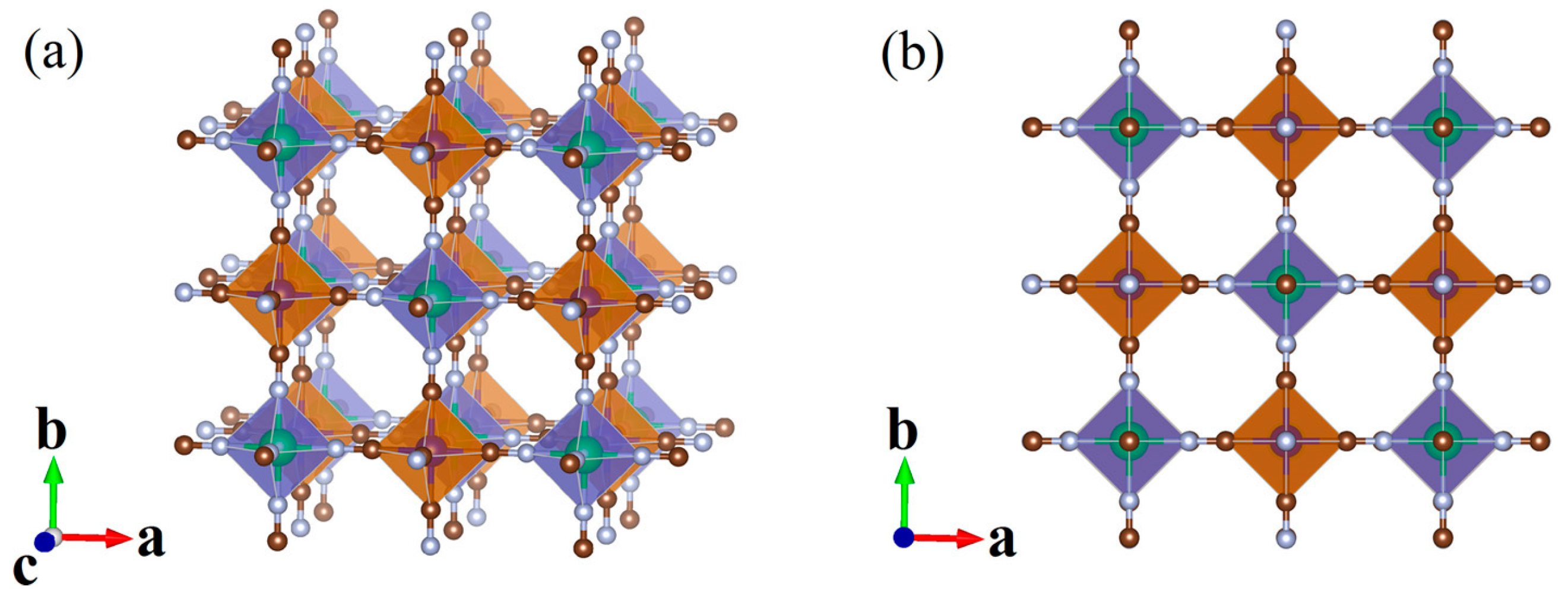
2. Materials and Methods
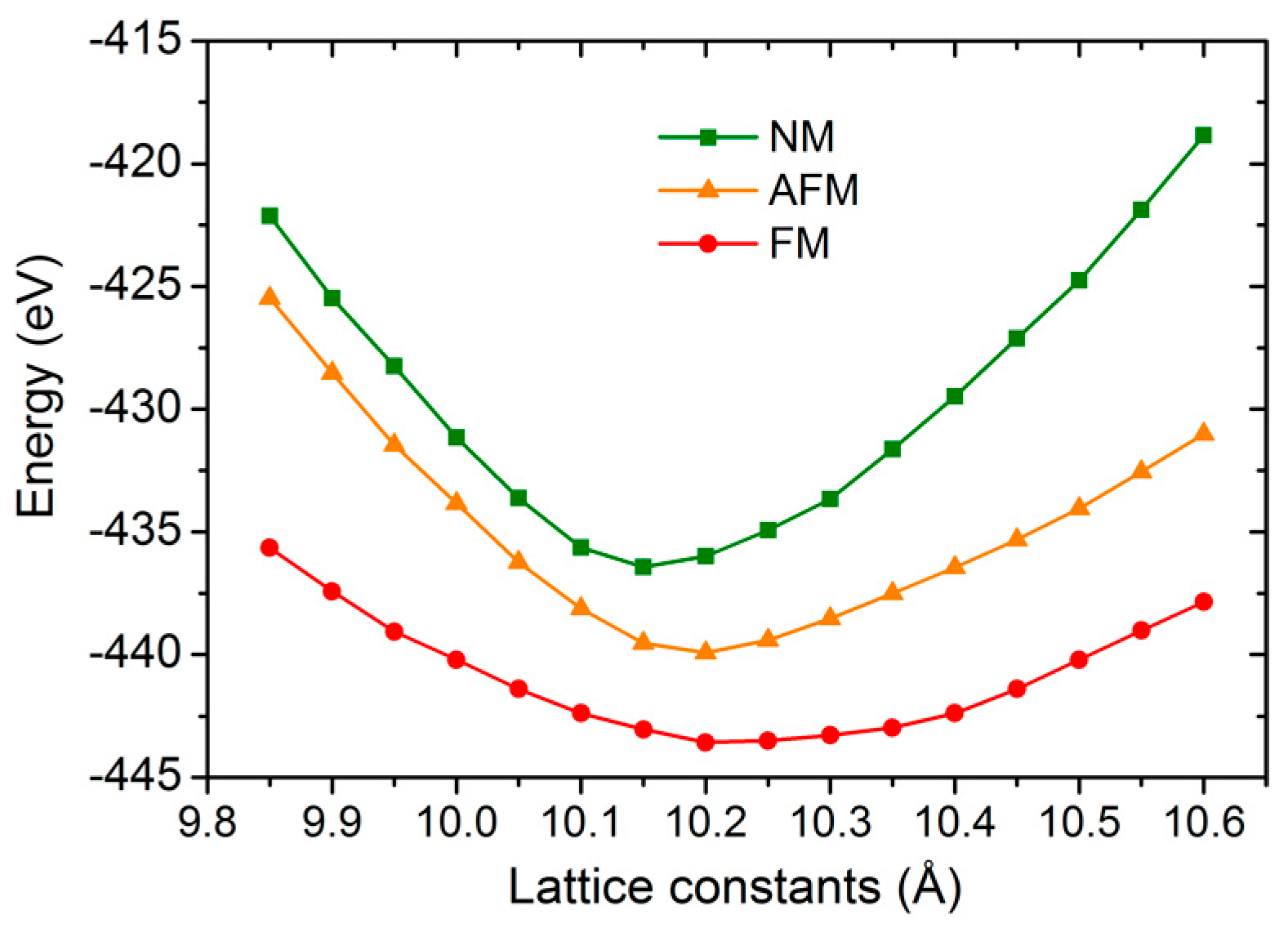
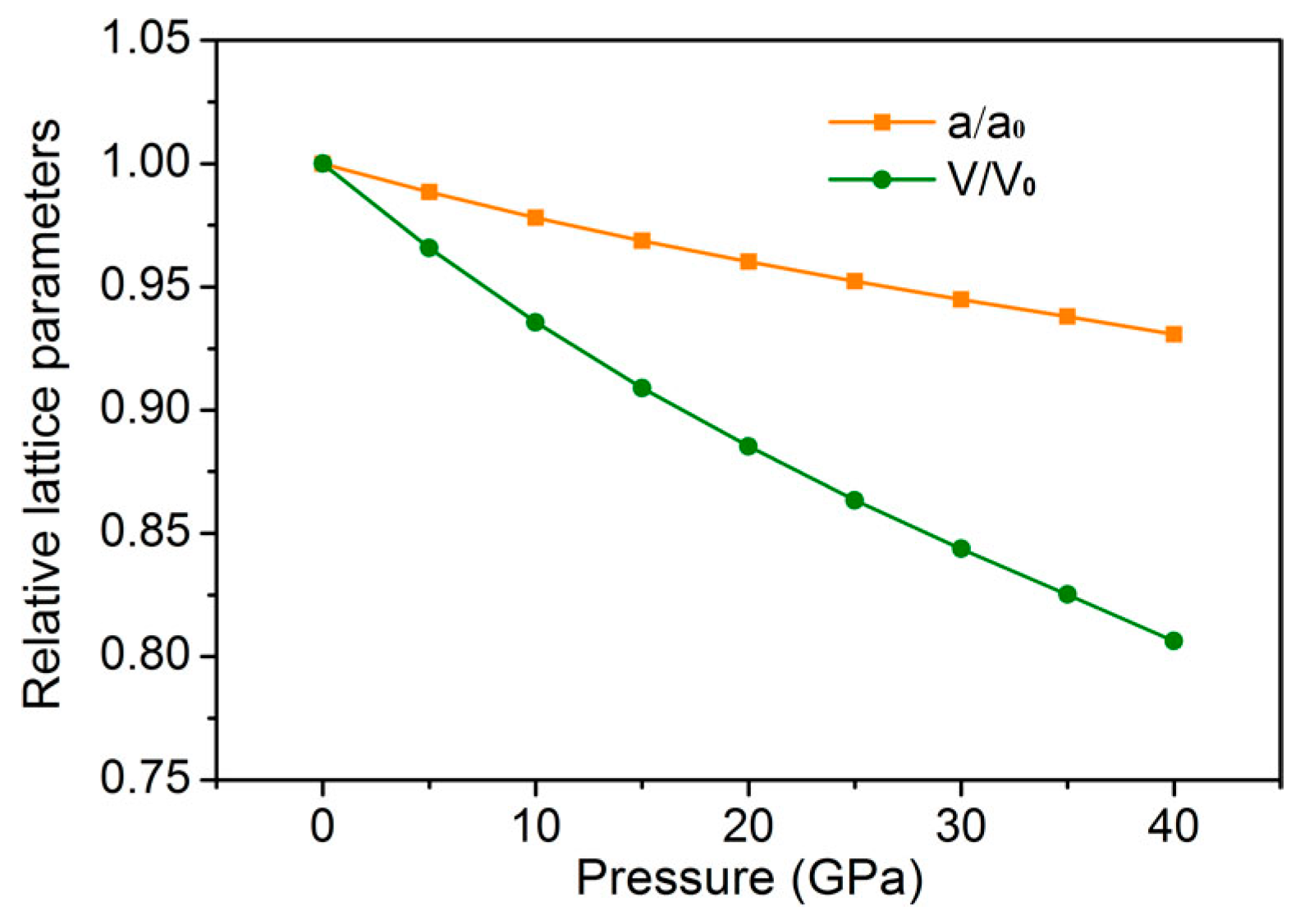
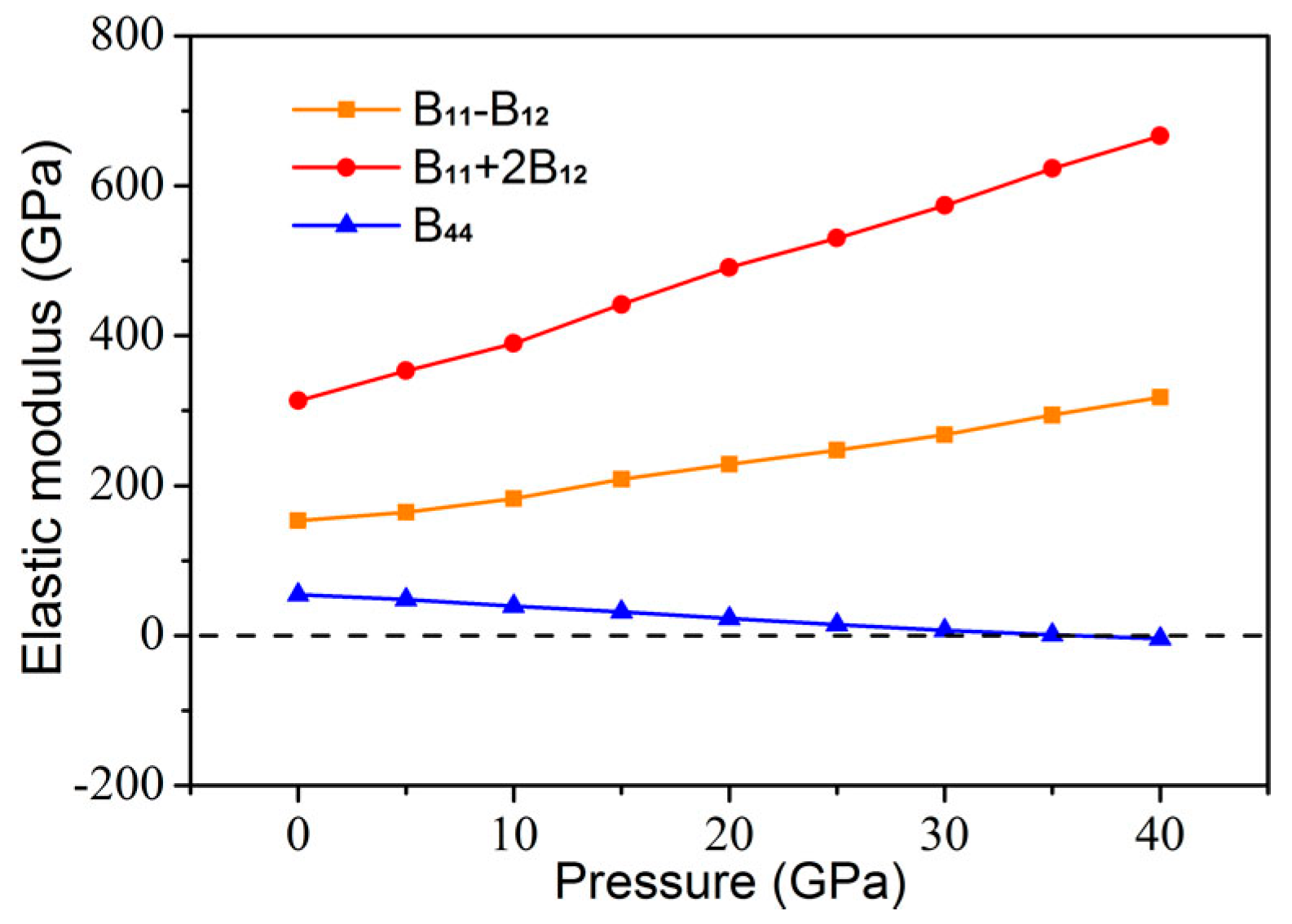
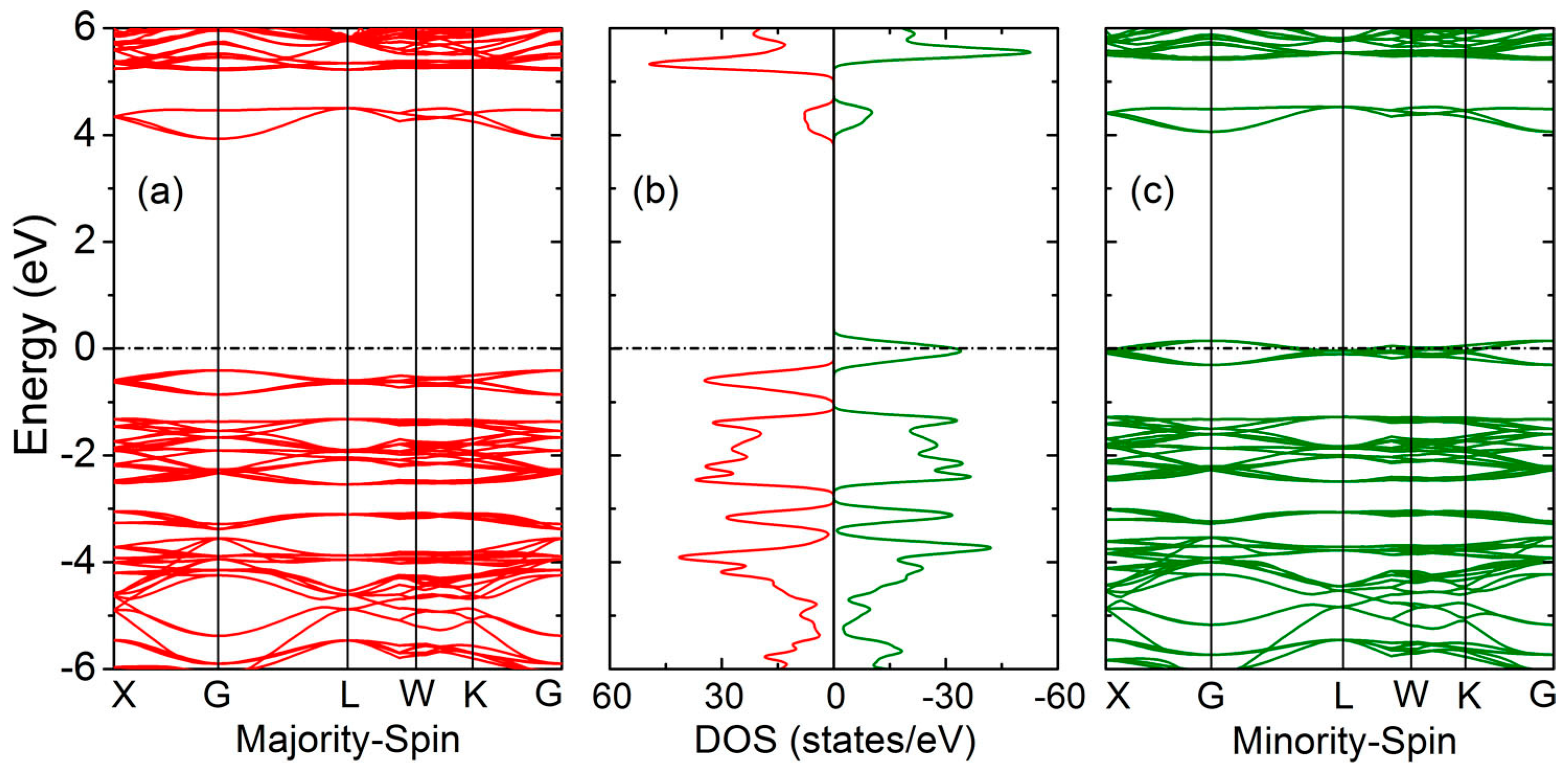
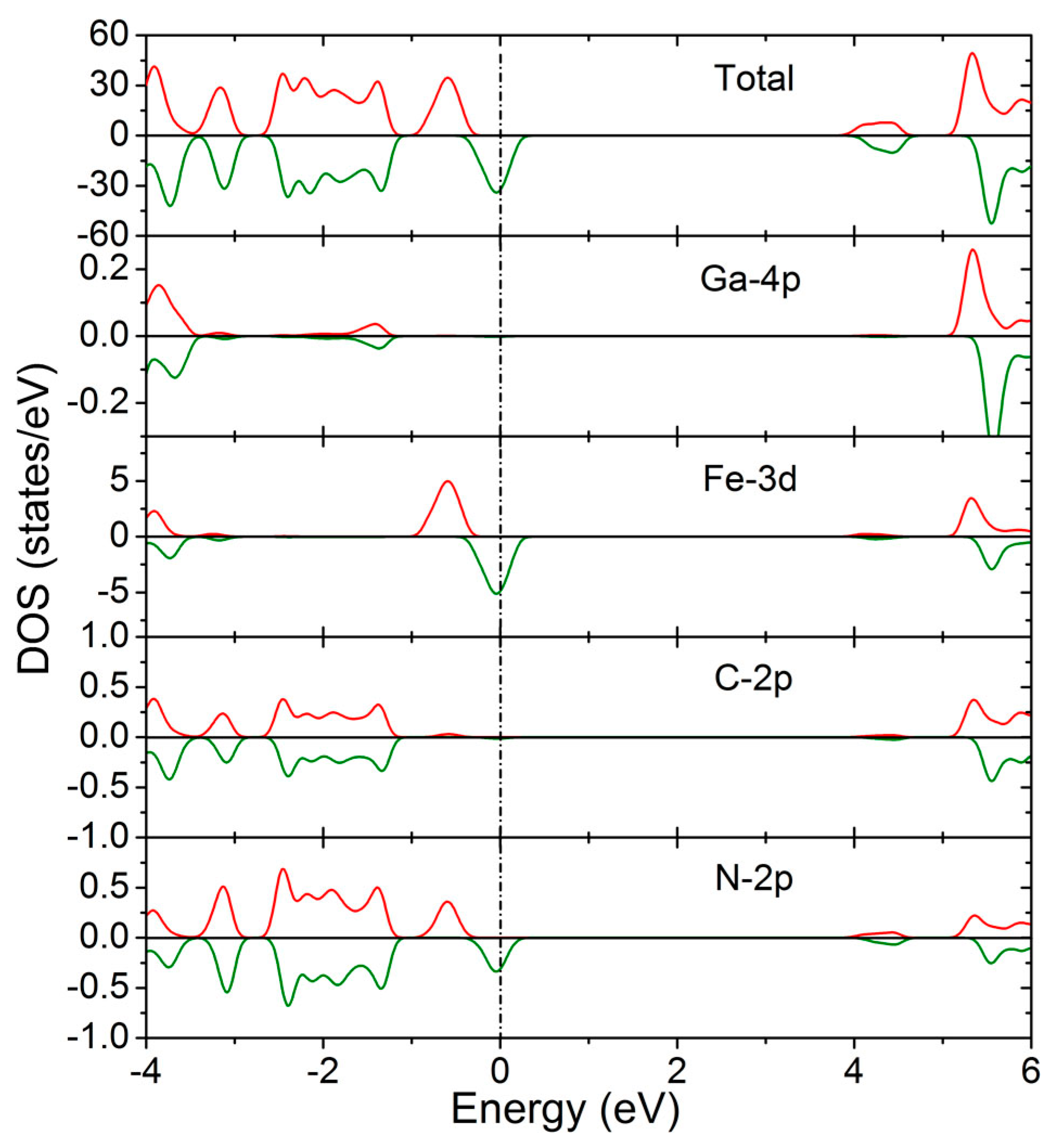
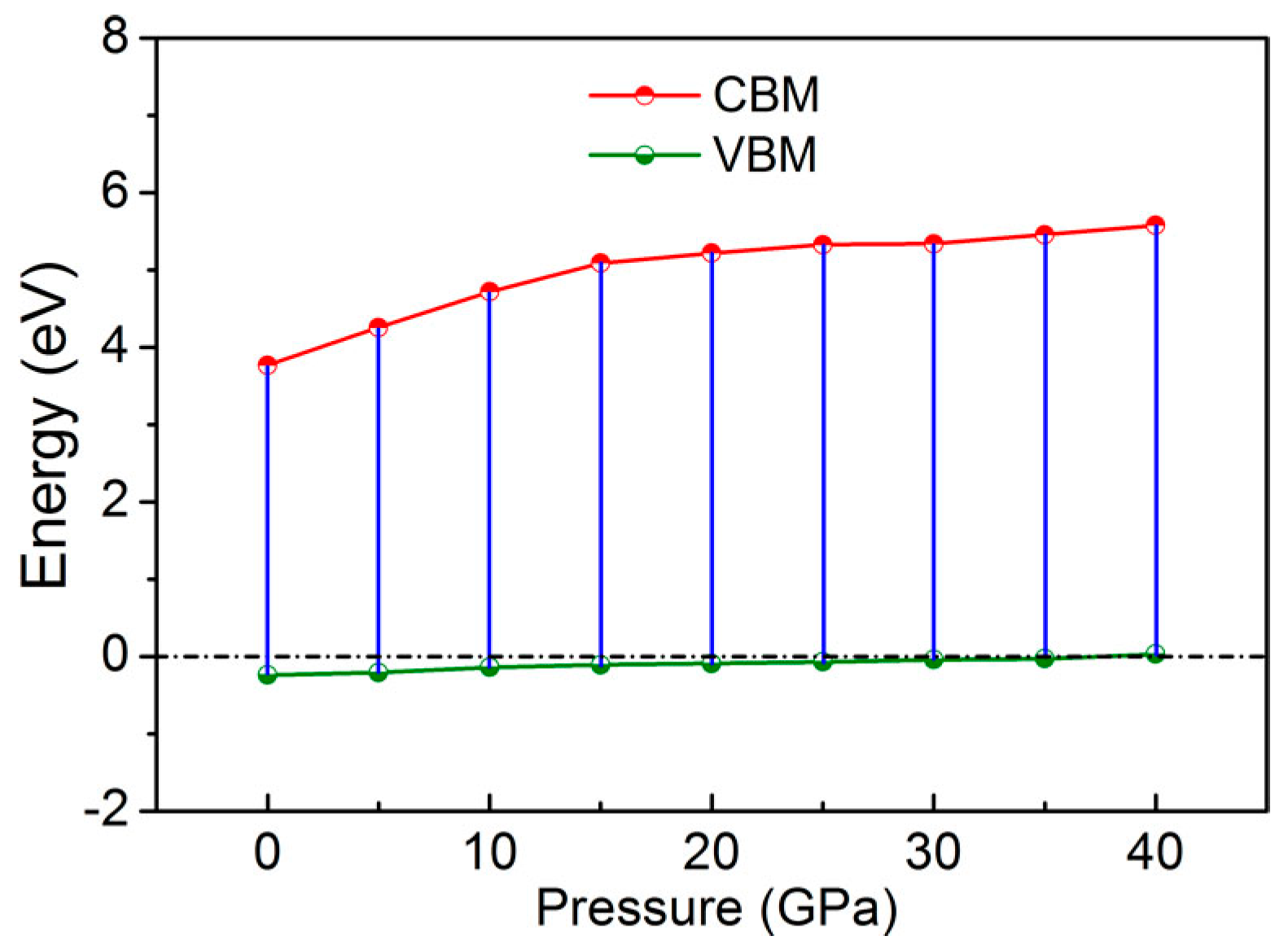
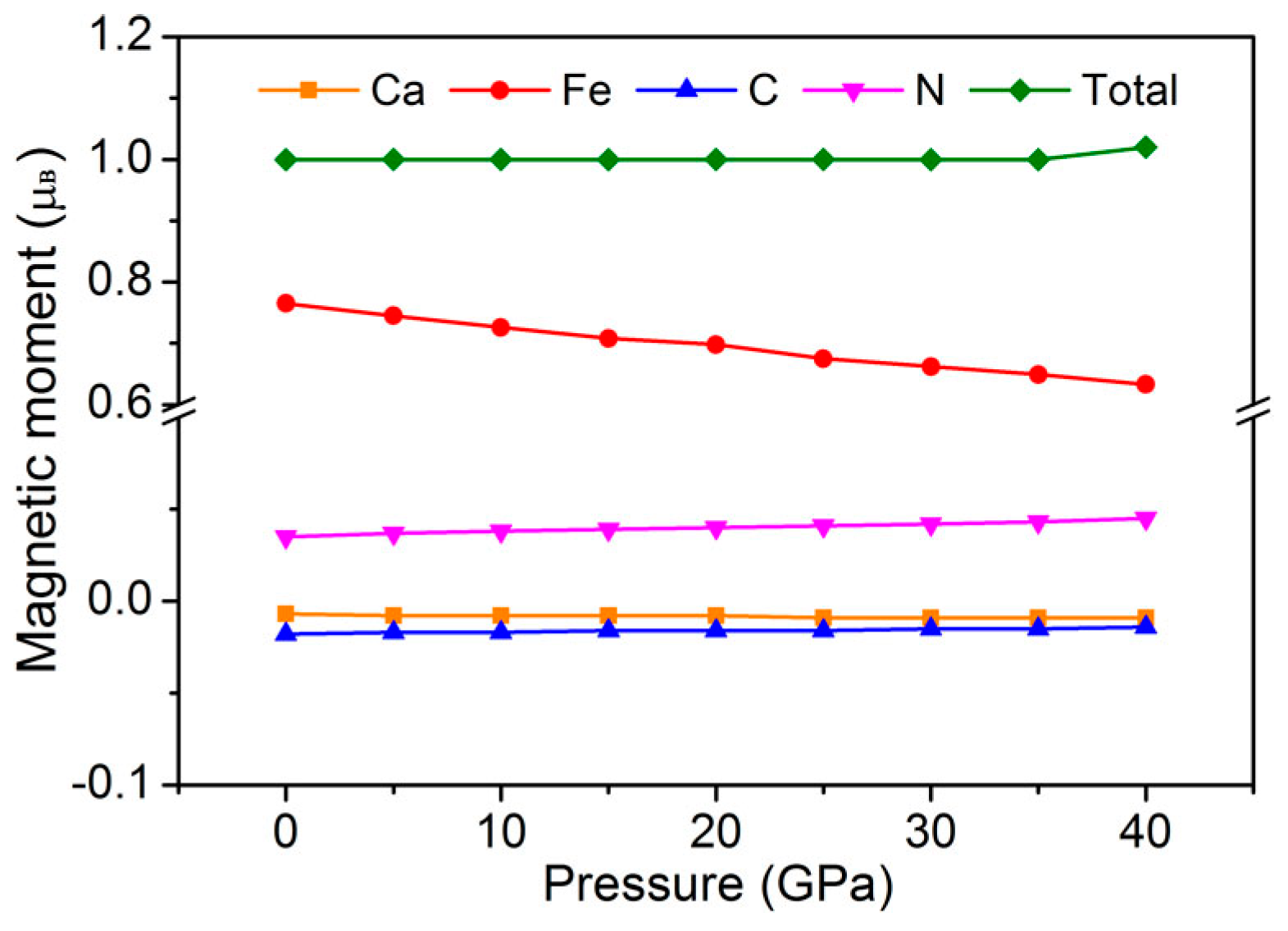
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wolf, S.A.; Awschalom, D.D.; Buhrman, R.A.; Daughton, J.M.; von Molnár, S.; Roukes, M.L.; Chtchelkanova, A.Y.; Treger, D.M. Spintronics: A spin-based electronics vision for the future. Science 2001, 294, 1488–1495. [Google Scholar] [CrossRef] [PubMed]
- Kioseoglou, G.; Hanbicki, A.T.; Sullivan, J.M.; Erve, O.M.J.; Li, C.H.; Erwin, S.C.; Mallory, R.; Yasar, M.; Petrou, A.; Jonker, B.T. Electrical spin injection from an n-type ferromagnetic semiconductor into a III–V device heterostructure. Nat. Mater. 2004, 3, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Jonker, B.T. Progress toward electrical injection of spin-polarized electrons into semiconductors. IEEE Proc. 2003, 91, 727–740. [Google Scholar] [CrossRef]
- Zutic, I.; Fabian, J.; Das Sarma, S. Spintronics: Fundamentals and applications. Rev. Mod. Phys. 2004, 76, 323–410. [Google Scholar] [CrossRef]
- Prinz, G.A. Magnetoelectronics. Science 1998, 282, 1660–1663. [Google Scholar] [CrossRef] [PubMed]
- Flatte, M.E.; Beyers, J.M. Spin diffusion in semiconductors. Phys. Rev. Lett. 2000, 84, 4220–4223. [Google Scholar] [CrossRef]
- Farshchi, R.; Ramsteiner, M. Spin injection from Heusler alloys into semiconductors: A materials perspective. J. Appl. Phys. 2013, 113, 191101. [Google Scholar] [CrossRef]
- Medvedeva, J.E.; Freeman, A.J.; Cui, X.Y.; Stampfl, C.; Newman, N. Half-Metallicity and Efficient Spin Injection in AlN/GaN:Cr (0001) Heterostructure. Phys. Rev. Lett. 2005, 94, 146602. [Google Scholar] [CrossRef]
- De Wijs, G.A.; De Groot, R.A. Towards 100% spin-polarized charge-injection: The half-metallic NiMnSb/CdS interface. Phys. Rev. B 2001, 64, 020402. [Google Scholar] [CrossRef]
- Li, J.; Gao, G.Y.; Min, Y.; Yao, K.L. Half-metallic YN2 monolayer: Dual spin filtering, dual spin diode and spin seebeck effects. Phys. Chem. Chem. Phys. 2016, 18, 28018–28023. [Google Scholar] [CrossRef]
- De Groot, R.A.; Mueller, F.M.; Van Engen, P.G.; Buschow, K.H.J. New Class of Materials: Half-Metallic Ferromagnets. Phys. Rev. Lett. 1983, 50, 2024. [Google Scholar] [CrossRef]
- Han, J.; Shen, J.; Gao, G. CrO2-based heterostructure and magnetic tunnel junction: Perfect spin filtering effect, spin diode effect and high tunnel magnetoresistance. RSC Adv. 2019, 9, 3550–3557. [Google Scholar] [CrossRef]
- Song, Y.; Dai, G. Spin filter and spin valve in ferromagnetic graphene. Appl. Phys. Lett. 2015, 106, 223104. [Google Scholar] [CrossRef]
- Everschor-Sitte, K.; Sitte, M.; MacDonald, A.H. Half-metallic magnetism and the search for better spin valves. J. Appl. Phys. 2014, 116, 083906. [Google Scholar] [CrossRef]
- Stefano, S. Molecular spintronics. Chem. Soc. Rev. 2011, 40, 3336–3355. [Google Scholar]
- Chen, Y.; Chen, S.; Wang, B.; Wu, B.; Huang, H.; Qin, X.; Li, D.; Yan, W. Half-Metallicity and Magnetism of the Quaternary Heusler Compound TiZrCoIn1-xGex from the First-Principles Calculations. Appl. Sci. 2019, 9, 620. [Google Scholar] [CrossRef]
- Huang, H.M.; Luo, S.J.; Yao, K.L. Half-metallicity and tetragonal distortion in semi-heusler alloy FeCrSe. J. Appl. Phys. 2014, 115, 043713. [Google Scholar] [CrossRef]
- Wang, X.T.; Dai, X.F.; Wang, L.Y.; Liu, X.F.; Wang, W.H.; Wu, G.H.; Tang, C.C.; Liu, G.D. Electronic structures and magnetism of Rh3Z (Z=Al, Ga, In, Si, Ge, Sn, Pb, Sb) with DO3 structures. J. Magn. Magn. Mater. 2015, 378, 16–23. [Google Scholar] [CrossRef]
- Feng, L.; Ma, J.; Yang, Y.; Lin, T.; Wang, L. The electronic, magnetic, half-metallic and mechanical properties of the equiatomic quaternary heusler compounds FeRhCrSi and FePdCrSi: A first-Principles Study. Appl. Sci. 2018, 8, 2370. [Google Scholar] [CrossRef]
- Schwarz, K. CrO2 predicted as a half-metallic ferromagnet. J. Phys. F Met. Phys. 1986, 16, L211–L2015. [Google Scholar] [CrossRef]
- Kim, W.; Kawaguchi, K.; Koshizaki, N. Fabrication and magnetoresistance of tunnel junctions using half-metallic Fe3O4. J. Appl. Phys. 2003, 93, 8032–8034. [Google Scholar] [CrossRef]
- Huang, H.M.; Jiang, Z.Y.; Lin, Y.M.; Zhou, B.; Zhang, C.K. Design of half-metal and spin gapless semiconductor for spintronics application viacation substitution in methylammonium lead iodide. Appl. Phys. Express 2017, 10, 123002. [Google Scholar] [CrossRef]
- Lv, S.H.; Li, H.P.; Liu, X.J.; Han, D.M.; Wu, Z.J.; Meng, J.A. new half-metallic ferromagnet La2NiFeO6: Predicted from first-principles calculations. J. Phys. Chem. C 2010, 114, 16710–16715. [Google Scholar] [CrossRef]
- Haneef, M.; Arif, S.; Akbar, J.; Abdul-Malik, A. Theoretical investigations of half-metallicity in Cr-substituted GaN, GaP, GaAs, GaSb material systems. J. Electron. Mater 2014, 43, 3169–3176. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, J.M. Effect of two identical 3d transition-metal atoms M doping (M=V, Cr, Mn, Fe, Co, and Ni) on the structural, electronic, and magnetic properties of ZnO. Phys. Status Solidi B 2017, 254, 1700098. [Google Scholar] [CrossRef]
- Gao, G.Y.; Yao, K.L.; Sasioglu, E.; Sandratskii, L.M.; Liu, Z.L.; Jiang, J.L. Half-metallic ferromagnetism in zinc-blende CaC, SrC, and BaC from first principles. Phys. Rev. B 2007, 75, 174442. [Google Scholar] [CrossRef]
- Xie, W.H.; Xu, Y.Q.; Liu, B.G.; Pettifor, D.G. Half-metallic ferromagnetism and structural stability of zincblende phases of the transition-metal chalcogenides. Phys. Rev. Lett. 2003, 91, 037204. [Google Scholar] [CrossRef]
- Yao, K.L.; Zhu, L.; Liu, Z.L. First-principles study of the ferromagnetic and half metallic properties of the fumarate-bridged polymer. Eur. Phys. J. B 2004, 39, 283–286. [Google Scholar] [CrossRef]
- Huang, H.M.; Luo, S.J.; Liu, G.Y.; Yao, K.L. First-principles study of electronic structure and half-metallicity of molecule-based ferromagnet Cr[N(CN)2]2. Chin. J. Chem. Phys. 2011, 24, 189–193. [Google Scholar] [CrossRef]
- Zheng, J.M.; He, R.J.; Wan, Y.; Zhao, P.J.; Guo, P.; Jiang, Z.Y. Half- metal state of a Ti2C monolayer by asymmetric surface decoration. Phys. Chem. Chem. Phys. 2019, 21, 3318–3326. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Li, S.S.; Zhang, C.W.; Zhang, S.F.; Ji, W.X.; Li, P.; Wang, P.J. High-temperature Dirac half-metal PdCl3: A promising candidate for realizing quantum anomalous Hall effect. J. Mater. Chem. C 2018, 6, 10284–10291. [Google Scholar] [CrossRef]
- Lv, P.; Tang, G.; Yang, C.; Deng, J.M.; Liu, Y.Y.; Wang, X.Y.; Wang, X.Q.; Hong, J.W. Half-metallicity in two-dimensional Co2Se3 monolayer with superior mechanical flexibility. 2D Mater. 2018, 5, 045026. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, H.L.; Zhang, L.F.; Hu, S.L.; Hu, Z.P. Computational study on the half-metallicity in transition metal-oxide-incorporated 2D g-C3N4 nanosheets. Front. Phys. 2018, 13, 138108. [Google Scholar] [CrossRef]
- Deng, L.; Yang, Z.; Tan, L.; Zeng, L.; Zhu, Y.; Guo, L. Investigation of the Prussian Blue Analog Co3[Co(CN)6]2 as an Anode Material for Nonaqueous Potassium-Ion Batteries. Adv. Mater. 2018, 30, 1802510. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Wang, H.; Deng, B.; Zhang, H.; Wang, L.; Wan, Q.; Yan, X.; Qu, M. A Mn Fe based Prussian blue Analogue@Reduced graphene oxide composite as high capacity and superior rate capability anode for lithium-ion batteries. Carbon 2019, 143, 706–713. [Google Scholar] [CrossRef]
- Gao, Q.; Shi, N.; Sanson, A.; Sun, Y.; Milazzo, R.; Olivi, L.; Zhu, H.; Lapidus, S.H.; Zheng, L.; Chen, J.; Xing, X. Tunable Thermal Expansion from Negative, Zero, to Positive in Cubic Prussian Blue Analogues of GaFe(CN)6. Inorg. Chem. 2018, 57, 14027–14030. [Google Scholar] [CrossRef] [PubMed]
- Xiong, P.; Zeng, G.; Zeng, L.; Wei, M. Prussian blue analogues Mn[Fe(CN)6]0.6667·nH2O cubes as an anode material for lithium-ion batteries. Dalton Trans. 2015, 44, 16746–16751. [Google Scholar] [CrossRef] [PubMed]
- Holmes, S.H.; Girolami, G.S. Sol-Gel Synthesis of KVII[CrIII(CN)6]·2H2O: A Crystalline Molecule-Based Magnet with a Magnetic Ordering Temperature above 100 °C. J. Am. Chem. Soc. 1999, 121, 5593–5594. [Google Scholar] [CrossRef]
- Sato, O.; Hayami, S.; Einaga, Y.; Gu, Z.Z. Control of the Magnetic and Optical Properties in Molecular Compounds by Electrochemical, Photochemical and Chemical Methods. Bull. Chem. Soc. Jpn. 2003, 76, 443–470. [Google Scholar] [CrossRef]
- Wojdel, J.C.; Moreira, I.P.R.; Bromleyac, S.T.; Illas, F. Prediction of half-metallic conductivity in Prussian Blue derivatives. J. Mater. Chem. 2009, 19, 2032–2036. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Lv, Y.; Liu, C.; Wang, F.; Jiang, W. Site preference of transition-metal elements additions on mechanical and electronic properties of B2DyCu-based alloys. Mater. Des. 2017, 133, 476–486. [Google Scholar] [CrossRef]
- Zhang, X.D.; Huang, W.Y.; Ma, H.; Yu, H.; Jiang, W. First-principles prediction of the physical properties of ThM2Al20 (M= Ti, V, Cr) intermetallics. Solid State Commun. 2018, 284–286, 75–83. [Google Scholar] [CrossRef]
- Mehmood, N.; Ahmad, R.; Murtaza, G. Ab initio investigations of structural, elastic, mechanical, electronic, magnetic, and optical properties of half-heusler compounds RhCrZ (Z = Si, Ge). J. Supercond. Novel Magn. 2017, 30, 2481–2488. [Google Scholar] [CrossRef]
- Huang, Y.C.; Guo, X.F.; Ma, Y.L.; Shao, H.B.; Xiao, Z.B. Stabilities, electronic and elastic properties of L12-Al3(Sc1-x,Zrx) with different Zr content: A first-principles study. Physica B 2018, 548, 27–33. [Google Scholar] [CrossRef]
- Huang, H.M.; Luo, S.J.; Xiong, Y.C. Pressure-induced electronic, magnetic, half-metallic, and mechanical properties of half-Heusler compound CoCrBi. J. Magn. Magn. Mater. 2017, 438, 5–11. [Google Scholar] [CrossRef]
- Anderson, O.L. A simplified method for calculating the debye temperature from elastic constants. J. Phys. Chem. Solids 1963, 24, 909–917. [Google Scholar] [CrossRef]
- Allali, D.; Bouhemadou, A.; Zerarga, F.; Ghebouli, M.A.; Bin-Omran, S. Prediction study of the elastic and thermodynamic properties of the SnMg2O4, SnZn2O4 and SnCd2O4 spinel oxides. Comput. Mater. Sci. 2012, 60, 217–223. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom | Exp. | Present | ||||
|---|---|---|---|---|---|---|
| x | y | z | x | y | z | |
| Ga | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Fe | 0.5 | 0.0 | 0.0 | 0.5 | 0.0 | 0.0 |
| C | 0.3043 | 0.0 | 0.0 | 0.3253 | 0.0 | 0.0 |
| N | 0.1883 | 0.0 | 0.0 | 0.2114 | 0.0 | 0.0 |
| Pressure | a (Å) | C-N(Å) | Ga-N(Å) | Fe-C(Å) | C(x,0,0) | N(x,0,0) |
|---|---|---|---|---|---|---|
| 0 | 10.1883 | 1.160 | 2.155 | 1.780 | 0.32533 | 0.21148 |
| 5 | 10.0706 | 1.156 | 2.118 | 1.762 | 0.32508 | 0.21029 |
| 10 | 9.9649 | 1.152 | 2.085 | 1.745 | 0.32492 | 0.20928 |
| 15 | 9.8695 | 1.149 | 2.057 | 1.729 | 0.32481 | 0.20843 |
| 20 | 9.7830 | 1.145 | 2.028 | 1.719 | 0.32430 | 0.20728 |
| 25 | 9.7015 | 1.142 | 2.008 | 1.701 | 0.32471 | 0.20701 |
| 30 | 9.6271 | 1.138 | 1.987 | 1.688 | 0.32461 | 0.20636 |
| 35 | 9.5563 | 1.135 | 1.967 | 1.676 | 0.32459 | 0.20579 |
| 40 | 9.4828 | 1.132 | 1.945 | 1.665 | 0.32447 | 0.20512 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Huang, H.; Luo, S. First Principles Study on the Effect of Pressure on the Structure, Elasticity, and Magnetic Properties of Cubic GaFe(CN)6 Prussian Blue Analogue. Appl. Sci. 2019, 9, 1607. https://doi.org/10.3390/app9081607
Zhang C, Huang H, Luo S. First Principles Study on the Effect of Pressure on the Structure, Elasticity, and Magnetic Properties of Cubic GaFe(CN)6 Prussian Blue Analogue. Applied Sciences. 2019; 9(8):1607. https://doi.org/10.3390/app9081607
Chicago/Turabian StyleZhang, Chuankun, Haiming Huang, and Shijun Luo. 2019. "First Principles Study on the Effect of Pressure on the Structure, Elasticity, and Magnetic Properties of Cubic GaFe(CN)6 Prussian Blue Analogue" Applied Sciences 9, no. 8: 1607. https://doi.org/10.3390/app9081607
APA StyleZhang, C., Huang, H., & Luo, S. (2019). First Principles Study on the Effect of Pressure on the Structure, Elasticity, and Magnetic Properties of Cubic GaFe(CN)6 Prussian Blue Analogue. Applied Sciences, 9(8), 1607. https://doi.org/10.3390/app9081607