From Symmetry Breaking via Charge Migration to Symmetry Restoration in Electronic Ground and Excited States: Quantum Control on the Attosecond Time Scale

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Model, Concept, Theory and Methods
2.1. Model and Basic Theory
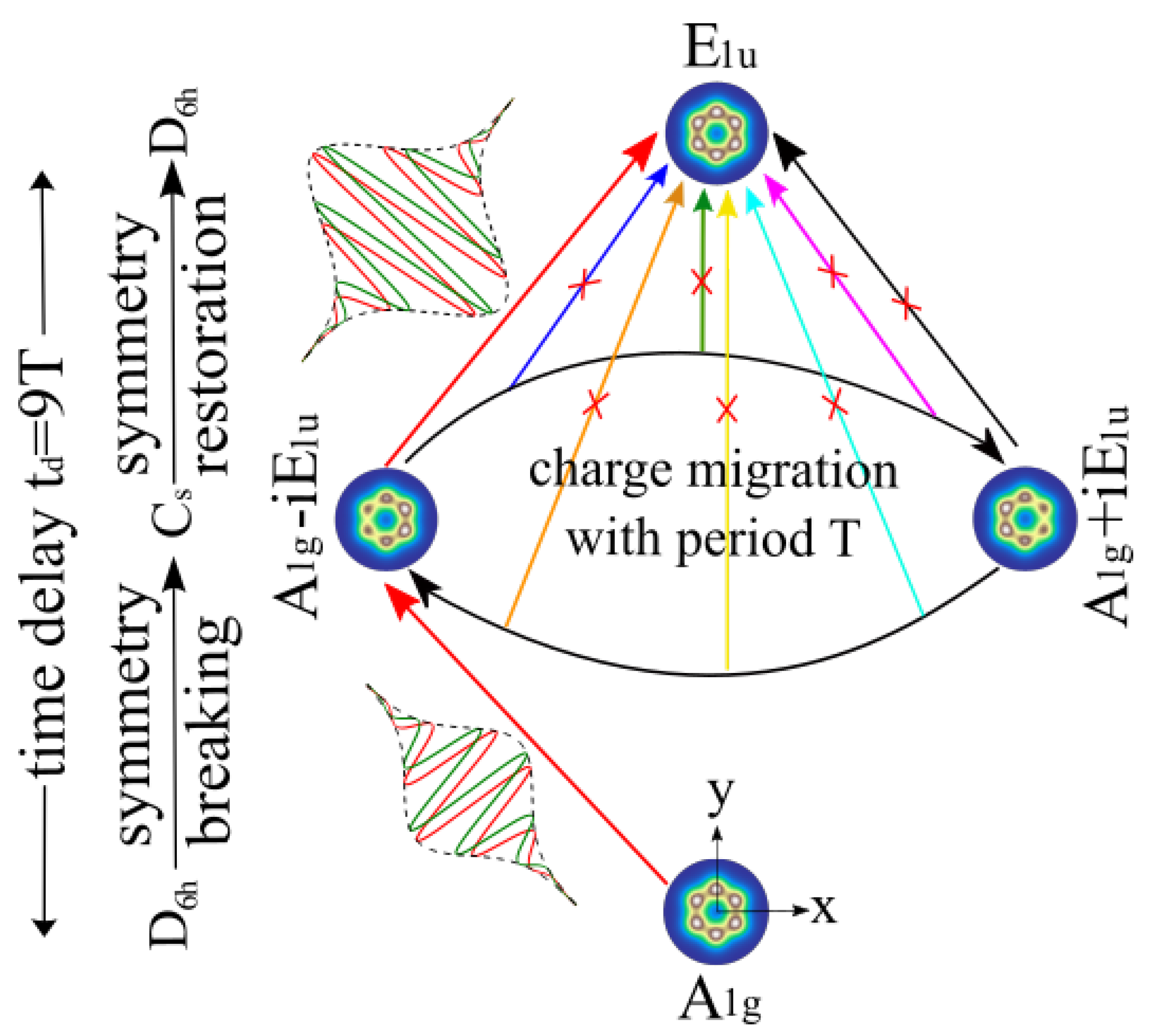
2.2. Conceptual Background for the New Symmetry Restoration Strategy
2.3. Extended Theory for the New Strategy
3. Results and Discussions
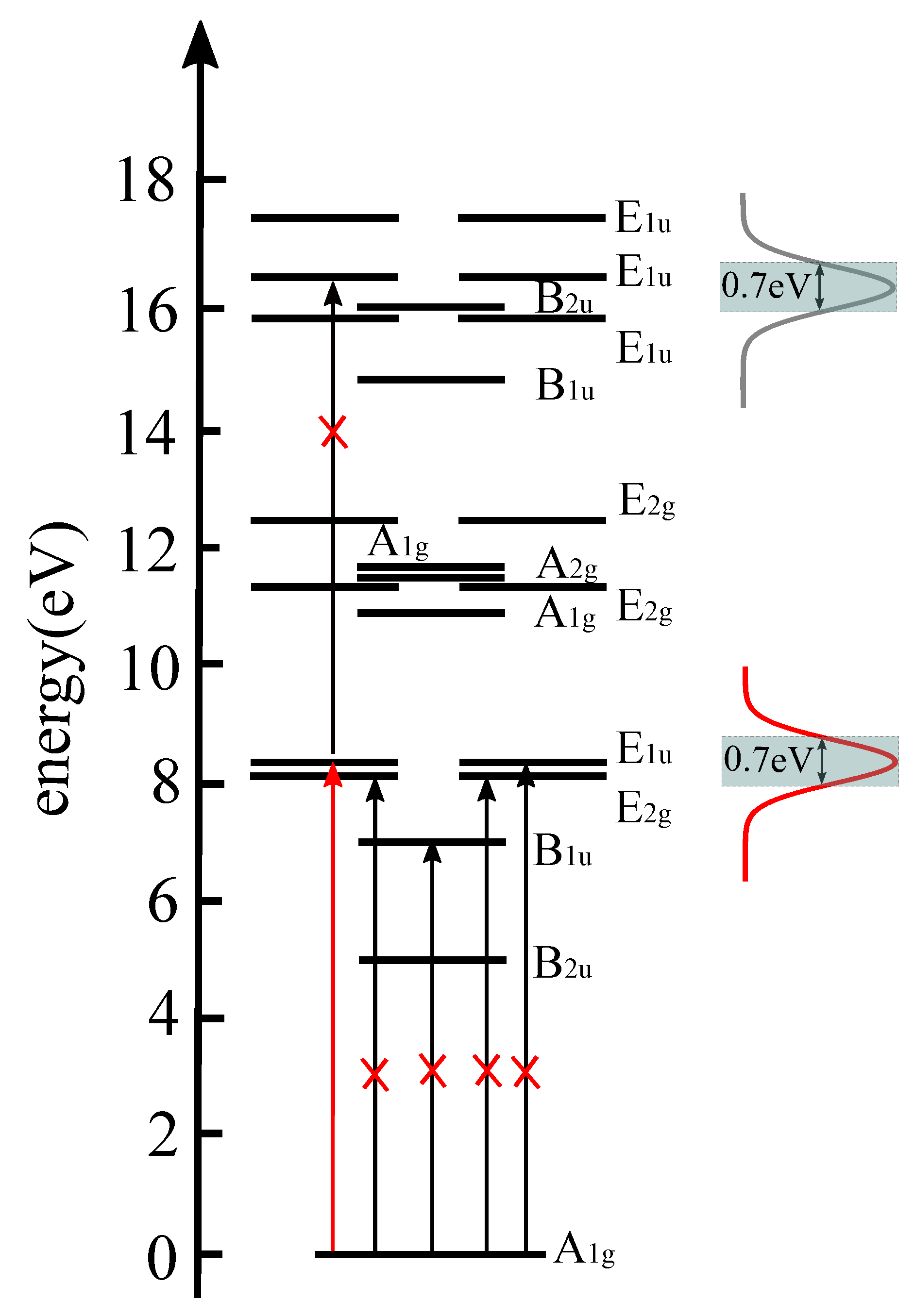
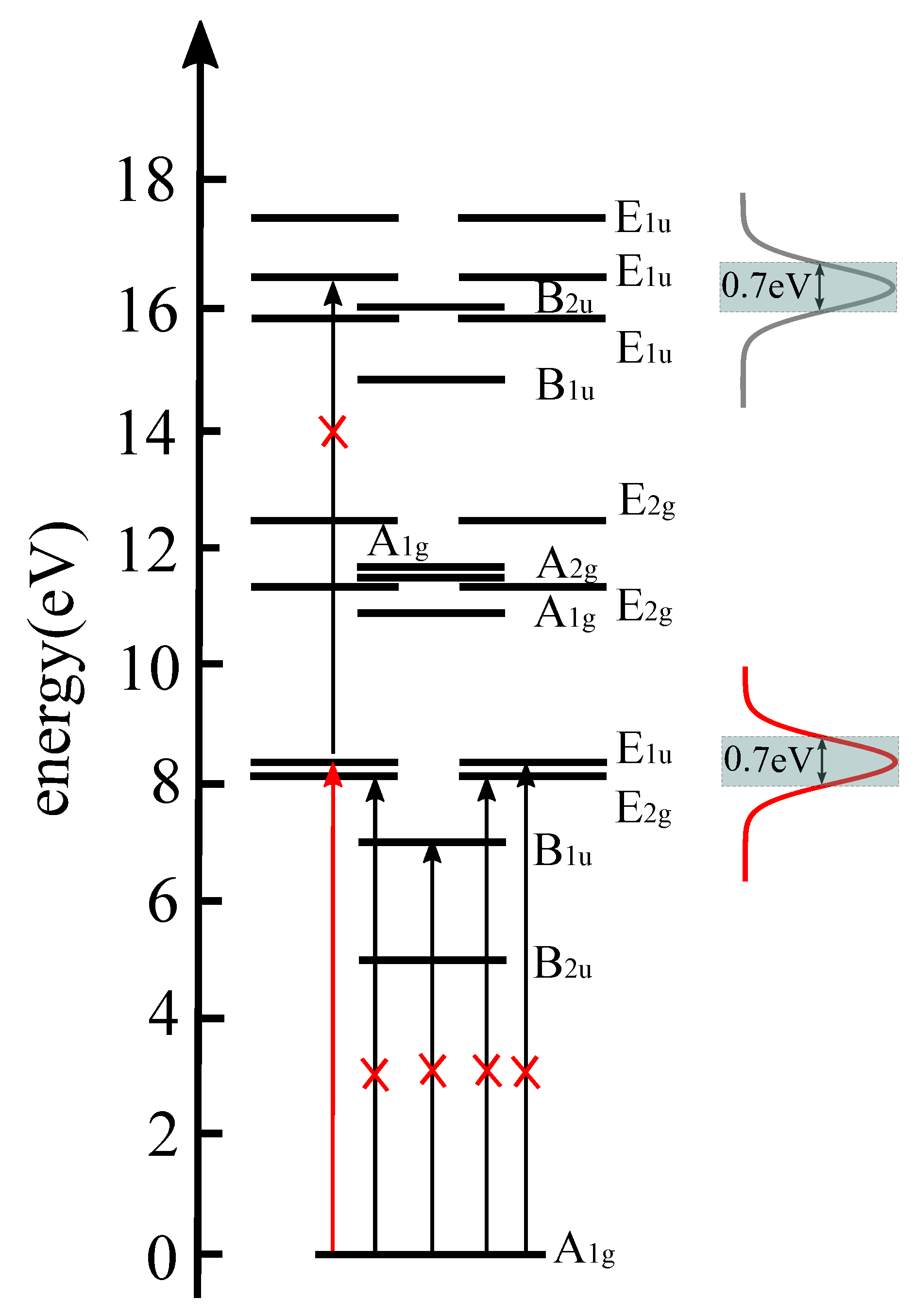
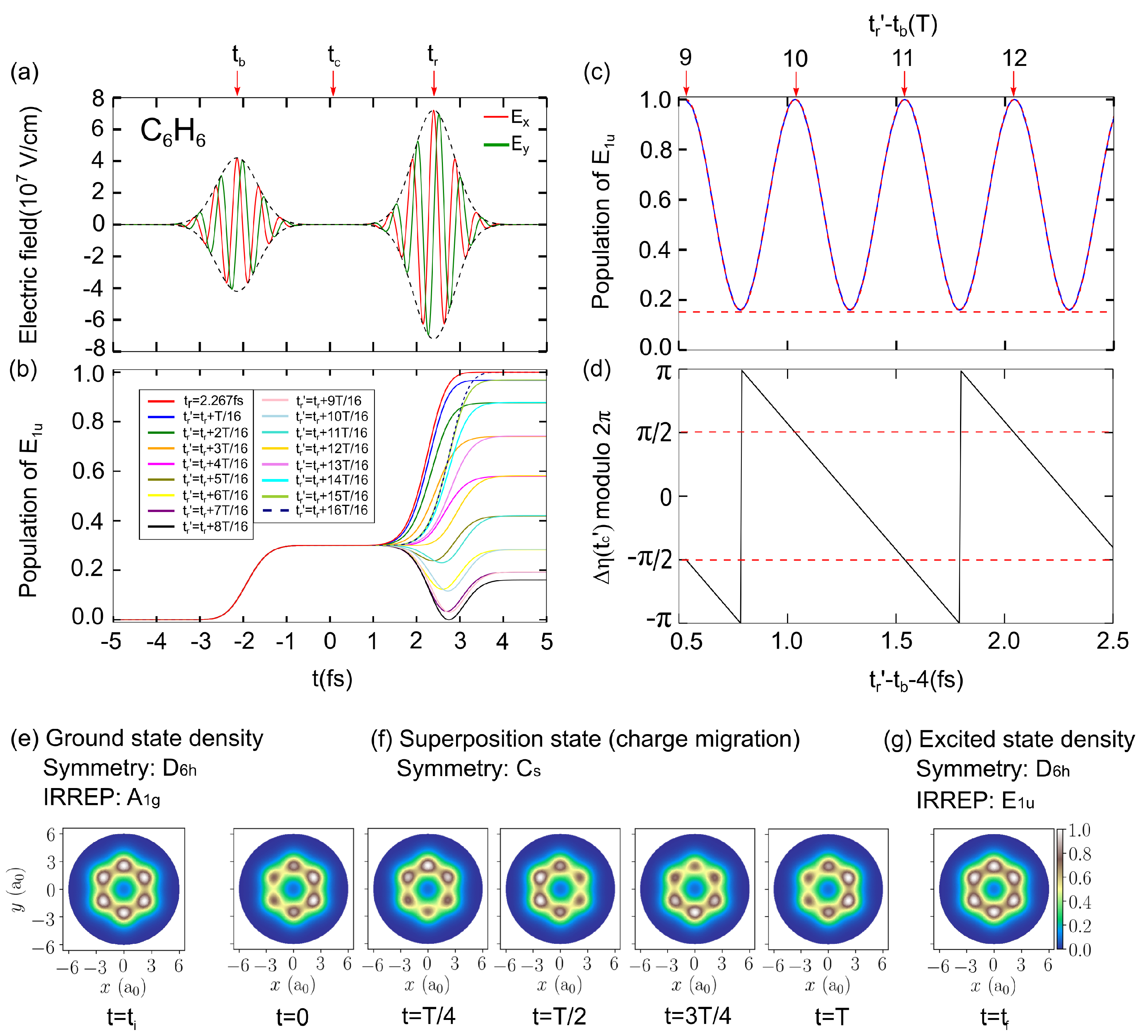
3.1. The Proof-of-Principle for Quantum Control of Symmetry Breaking and Restoration of Molecules in Electronic Ground and Excited States with Different
3.2. The Requirement of Attosecond Precision for the Proper Time Delay Between the Laser Pulses for Electronic Structure Symmetry Breaking and Restoration
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ivanov, M.; Corkum, P.B.; Dietrich, P. Coherent Control and Collapse of Symmetry in a Two-Level System in an Intense Laser Field. Laser Phys. 1993, 3, 375–380. [Google Scholar]
- Weinacht, T.C.; Ahn, J.; Bucksbaum, P.H. Measurement of the Amplitude and Phase of a Sculpted Rydberg Wave Packet. Phys. Rev. Lett. 1998, 80, 5508–5511. [Google Scholar] [CrossRef]
- Elghobashi, N.; González, L.; Manz, J. Quantum model simulations of symmetry breaking and control of bond selective dissociation of FHF− using IR+UV laser pulses. J. Chem. Phys. 2004, 120, 8002–8014. [Google Scholar] [CrossRef] [PubMed]
- Arasaki, Y.; Takatsuka, K. Optical conversion of conical intersection to avoided crossing. Phys. Chem. Chem. Phys. 2010, 12, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Arasaki, Y.; Wang, K.; McKoy, V.; Takatsuka, K. Monitoring the effect of a control pulse on a conical intersection by time-resolved photoelectron spectroscopy. Phys. Chem. Chem. Phys. 2011, 13, 8681–8689. [Google Scholar] [CrossRef] [PubMed]
- Alnasser, A.S.; Kübel, M.; Siemering, R.; Bergues, B.; Kling, N.G.; Betsch, K.J.; Deng, Y.; Schmidt, J.; Alahmed, Z.A.; Azzeer, A.M.; et al. Subfemtosecond steering of hydrocarbon deprotonation through superposition of vibrational modes. Nat. Commun. 2014, 5, 3800. [Google Scholar] [CrossRef]
- Pertot, Y.; Schmidt, C.; Matthews, M.; Chauvet, A.; Huppert, M.; Svoboda, V.; Conta, A.; Tehlar, A.; Baykusheva, D.; Wolf, J.-P.; et al. Time-resolved x-ray absorption spectroscopy with a water window high-harmonic source. Science 2017, 355, 264–267. [Google Scholar] [CrossRef]
- Cederbaum, L.S.; Zobeley, J. Ultrafast charge migration by electron correlation. Chem. Phys. Lett. 1999, 307, 205–210. [Google Scholar] [CrossRef]
- Remacle, F.; Levine, R.D. An electronic time scale in chemistry. Proc. Natl. Acad. Sci. USA 2006, 103, 6793–6798. [Google Scholar] [CrossRef]
- Chelkowski, S.; Yudin, G.L.; Bandrauk, A.D. Observing electron motion in molecules. J. Phys. B 2006, 39, S409–S417. [Google Scholar] [CrossRef]
- Barth, I.; Manz, J. Periodic Electron Circulation Induced by Circularly Polarized Laser Pulses: Quantum Model Simulations for Mg Porphyrin. Angew. Chem. Int. Ed. 2006, 45, 2962–2965. [Google Scholar] [CrossRef] [PubMed]
- Kanno, M.; Kono, H.; Fujimura, Y. Control of π-Electron Rotation in Chiral Aromatic Molecules by Nonhelical Laser Pulses. Angew. Chem. Int. Ed. 2006, 45, 7995–7998. [Google Scholar] [CrossRef] [PubMed]
- Mineo, H.; Lin, S.H.; Fujimura, Y. Vibrational effects on UV/Vis laser-driven π-electron ring currents in aromatic ring molecules. Chem. Phys. 2014, 442, 103–110. [Google Scholar] [CrossRef]
- Despré, V.; Marciniak, A.; Loriot, V.; Galbraith, M.C.E.; Rouzée, A.; Vrakking, M.J.J.; Lépine, F.; Kuleff, A.I. Attosecond Hole Migration in Benzene Molecules Surviving Nuclear Motion. J. Phys. Chem. Lett. 2015, 6, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Kraus, P.M.; Mignolet, B.; Baykusheva, D.; Rupenyan, A.; Horny, L.; Penka, E.F.; Grassi, G.; Tolstikhin, O.I.; Schneider, J.; Jensen, F.; et al. Measurement and laser control of attosecond charge migration in ionized iodoacetylene. Science 2015, 350, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K. To catch and smash charge on the hop. Science 2015, 350, 740–741. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Manz, J.; Paulus, B.; Pohl, V.; Tremblay, J.C.; Yang, Y. Quantum control of electronic fluxes during adiabatic attosecond charge migration in degenerate superposition states of benzene. Chem. Phys. 2017, 482, 146–159. [Google Scholar] [CrossRef]
- Golubev, N.V.; Despré, V.; Kuleff, A. Quantum control with smoothly varying pulses: General theory and application to charge migration. J. Mod. Opt. 2017, 64, 1031–1041. [Google Scholar] [CrossRef]
- Liu, C.; Manz, J.; Ohmori, K.; Sommer, C.; Takei, N.; Tremblay, J.C.; Zhang, Y. Attosecond Control of Restoration of Electronic Structure Symmetry. Phys. Rev. Lett. 2018, 121, 173201. [Google Scholar] [CrossRef]
- Liu, C.; Manz, J.; Tremblay, J.C. Progress in Ultrafast Intense Laser Science XIV; Yamanouchi, K., Martin, P., Sentis, M., Li, R., Normand, D., Eds.; Springer International Publishing: Cham, Switzerland, 2018; Volume 14, Chapter 7; pp. 117–141. ISBN 9783030037857. [Google Scholar]
- Bacic, Z.; Benoit, D.; Biczysko, M.; Bowman, J.; Bradforth, S.; Burd, T.; Chambaud, G.; Clary, D.; Crépin, C.; Dracinsky, M.; et al. Molecules in confinement in clusters, quantum solvents and matrices: General discussion. Faraday Discuss. 2018, 212, 569–601. [Google Scholar] [CrossRef]
- Bandrauk, A.D.; Chelkowski, S.; Corkum, P.B.; Manz, J.; Yudin, G.L. Attosecond Photoionization of a Coherent Superposition of Bound and Dissociative Molecular States. J. Phys. B 2009, 42, 134001. [Google Scholar] [CrossRef]
- Mendive-Tapia, D.; Vacher, M.; Bearpark, M.J.; Robb, M.A. Coupled electron-nuclear dynamics: Charge migration and charge transfer initiated near a conical intersection. J. Chem. Phys. 2013, 139, 044110. [Google Scholar] [CrossRef] [PubMed]
- Ulusoy, I.; Nest, M. Correlated Electron Dynamics: How Aromaticity Can Be Controlled. J. Am. Chem. Soc. 2011, 133, 20230–20236. [Google Scholar] [CrossRef] [PubMed]
- Hermann, G.; Liu, C.; Manz, J.; Paulus, B.; Pérez-Torres, J.F.; Pohl, V.; Tremblay, J.C. Multidirectional Angular Electronic Flux during Adiabatic Attosecond Charge Migration in Excited Benzene. J. Phys. Chem. A 2016, 120, 5360–5369. [Google Scholar] [CrossRef] [PubMed]
- Hermann, G.; Liu, C.; Manz, J.; Paulus, B.; Pohl, V.; Tremblay, J.C. Attosecond Angular Flux of Partial Charges on the Carbon Atoms of Benzene in Non-Aromatic Excited States. Chem. Phys. Lett. 2017, 683, 553–558. [Google Scholar] [CrossRef]
- Alber, G.; Ritsch, H.; Zoller, P. Generation and detection of Rydberg wave packets by short laser pulses. Phys. Rev. A 1986, 34, 1058–1064. [Google Scholar] [CrossRef]
- Noordam, L.D.; Duncam, D.I.; Gallagher, T.F. Ramsey fringes in atomic Rydberg wave packets. Phys. Rev. A. 1992, 45, 4734–4737. [Google Scholar] [CrossRef]
- Corkum, P.B. Plasma perspective on strong-field multiphoton ionization. Phys. Rev. Lett. 1993, 71, 1994–1997. [Google Scholar] [CrossRef]
- Corkum, P.B.; Krausz, F. Attosecond science. Nat. Phys. 2007, 3, 381–387. [Google Scholar] [CrossRef]
- Krausz, F.; Ivanov, M. Attosecond physics. Rev. Mod. Phys. 2009, 81, 163–234. [Google Scholar] [CrossRef]
- Chelkowski, S.; Bredtmann, T.; Bandrauk, A.D. High-order-harmonic generation from coherent electron wave packets in atoms and molecules as a tool for monitoring attosecond electrons. Phys. Rev. A 2012, 85, 033404. [Google Scholar] [CrossRef]
- Schultz, T.; Vrakking, M. Attosecond and XUV Physics; Wiley-VCH: Weinheim, Germany, 2014; ISBN 9783527411245. [Google Scholar]
- Woodward, R.; Hoffmann, R. Die Erhaltung der Orbitalsymmetrie; Verlag Chemie: Weinheim, Germany, 1972; ISBN 3527253238. [Google Scholar]
- Halevi, E. Orbital Symmetry and Reaction Mechanism: The OCAMS View; Springer: Berlin, Germany, 1992; ISBN 9783642835681. [Google Scholar]
- Campbell, E.E.B.; Schmidt, H.; Hertel, I.V. Symmetry and Angular Momentum in Collisions with Laser Excited Polarized Atoms. Adv. Chem. Phys. 1988, 72, 37–114. [Google Scholar] [CrossRef]
- Al-Jabour, S.; Leibscher, M. Effects of Molecular Symmetry on Quantum Reaction Dynamics: Novel Aspects of Photoinduced Nonadiabatic Dynamics. J. Phys. Chem. A 2015, 119, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Schleich, W.P.; Walther, H. Elements of Quantum Information; Wiley-VCH: Weinheim, Germany, 2007; ISBN 9783527407255. [Google Scholar]
- Brif, C.; Chakrabarti, R.; Rabitz, H. Control of quantum phenomena: Past, present and future. New. J. Phys. 2010, 12, 075008. [Google Scholar] [CrossRef]
- Shapiro, M.; Brumer, P. Quantum Control of Molecular Processes, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2012; ISBN 9783527409044. [Google Scholar]
- Tannor, D.J. Introduction to Quantum Mechanics: A Time-Dependent Perspective; University Science Books: Sausalito, CA, USA, 2012; ISBN 10:1-891389-23-8. [Google Scholar]
- Barth, I.; Manz, J. Unidirectional Electronic Ring Current driven by a Few Cycle Circularly Polarized Pulse: Quantum Model Simulations for Mg-Porphyrine. J. Am. Chem. Soc. 2006, 128, 7043–7049. [Google Scholar] [CrossRef] [PubMed]
- Hermann, G.; Pohl, V.; Tremblay, J.C.; Paulus, B.; Hege, H.-C.; Schild, A. ORBKIT—A Modular Python Toolbox for Cross-Platform Post-Processing of Quantum Chemical Wavefunction Data. J. Comput. Chem. 2016, 37, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Pohl, V.; Hermann, G.; Tremblay, J.C. An Open-Source Framework for Analyzing N-Electron Dynamics: I. Multi-Determinantal Wave Functions. J. Comput. Chem. 2017, 38, 1515–1527. [Google Scholar] [CrossRef]
- Hermann, G.; Pohl, V.; Tremblay, J.C. An Open-Source Framework for N-Electron Dynamics: II. Hybrid Density Functional Theory Configuration Interaction Methodology. J. Comput. Chem. 2017, 38, 2378–2387. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Liu, C.; Manz, J.; Yang, Y. Laser Sculpting of Atomic sp, sp2, and sp3 Hybrid Orbitals. Chem. Phys. Chem. 2015, 16, 191–196. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Manz, J.; Tremblay, J.C. From Symmetry Breaking via Charge Migration to Symmetry Restoration in Electronic Ground and Excited States: Quantum Control on the Attosecond Time Scale. Appl. Sci. 2019, 9, 953. https://doi.org/10.3390/app9050953
Liu C, Manz J, Tremblay JC. From Symmetry Breaking via Charge Migration to Symmetry Restoration in Electronic Ground and Excited States: Quantum Control on the Attosecond Time Scale. Applied Sciences. 2019; 9(5):953. https://doi.org/10.3390/app9050953
Chicago/Turabian StyleLiu, ChunMei, Jörn Manz, and Jean Christophe Tremblay. 2019. "From Symmetry Breaking via Charge Migration to Symmetry Restoration in Electronic Ground and Excited States: Quantum Control on the Attosecond Time Scale" Applied Sciences 9, no. 5: 953. https://doi.org/10.3390/app9050953
APA StyleLiu, C., Manz, J., & Tremblay, J. C. (2019). From Symmetry Breaking via Charge Migration to Symmetry Restoration in Electronic Ground and Excited States: Quantum Control on the Attosecond Time Scale. Applied Sciences, 9(5), 953. https://doi.org/10.3390/app9050953