Advances in Engineering and Application of Optogenetic Indicators for Neuroscience


Abstract
:1. Introduction
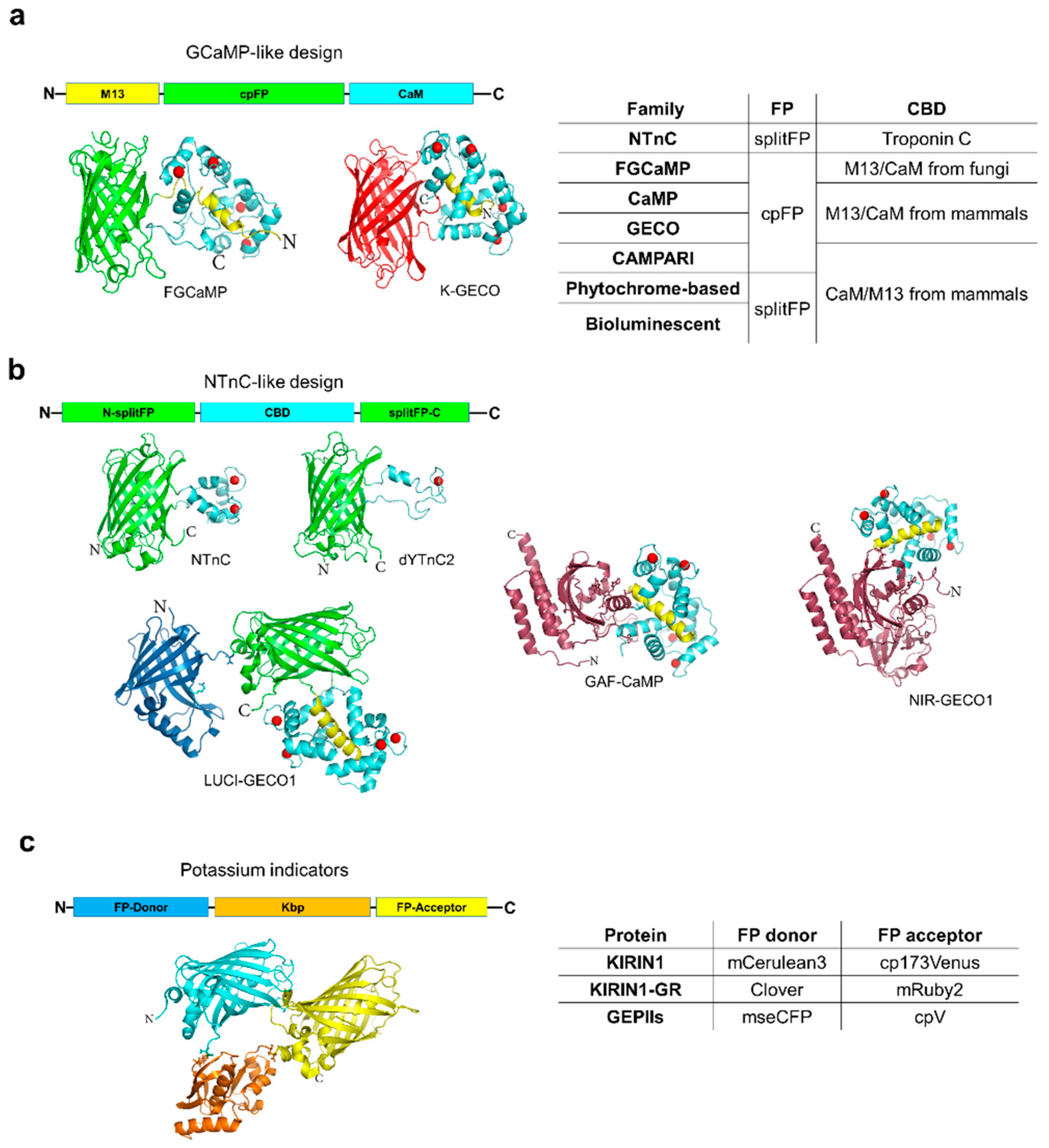
2. Calcium Indicators
3. Potassium Indicators
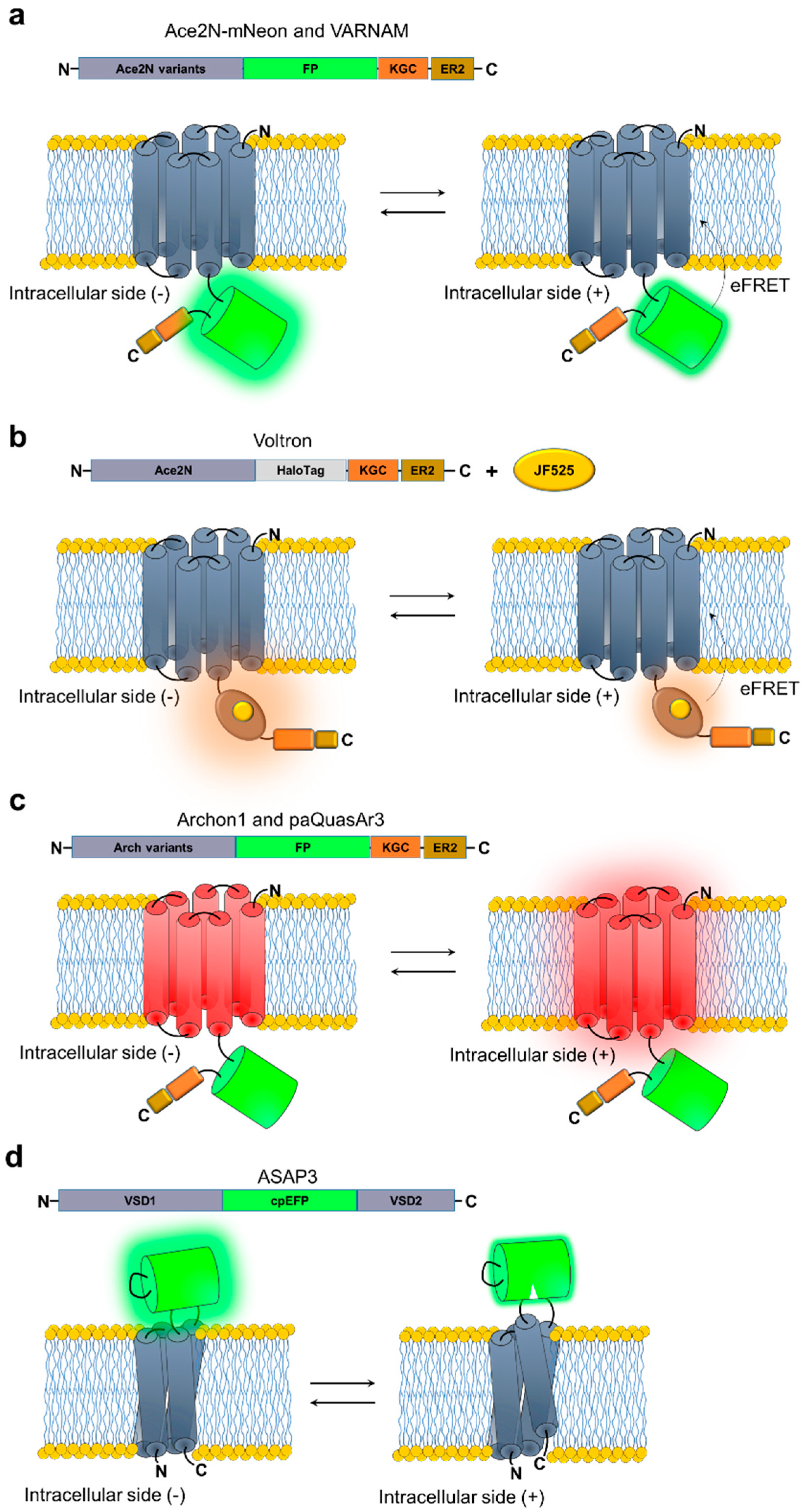
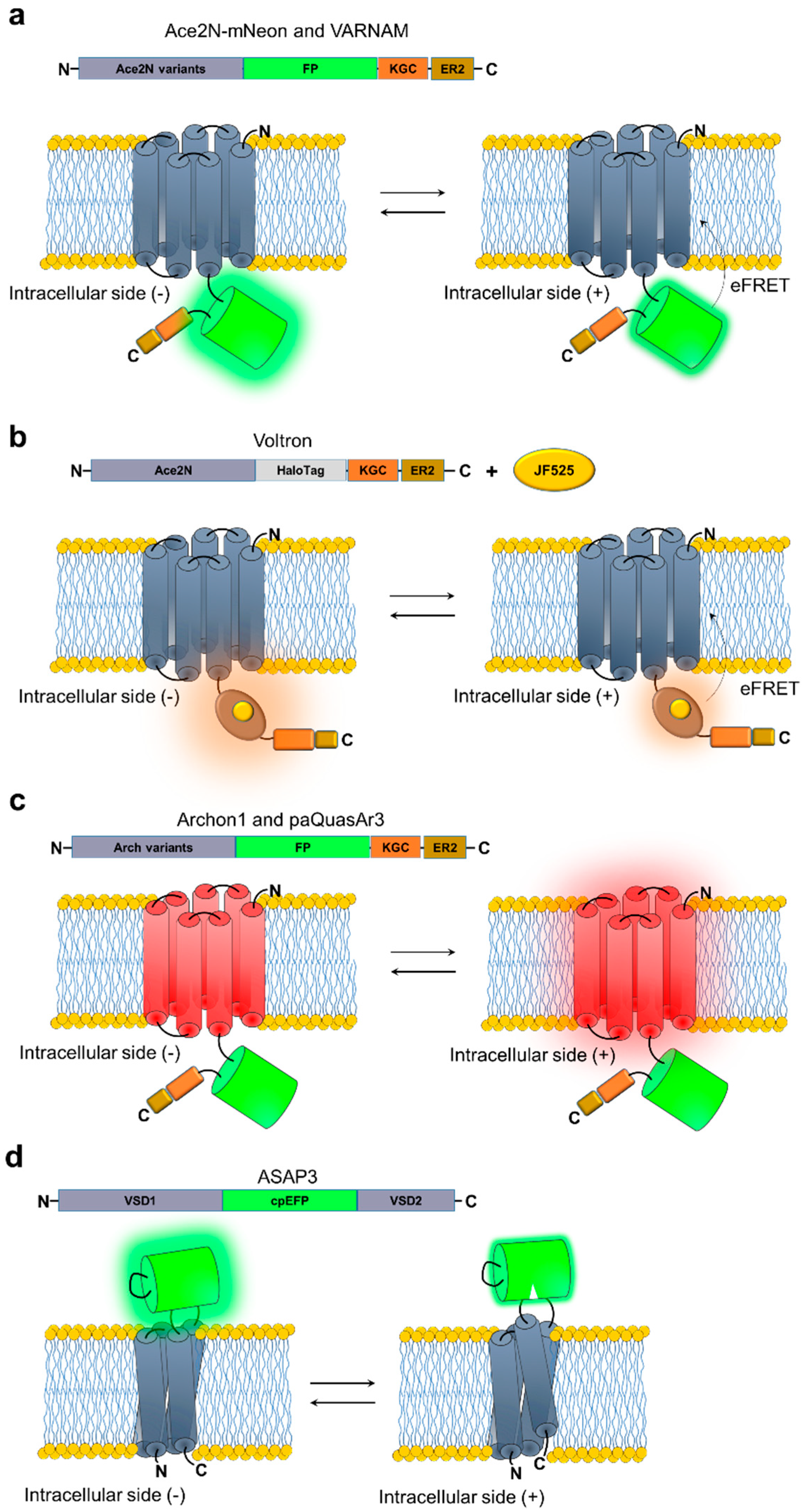
4. Voltage Sensors
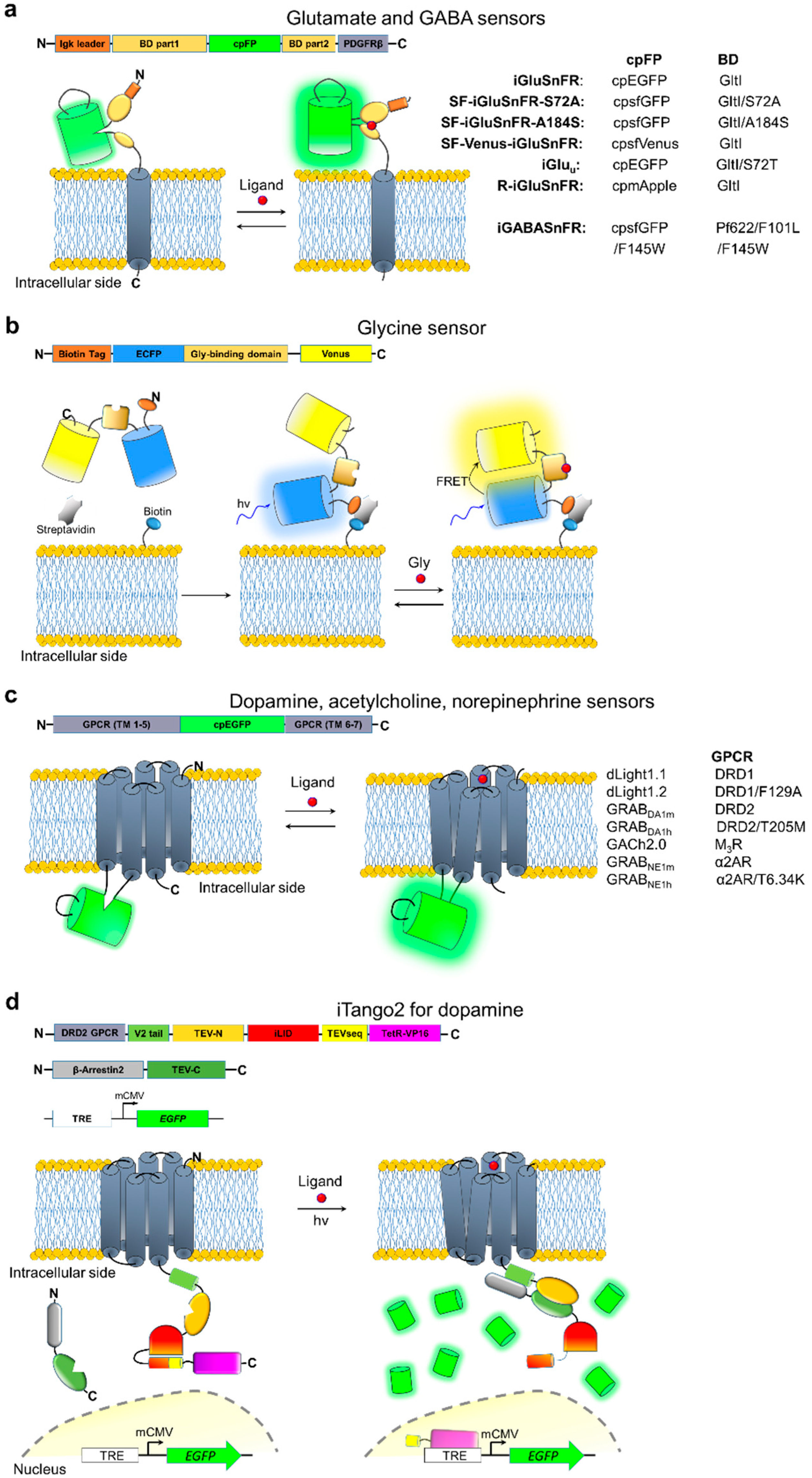
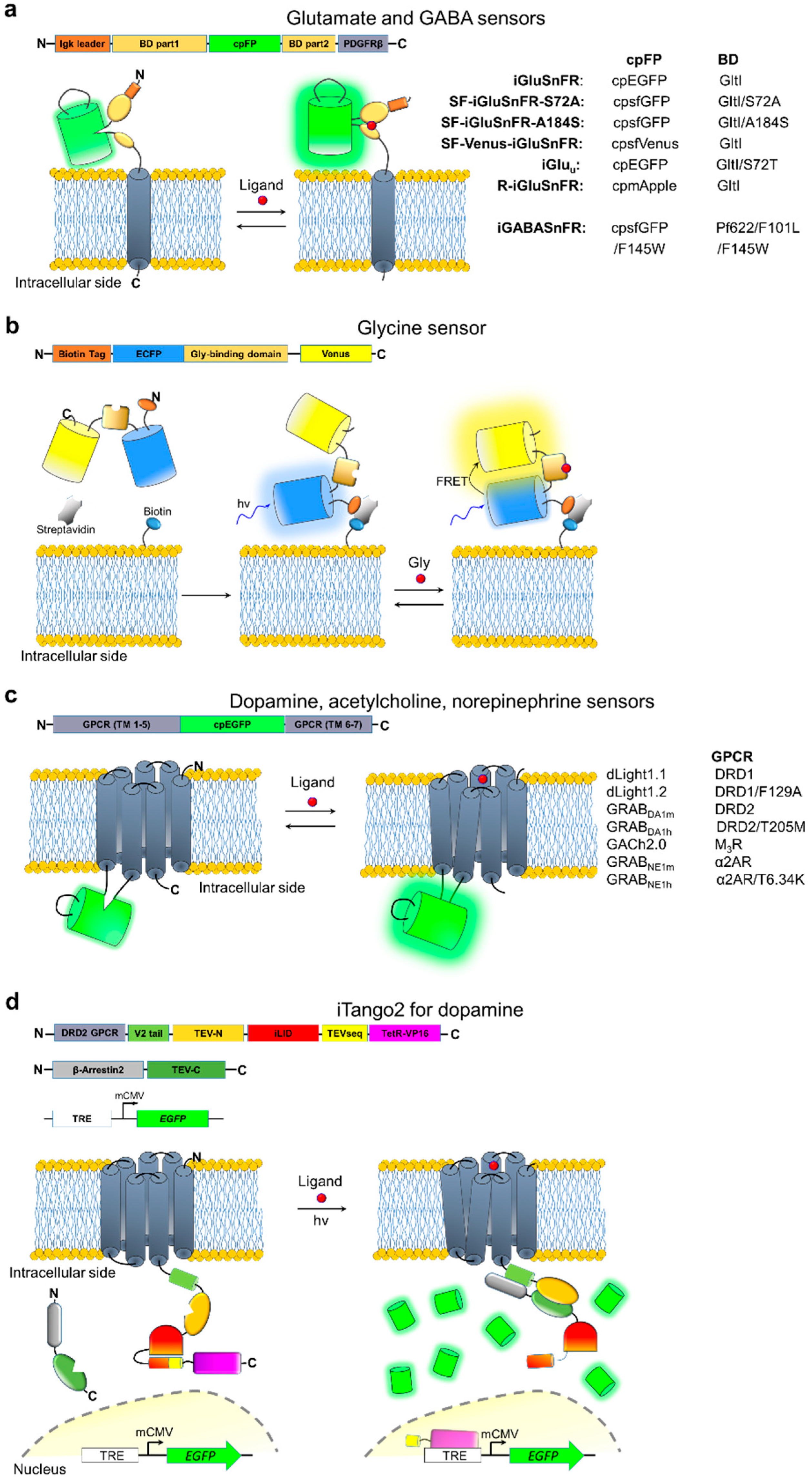
5. Neurotransmitter Indicators
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Deisseroth, K.; Feng, G.; Majewska, A.K.; Miesenbock, G.; Ting, A.; Schnitzer, M.J. Next-Generation Optical Technologies for Illuminating Genetically Targeted Brain Circuits. J. Neurosci. 2006, 26, 10380–10386. [Google Scholar] [CrossRef] [PubMed]
- Knopfel, T.; Lin, M.Z.; Levskaya, A.; Tian, L.; Lin, J.Y.; Boyden, E.S. Toward the Second Generation of Optogenetic Tools. J. Neurosci. 2010, 30, 14998–15004. [Google Scholar] [CrossRef] [PubMed]
- Kügler, S.; Kilic, E.; Bähr, M. Human synapsin 1 gene promoter confers highly neuron-specific long-term transgene expression from an adenoviral vector in the adult rat brain depending on the transduced area. Gene Ther. 2003, 10, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Sauer, B. Inducible gene targeting in mice using the Cre/lox system. Methods 1998, 14, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Madisen, L.; Garner, A.R.; Shimaoka, D.; Chuong, A.S.; Klapoetke, N.C.; Li, L.; Van der Bourg, A.; Niino, Y.; Egolf, L.; Monetti, C. Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 2015, 85, 942–958. [Google Scholar] [CrossRef] [PubMed]
- Daigle, T.L.; Madisen, L.; Hage, T.A.; Valley, M.T.; Knoblich, U.; Larsen, R.S.; Takeno, M.M.; Huang, L.; Gu, H.; Larsen, R. A Suite of Transgenic Driver and Reporter Mouse Lines with Enhanced Brain-Cell-Type Targeting and Functionality. Cell 2018, 174, 465–480. [Google Scholar] [CrossRef]
- Margolis, D.J.; Lütcke, H.; Schulz, K.; Haiss, F.; Weber, B.; Kügler, S.; Hasan, M.T.; Helmchen, F. Reorganization of cortical population activity imaged throughout long-term sensory deprivation. Nat. Neurosci. 2012, 15, 1539–1546. [Google Scholar] [CrossRef]
- Huber, D.; Gutnisky, D.A.; Peron, S.; O’Connor, D.H.; Wiegert, J.S.; Tian, L.; Oertner, T.G.; Looger, L.L.; Svoboda, K. Multiple dynamic representations in the motor cortex during sensorimotor learning. Nature 2012, 484, 473–478. [Google Scholar] [CrossRef]
- Dana, H.; Mohar, B.; Sun, Y.; Narayan, S.; Gordus, A.; Hasseman, J.P.; Tsegaye, G.; Holt, G.T.; Hu, A.; Walpita, D. Sensitive red protein calcium indicators for imaging neural activity. Elife 2016, 5, 1–24. [Google Scholar] [CrossRef]
- Zucker, R.S. Calcium- and activity-dependent synaptic plasticity. Curr. Opin. Neurobiol. 1999, 9, 305–313. [Google Scholar] [CrossRef]
- Grienberger, C.; Konnerth, A. Imaging Calcium in Neurons. Neuron 2012, 73, 862–885. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Helmchen, F.; Borst, J.G.; Sakmann, B. Calcium dynamics associated with a single action potential in a CNS presynaptic terminal. Biophys. J. 1997, 72, 1458–1471. [Google Scholar] [CrossRef]
- Koester, H.J.; Sakmann, B. Calcium dynamics associated with action potentials in single nerve terminals of pyramidal cells in layer 2/3 of the young rat neocortex. J. Physiol. 2000, 529, 625–646. [Google Scholar] [CrossRef] [PubMed]
- Broussard, G.J.; Liang, R.; Tian, L. Monitoring activity in neural circuits with genetically encoded indicators. Front. Mol. Neurosci. 2014, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.Z.; Schnitzer, M.J. Genetically encoded indicators of neuronal activity. Nat. Neurosci. 2016, 19, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A.; Llopis, J.; Heim, R.; Michael McCaffery, J.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997. [Google Scholar] [CrossRef]
- Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1999. [Google Scholar] [CrossRef]
- Nagai, T.; Sawano, A.; Park, E.S.; Miyawaki, A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc. Natl. Acad. Sci. USA 2001. [Google Scholar] [CrossRef]
- Nakai, J.; Ohkura, M.; Imoto, K. A high signal-to-noise Ca2+ probe composed of a single green fluorescent protein. Nat. Biotechnol. 2001. [Google Scholar] [CrossRef]
- Pérez Koldenkova, V.; Nagai, T. Genetically encoded Ca2+ indicators: Properties and evaluation. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, E.C.; Mehta, S.; Zhang, J. Genetically Encoded Fluorescent Biosensors Illuminate the Spatiotemporal Regulation of Signaling Networks. Chem. Rev. 2018, 118, 11707–11794. [Google Scholar] [CrossRef] [PubMed]
- Ziv, Y.; Burns, L.D.; Cocker, E.D.; Hamel, E.O.; Ghosh, K.K.; Kitch, L.J.; El Gamal, A.; Schnitzer, M.J. Long-term dynamics of CA1 hippocampal place codes. Nat. Neurosci. 2013. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, M.B.; Orger, M.B.; Robson, D.N.; Li, J.M.; Keller, P.J. Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat. Methods 2013. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, M.E.J.; Dombeck, D.A. Calcium transient prevalence across the dendritic arbour predicts place field properties. Nature 2014. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.Z.; Nehme, R.; Adam, Y.; Jung, E.S.; Wu, H.; Eggan, K.; Arnold, D.B.; Cohen, A.E. All-optical synaptic electrophysiology probes mechanism of ketamine-induced disinhibition. Nat. Methods 2018, 15, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Broussard, G.J.; Liang, Y.; Fridman, M.; Unger, E.K.; Meng, G.; Xiao, X.; Na, J.; Leopoldo, P.; Tian, L. In vivo measurement of afferent activity with axon-specific calcium imaging. Nat. Neurosci. 2018. [Google Scholar] [CrossRef]
- Steinmetz, N.A.; Buetfering, C.; Lecoq, J.; Lee, C.R.; Peters, A.J.; Jacobs, E.A.K.; Coen, P.; Ollerenshaw, D.R.; Valley, M.T.; de Vries, S.E.J. Aberrant Cortical Activity in Multiple GCaMP6-Expressing Transgenic Mouse Lines. Eneuro 2017, 4, ENEURO.0207-17. [Google Scholar] [CrossRef]
- McMahon, S.M.; Jackson, M.B. An Inconvenient Truth: Calcium Sensors Are Calcium Buffers. Trends Neurosci. 2018. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, N.; He, Y.; Liu, Y.; Ge, L.; Zou, L.; Song, S.; Xiong, W.; Liu, X. Improved calcium sensor GCaMP-X overcomes the calcium channel perturbations induced by the calmodulin in GCaMP. Nat. Commun. 2018, 9, 1504. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Hires, S.A.; Mao, T.; Huber, D.; Chiappe, M.E.; Chalasani, S.H.; Petreanu, L.; Akerboom, J.; McKinney, S.A.; Schreiter, E.R. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 2009. [Google Scholar] [CrossRef] [PubMed]
- Barykina, N.V.; Subach, O.M.; Piatkevich, K.D.; Jung, E.E.; Malyshev, A.Y.; Smirnov, I.V.; Bogorodskiy, A.O.; Borshchevskiy, V.; Varizhuk, A.; Pozmogova, G.E. Green fluorescent genetically encoded calcium indicator based on calmodulin/M13-peptide from fungi. PLoS ONE 2017, 12, e0183757. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Araki, S.; Wu, J.; Teramoto, T.; Chang, Y.F.; Nakano, M.; Abdelfattah, A.S.; Fujiwara, M.; Ishihara, T.; Nagai, T. An Expanded Palette of Genetically Encoded Ca2+ Indicators -Supplementay Info. Science 2011. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Abdelfattah, A.S.; Zhao, Y.; Ruangkittisakul, A.; Ballanyi, K.; Campbell, R.E.; Harrison, D.J. Microfluidic cell sorter-aided directed evolution of a protein-based calcium ion indicator with an inverted fluorescent response. Integr. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.R.; Badura, A.; Pacheco, D.A.; Lynch, L.A.; Schneider, E.R.; Taylor, M.P.; Hogue, I.B.; Enquist, L.W.; Murthy, M.; Wang, S.S.H. Fast GCaMPs for improved tracking of neuronal activity. Nat. Commun. 2013. [Google Scholar] [CrossRef] [PubMed]
- Dana, H.; Sun, Y.; Mohar, B.; Hulse, B.; Hasseman, J.P.; Tsegaye, G.; Tsang, A.; Wong, A.; Patel, R.; Macklin, J.J. High-performance GFP-based calcium indicators for imaging activity in neuronal populations and microcompartments. bioRxiv 2018. [Google Scholar] [CrossRef]
- Barykina, N.V.; Subach, O.M.; Doronin, D.A.; Sotskov, V.P.; Roshchina, M.A.; Kunitsyna, T.A.; Malyshev, A.Y.; Smirnov, I.V.; Azieva, A.M.; Sokolov, I.S. A new design for a green calcium indicator with a smaller size and a reduced number of calcium-binding sites. Sci. Rep. 2016, 6, 34447. [Google Scholar] [CrossRef]
- Barykina, N.V.; Doronin, D.A.; Subach, O.M.; Sotskov, V.P.; Plusnin, V.V.; Ivleva, O.A.; Gruzdeva, A.M.; Kunitsyna, T.A.; Ivashkina, O.I.; Lazutkin, A.A. NTnC-like genetically encoded calcium indicator with a positive and enhanced response and fast kinetics. Sci. Rep. 2018. [Google Scholar] [CrossRef]
- Moeyaert, B.; Holt, G.; Madangopal, R.; Perez-Alvarez, A.; Fearey, B.C.; Trojanowski, N.F.; Ledderose, J.; Zolnik, T.A.; Das, A.; Patel, D. Improved methods for marking active neuron populations. Nat. Commun. 2018. [Google Scholar] [CrossRef]
- Doronin, D.A.; Barykina, N.V.; Subach, O.M.; Sotskov, V.P.; Plusnin, V.V.; Ivleva, O.A.; Isaakova, E.A.; Varizhuk, A.M.; Pozmogova, G.E.; Malyshev, A.Y. Genetically encoded calcium indicator with NTnC-like design and enhanced fluorescence contrast and kinetics. BMC Biotechnol. 2018, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, M.; Sasaki, T.; Kobayashi, C.; Ikegaya, Y.; Nakai, J. An improved genetically encoded red fluorescent Ca2+ indicator for detecting optically evoked action potentials. PLoS ONE 2012, 7, e1796. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Dana, H.; Abdelfattah, A.S.; Patel, R.; Shea, J.; Molina, R.S.; Rawal, B.; Rancic, V.; Chang, Y.F.; Wu, L. A genetically encoded Ca2+ indicator based on circularly permutated sea anemone red fluorescent protein eqFP578. BMC Biol. 2018, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lanin, A.A.; Chebotarev, A.S.; Barykina, N.V.; Subach, F.V.; Zheltikov, A.M. The whither of bacteriophytochrome-based near-infrared fluorescent proteins: Insights from two-photon absorption spectroscopy. J. Biophotonics 2019, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Piatkevich, K.D.; Larney, B.; Abdelfattah, A.S.; Sohum, M.; Murdock, M.H.; Gottschalk, S.; Molina, R.S.; Zhang, W.; Chen, Y. A genetically encoded near-infrared fluorescent calcium ion indicator. Nat. Methods 2019, 1, 1–7. [Google Scholar] [CrossRef]
- Qian, Y.; Rancic, V.; Wu, J.; Ballanyi, K.; Campbell, R.E. A bioluminescent Ca2+ indicator based on a topological variant of GCaMP6s. ChemBioChem 2018, 1–6. [Google Scholar] [CrossRef]
- Shen, Y.; Wu, S.-Y.; Rancic, V.; Aggarwal, A.; Qian, Y.; Miyashita, S.-I.; Ballanyi, K.; Campbell, R.E.; Dong, M. Genetically encoded fluorescent indicators for imaging intracellular potassium ion concentration. Commun. Biol. 2019, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Bischof, H.; Rehberg, M.; Stryeck, S.; Artinger, K.; Eroglu, E.; Waldeck-Weiermair, M.; Gottschalk, B.; Rost, R.; Deak, A.T.; Niedrist, T. Novel genetically encoded fluorescent probes enable real-Time detection of potassium in vitro and in vivo. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Klappa, P.; Kietzmann, D.T. The Endoplasmic Reticulum: Folding, Calcium Homeostasis, Signaling, and Redox Control. Antioxid. Redox Signal. 2006. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018. [Google Scholar] [CrossRef]
- Giorgi, C.; Agnoletto, C.; Bononi, A.; Bonora, M.; de Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Poletti, F.; Rimessi, A. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 2012. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta Mol. Cell Res. 2013. [Google Scholar] [CrossRef] [PubMed]
- Shui, B.; Wang, Q.; Lee, F.; Byrnes, L.J.; Chudakov, D.M.; Lukyanov, S.A.; Sonderman, H.; Kotlikoff, M.I. Circular permutation of red fluorescent proteins. PLoS ONE 2011. [Google Scholar] [CrossRef] [PubMed]
- Akerboom, J.; Carreras Calderón, N.; Tian, L.; Wabnig, S.; Prigge, M.; Tolö, J.; Gordus, A.; Orger, M.B.; Severi, K.E.; Macklin, J.J. Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front. Mol. Neurosci. 2013, 6, 1–29. [Google Scholar] [CrossRef]
- Shen, Y.; Wiens, M.D.; Campbell, R.E. A photochromic and thermochromic fluorescent protein. RSC Adv. 2014. [Google Scholar] [CrossRef]
- Wu, J.; Liu, L.; Matsuda, T.; Zhao, Y.; Rebane, A.; Drobizhev, M.; Chang, Y.-F.; Araki, S.; Arai, Y.; March, K. Improved orange and red Ca2+ indicators and photophysical considerations for optogenetic applications. ACS Chem. Neurosci. 2013. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Abdelfattah, A.S.; Miraucourt, L.S.; Kutsarova, E.; Ruangkittisakul, A.; Zhou, H.; Ballanyi, K.; Wicks, G.; Drobizhev, M.; Rebane, A. A long Stokes shift red fluorescent Ca2+ indicator protein for two-photon and ratiometric imaging. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Takeuchi, A.; Horigane, S.I.; Ohkura, M.; Gengyo-Ando, K.; Fujii, H.; Kamijo, S.; Takemoto-Kimura, S.; Kano, M.; Nakai, J. Rational design of a high-affinity, fast, red calcium indicator R-CaMP2. Nat. Methods 2014. [Google Scholar] [CrossRef]
- Hochbaum, D.R.; Zhao, Y.; Farhi, S.L.; Klapoetke, N.; Werley, C.A.; Kapoor, V.; Zou, P.; Kralj, J.M.; Maclaurin, D.; Smedemark-Margulies, N. All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat. Methods 2014, 11, 825–833. [Google Scholar] [CrossRef]
- Adam, Y.; Kim, J.J.; Lou, S.; Cohen, A.E. All-optical electrophysiology reveals brain-state dependent changes in hippocampal subthreshold dynamics and excitability. bioRxiv 2018. [Google Scholar] [CrossRef]
- Klapoetke, N.C.; Murata, Y.; Kim, S.S.; Pulver, S.R.; Birdsey-Benson, A.; Cho, Y.K.; Morimoto, T.K.; Chuong, A.S.; Carpenter, E.J.; Tian, Z. Independent optical excitation of distinct neural populations. Nat. Methods 2014, 11, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Trojanowski, N.F.; Nelson, M.D.; Flavell, S.W.; Fang-Yen, C.; Raizen, D.M. Distinct Mechanisms Underlie Quiescence during Two Caenorhabditis elegans Sleep-Like States. J. Neurosci. 2015, 35, 14571–14584. [Google Scholar] [CrossRef] [PubMed]
- Piatkevich, K.D.; Subach, F.V.; Verkhusha, V.V. Engineering of bacterial phytochromes for near-infrared imaging, sensing, and light-control in mammals. Chem. Soc. Rev. 2013, 42, 3441–3452. [Google Scholar] [CrossRef] [PubMed]
- Piatkevich, K.D.; Suk, H.-J.; Kodandaramaiah, S.B.; Yoshida, F.; DeGennaro, E.M.; Drobizhev, M.; Hughes, T.E.; Desimone, R.; Boyden, E.S.; Verkhusha, V.V. Near-Infrared Fluorescent Proteins Engineered from Bacterial Phytochromes in Neuroimaging. Biophys. J. 2017, 113, 2299–2309. [Google Scholar] [CrossRef]
- Piatkevich, K.D.; Jung, E.E.; Straub, C.; Linghu, C.; Park, D.; Suk, H.-J.; Hochbaum, D.R.; Goodwin, D.; Pnevmatikakis, E.; Pak, N. A robotic multidimensional directed evolution approach applied to fluorescent voltage reporters. Nat. Chem. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Baird, M.A.; Allen, J.R.; Howe, E.S.; Klassen, M.P.; Reade, A.; Makhijani, K.; Song, Y.; Liu, S.; Murthy, Z. A naturally monomeric infrared fluorescent protein for protein labeling in vivo. Nat. Methods 2015, 12, 763–765. [Google Scholar] [CrossRef] [PubMed]
- Filonov, G.S.; Piatkevich, K.D.; Ting, L.-M.; Zhang, J.; Kim, K.; Verkhusha, V.V. Bright and stable near-infrared fluorescent protein for in vivo imaging. Nat. Biotechnol. 2011, 29, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Piatkevich, K.D.; Subach, F.V.; Verkhusha, V.V. Far-red light photoactivatable near-infrared fluorescent proteins engineered from a bacterial phytochrome. Nat. Commun. 2013, 4, 2153. [Google Scholar] [CrossRef]
- Rumyantsev, K.A.; Shcherbakova, D.M.; Zakharova, N.I.; Emelyanov, A.V.; Turoverov, K.K.; Verkhusha, V.V. Minimal domain of bacterial phytochrome required for chromophore binding and fluorescence. Sci. Rep. 2015, 5, 18348. [Google Scholar] [CrossRef]
- Fosque, B.F.; Sun, Y.; Dana, H.; Yang, C.T.; Ohyama, T.; Tadross, M.R.; Patel, R.; Zlatic, M.; Kim, D.S.; Ahrens, M.B. Labeling of active neural circuits in vivo with designed calcium integrators. Science 2015. [Google Scholar] [CrossRef]
- Stringer, C.; Pachitariu, M.; Steinmetz, N.; Reddy, C.; Carandini, M.; Harris, K.D. Spontaneous behaviors drive multidimensional, brain-wide neural activity. bioRxiv 2018. [Google Scholar] [CrossRef]
- Saito, K.; Chang, Y.F.; Horikawa, K.; Hatsugai, N.; Higuchi, Y.; Hashida, M.; Yoshida, Y.; Matsuda, T.; Arai, Y.; Nagai, T. Luminescent proteins for high-speed single-cell and whole-body imaging. Nat. Commun. 2012, 3, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.M.; Valdes, R. Understanding the sodium pump and its relevance to disease. Clin. Chem. 1994, 40, 1674–1685. [Google Scholar] [PubMed]
- Rose, C.R.; Konnerth, A. NMDA receptor-mediated Na+ signals in spines and dendrites. J. Neurosci. 2001, 21, 4207–4214. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.R. Book Review: Na+ Signals at Central Synapses. Neurosci. 2002. [Google Scholar] [CrossRef] [PubMed]
- Llinás, R.R. The intrinsic electrophysiological properties of mammalian neurons: Insights into central nervous system function. Science 1988, 242, 1654–1664. [Google Scholar] [CrossRef] [PubMed]
- Bean, B.P. The action potential in mammalian central neurons. Nat. Rev. Neurosci. 2007, 8, 451–465. [Google Scholar] [CrossRef]
- Stuart, G.J.; Spruston, N. Dendritic integration: 60 years of progress. Nat. Neurosci. 2015, 18, 1713–1721. [Google Scholar] [CrossRef]
- Peterka, D.S.; Takahashi, H.; Yuste, R. Imaging voltage in neurons. Neuron 2011, 69, 9–21. [Google Scholar] [CrossRef]
- Kannan, M.; Vasan, G.; Huang, C.; Haziza, S.; Li, J.Z.; Inan, H.; Schnitzer, M.J.; Pieribone, V.A. Fast, in vivo voltage imaging using a red fluorescent indicator. Nat. Methods 2018, 15, 1108–1116. [Google Scholar] [CrossRef]
- Chavarha, M.; Villette, V.; Dimov, I.K.; Pradhan, L.; Evans, S.W.; Shi, D.; Yang, R.; Chamberland, S.; Bradley, J.; Mathieu, B.; et al. Fast two-photon volumetric imaging of an improved voltage indicator reveals electrical activity in deeply located neurons in the awake brain. bioRxiv 2018. [Google Scholar] [CrossRef]
- Abdelfattah, A.S.; Kawashima, T.; Singh, A.; Novak, O.; Mensh, B.D.; Paninski, L.; Macklin, J.J.; Podgorski, K.; Lin, B.J.; Chen, T.W. Bright and photostable chemigenetic indicators for extended in vivo voltage imaging. bioRxiv 2018. [Google Scholar] [CrossRef]
- Siegel, M.S.; Isacoff, E.Y. A genetically encoded optical probe of membrane voltage. Neuron 1997. [Google Scholar] [CrossRef]
- Gong, Y.; Huang, C.; Li, J.Z.; Grewe, B.F.; Zhang, Y.; Eismann, S.; Schnitzer, M.J. High-speed recording of neural spikes in awake mice and flies with a fluorescent voltage sensor. Science 2015, 350, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Yizhar, O.; Fenno, L.; Zhang, F.; Hegemann, P.; Diesseroth, K. Microbial Opsins: A Family of Single-Component Tools for Optical Control of Neural Activity Topic Introduction Microbial Opsins: A Family of Single-Component Tools for Optical Control of Neural Activity. Cold Spring Harb. Protoc. 2011, 273–283. [Google Scholar] [CrossRef]
- Mattis, J.; Tye, K.M.; Ferenczi, E.A.; Ramakrishnan, C.; Shea, D.J.O.; Prakash, R.; Gunaydin, L.A.; Hyun, M.; Fenno, L.E.; Gradinaru, V.P. rinciples for applying optogenetic tools derived from direct comparative analysis of microbial opsins. Nat. Methods 2011, 9, 159–172. [Google Scholar] [CrossRef]
- Nagel, G.; Szellas, T.; Huhn, W.; Kateriya, S.; Adeishvili, N.; Berthold, P.; Ollig, O.; Hegemann, P.; Bamberg, E. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. USA 2003, 100, 13940–13945. [Google Scholar] [CrossRef] [PubMed]
- Gradinaru, V.; Thompson, K.R.; Deisseroth, K. eNpHR: A Natronomonas halorhodopsin enhanced for optogenetic applications. Brain Cell Biol. 2008, 36, 129–139. [Google Scholar] [CrossRef]
- Kralj, J.M.; Hochbaum, D.R.; Douglass, A.D.; Cohen, A.E. Electrical spiking in Escherichia coli probed with a fluorescent voltage-indicating protein. Science 2011. [Google Scholar] [CrossRef]
- Kralj, J.M.; Douglass, A.D.; Hochbaum, D.R.; MacLaurin, D.; Cohen, A.E. Optical recording of action potentials in mammalian neurons using a microbial rhodopsin. Nat. Methods 2012. [Google Scholar] [CrossRef]
- Lanyi, J. Bacteriorhodopsin. Annu. Rev. Physiol. 2004, 66, 665–688. [Google Scholar] [CrossRef] [PubMed]
- Maclaurin, D.; Venkatachalam, V.; Lee, H.; Cohen, A.E. Mechanism of voltage-sensitive fluorescence in a microbial rhodopsin. Proc. Natl. Acad. Sci. USA 2013, 110, 5939–5944. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, S.; Zauner, G.; Aartsma, T.J.; Engelkamp, H.; Hatzakis, N.; Rowan, A.E.; Nolte, R.J.M.; Christianen, P.C.M.; Canters, G.W. The enzyme mechanism of nitrite reductase studied at single-molecule level. Proc. Natl. Acad. Sci. USA 2008, 105, 3250–3255. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, H.; Fields, A.P.; Kralj, J.M.; Spudich, J.L.; Rothschild, K.J.; Cohen, A.E. Ultrasensitive measurements of microbial rhodopsin photocycles using photochromic FRET. Photochem. Photobiol. 2012, 88, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Wagner, M.J.; Li, J.Z.; Schnitzer, M.J. Imaging neural spiking in brain tissue using FRET-opsin protein voltage sensors. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Zhao, Y.; Douglass, A.D.; Hochbaum, D.R.; Brinks, D.; Werley, C.A.; Harrison, D.J.; Campbell, R.E.; Cohen, A.E. Bright and fast multicoloured voltage reporters via electrochromic FRET. Nat. Commun. 2014, 5, 4625. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.Y.; Han, X.; Dobry, A.S.; Qian, X.; Chuong, A.S.; Li, M.; Henninger, M.A.; Belfort, G.M.; Lin, Y.; Monahan, P.E. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature 2010, 463, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, S.P.; Ewers, D.; Gazzarrini, S.; Moroni, A.; Gradmann, D.; Hegemann, P. H+-pumping rhodopsin from the marine alga Acetabularia. Biophys. J. 2006, 91, 1471–1479. [Google Scholar] [CrossRef]
- Lou, S.; Adam, Y.; Weinstein, E.N.; Williams, E.; Williams, K.; Parot, V.; Kavokine, N.; Liberles, S.; Madisen, S.; Zeng, H. Genetically Targeted All-Optical Electrophysiology with a Transgenic Cre-Dependent Optopatch Mouse. J. Neurosci. 2016, 36, 11059–11073. [Google Scholar] [CrossRef]
- Grimm, J.B.; English, B.P.; Chen, J.; Slaughter, J.P.; Zhang, Z.; Revyakin, A.; Patel, R.; Macklin, J.J.; Normanno, D.A.; Singer, R.H. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Methods 2015. [Google Scholar] [CrossRef]
- Lim, S.T.; Antonucci, D.E.; Scannevin, R.H.; Trimmer, J.S. A Novel Targeting Signal for Proximal Clustering of the Kv2.1 K+ Channel in Hippocampal Neurons. Neuron 2000, 25, 385–397. [Google Scholar] [CrossRef]
- Baker, C.A.; Elyada, Y.M.; Parra, A.; Bolton, M.M.L. Cellular resolution circuit mapping with temporal-focused excitation of soma-targeted channelrhodopsin. Elife 2016, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, O.; Tanese, D.; Zampini, V.; Changyang, L.; Kiryln, P.; Ronzitti, E.; Papagiakoumou, E.; Boyden, E.S.; Emiliani, V. Temporally precise single-cell resolution optogenetics. Nat. Neurosci. 2017, 20, 1796. [Google Scholar] [CrossRef] [PubMed]
- Chien, M.-P.; Brinks, D.; Adam, Y.; Bloxham, W.; Kheifets, S.; Cohen, A.E. Two-photon photoactivated voltage imaging in tissue with an Archaerhodopsin-derived reporter. bioRxiv 2017. [Google Scholar] [CrossRef]
- Cao, G.; Platisa, J.; Pieribone, V.A.; Raccuglia, D.; Kunst, M.; Nitabach, M.N. Genetically targeted optical electrophysiology in intact neural circuits. Cell 2013, 154, 904–913. [Google Scholar] [CrossRef]
- Yang, H.H.H.; St-Pierre, F.; Sun, X.; Ding, X.; Lin, M.Z.Z.; Clandinin, T.R.R. Subcellular Imaging of Voltage and Calcium Signals Reveals Neural Processing In Vivo. Cell 2016, 166, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Chamberland, S.; Yang, H.H.; Pan, M.M.; Evans, S.W.; Guan, S.; Chavarha, M.; Yang, Y.; Salesse, C.; Wu, H.; Wu, J.C. Fast two-photon imaging of subcellular voltage dynamics in neuronal tissue with genetically encoded indicators. Elife 2017, 6, 1–35. [Google Scholar] [CrossRef]
- Roome, C.J.; Kuhn, B. Simultaneous dendritic voltage and calcium imaging and somatic recording from Purkinje neurons in awake mice. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Kulkarni, R.U.; Vandenberghe, M.; Thunemann, M.; James, F.; Andreassen, O.A.; Djurovic, S.; Devor, A.; Miller, E.W. In Vivo Two-Photon Voltage Imaging with Sulfonated Rhodamine Dyes. ACS Cent. Sci. 2018, 4, 1371–1378. [Google Scholar] [CrossRef]
- St-Pierre, F.; Marshall, J.D.; Yang, Y.; Gong, Y.; Schnitzer, M.J.; Lin, M.Z. High-fidelity optical reporting of neuronal electrical activity with an ultrafast fluorescent voltage sensor. Nat. Neurosci. 2014, 17, 884–889. [Google Scholar] [CrossRef]
- Südhof, T.C. Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron 2013. [Google Scholar] [CrossRef] [PubMed]
- Scimemi, A.; Beato, M. Determining the neurotransmitter concentration profile at active synapses. Mol. Neurobiol. 2009. [Google Scholar] [CrossRef]
- Lüscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Von Gersdorff, H.; Sakaba, T.; Berglund, K.; Tachibana, M. Submillisecond kinetics of glutamate release from a sensory synapse. Neuron 1998. [Google Scholar] [CrossRef]
- Meldrum, B.S. Glutamate as a Neurotransmitter in the Brain: Review of Physiology and Pathology. J. Nutr. 2000, 130, 1007S–1015S. [Google Scholar] [CrossRef] [PubMed]
- McCormick, D.A. GABA as an inhibitory neurotransmitter in human cerebral cortex. J. Neurophysiol. 1989, 62, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, G. Neurotransmitter Transporters: Structure, Function, and Regulation; Reith, M.E.A., Ed.; Humana Press: New York, NY, USA, 2002; pp. 25–52. [Google Scholar]
- Snyder, S.H. Brain peptides as neurotransmitters. Science 1980, 209, 976–983. [Google Scholar] [CrossRef]
- Kreitzer, A.C. Neurotransmission: Emerging Roles of Endocannabinoids. Curr. Biol. 2005, 15, R549–R551. [Google Scholar] [CrossRef]
- Dawson, T.M.; Snyder, S.H. Gases as biological messengers: Nitric oxide and carbon monoxide in the brain. J. Neurosci. 1994, 14, 5147–5159. [Google Scholar] [CrossRef]
- Ames, G.F.L. Bacterial Periplasmic Transport Systems: Structure, Mechanism, and Evolution. Annu. Rev. Biochem. 1986. [Google Scholar] [CrossRef]
- De Lorimier, R.M.; Smith, J.J.; Dwyer, M.A.; Looger, L.L.; Sali, K.M.; Paavola, C.D.; Rizk, S.S.; Sadigov, S.; Conrad, D.W.; Loew, L. Construction of a fluorescent biosensor family. Protein Sci. 2002, 11, 2655–2675. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, M.A.; Hellinga, H.W. Periplasmic binding proteins: A versatile superfamily for protein engineering. Curr. Opin. Struct. Biol. 2004, 14, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Deuschle, K.; Okumoto, S.; Fehr, M.; Looger, L.L.; Kozhukh, L.; Frommer, W.B. Construction and optimization of a family of genetically encoded metabolite sensors by semirational protein engineering. Protein Sci. 2005, 14, 2304–2314. [Google Scholar] [CrossRef] [PubMed]
- Okumoto, S.; Looger, L.L.; Micheva, K.D.; Reimer, R.J.; Smith, S.J.; Frommer, W.B. Detection of glutamate release from neurons by genetically encoded surface-displayed FRET nanosensors. Proc. Natl. Acad. Sci. USA 2005, 102, 8740–8745. [Google Scholar] [CrossRef] [PubMed]
- Hires, S.A.; Zhu, Y.; Tsien, R.Y. Optical measurement of synaptic glutamate spillover and reuptake by linker optimized glutamate-sensitive fluorescent reporters. Proc. Natl. Acad. Sci. USA 2008, 105, 4411–4416. [Google Scholar] [CrossRef] [PubMed]
- Marvin, J.S.; Borghuis, B.G.; Tian, L.; Cichon, J.; Harnett, M.T.; Akerboom, J.; Gordus, A.; Renninger, S.L.; Chen, T.W.; Bargmann, C.I. An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat. Methods 2013, 10, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Taschenberger, H.; Woehler, A.; Neher, E. Superpriming of synaptic vesicles as a common basis for intersynapse variability and modulation of synaptic strength. Proc. Natl. Acad. Sci. USA 2016. [Google Scholar] [CrossRef]
- Helassa, N.; Dürst, C.D.; Coates, C.; Kerruth, S.; Arif, U.; Schulze, C.; Wiegert, J.S.; Geeves, M.; Oertner, T.G.; Török, K. Ultrafast glutamate sensors resolve high-frequency release at Schaffer collateral synapses. Proc. Natl. Acad. Sci. USA 2018, 115, 5594–5599. [Google Scholar] [CrossRef]
- Marvin, J.S.; Scholl, B.; Wilson, D.E.; Podgorski, K.; Kazemipour, A.; Müller, J.A.; Schoch, S.; Quiroz, F.J.U.; Rebola, N.; Bao, H. Stability, affinity, and chromatic variants of the glutamate sensor iGluSnFR. Nat. Methods 2018, 15, 936–939. [Google Scholar] [CrossRef]
- Wu, J.; Abdelfattah, A.S.; Zhou, H.; Ruangkittisakul, A.; Qian, Y.; Ballanyi, K.; Campbell, R.E. Genetically Encoded Glutamate Indicators with Altered Color and Topology. ACS Chem. Biol. 2018, 13, 1832–1837. [Google Scholar] [CrossRef]
- Looger, L.L.; Marvin, J.S.; Shimoda, Y.; Magloire, V.; Leite, M.; Kawashima, T.; Jensen, T.P.; Knott, E.L.; Novak, O.; Podgorski, K. A genetically encoded fluorescent sensor for in vivo imaging of GABA. bioRxiv 2018. [Google Scholar] [CrossRef]
- Zhang, W.H.; Herde, M.K.; Mitchell, J.A.; Whitfield, J.H.; Wulff, A.B.; Vongsouthi, V.; Sanchez-Romero, I.; Gulakova, P.E.; Minge, D.; Breithausen, B. Monitoring hippocampal glycine with the computationally designed optical sensor GlyFS. Nat. Chem. Biol. 2018, 14, 861–869. [Google Scholar] [CrossRef]
- Patriarchi, T.; Cho, J.R.; Merten, K.; Howe, M.W.; Marley, A.; Xiong, W.H.; Folk, R.W.; Broussard, G.J.; Liang, R.; Jang, M.J. Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science 2018, 360, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Zeng, J.; Jing, M.; Zhou, J.; Feng, J.; Owen, S.F.; Luo, Y.; Li, F.; Wang, H.; Yamaguchi, T. A Genetically Encoded Fluorescent Sensor Enables Rapid and Specific Detection of Dopamine in Flies, Fish, and Mice. Cell 2018, 174, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Jing, M.; Zhang, P.; Wang, G.; Feng, J.; Mesik, L.; Zeng, J.; Jiang, H.; Wang, S.; Looby, J.C.; Guagliardo, N.A. A genetically encoded fluorescent acetylcholine indicator for in vitro and in vivo studies. Nat. Biotechnol. 2018, 36, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhang, C.; Lischinsky, J.; Jing, M.; Zhou, J.; Wang, H.; Zhang, Y.; Dong, A.; Wu, Z.; Wu, H.; et al. A genetically encoded fluorescent sensor for rapid and specific in vivo detection of norepinephrine. bioRxiv 2018. [Google Scholar] [CrossRef]
- Lee, D.; Creed, M.; Jung, K.; Stefanelli, T.; Wendler, D.J.; Oh, W.C.; Mignocchi, N.L.; Lüscher, C.; Kwon, H.B. Temporally precise labeling and control of neuromodulatory circuits in the mammalian brain. Nat. Methods 2017, 14, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, G.D.; Nicholson-Guthrie, C.S. gamma-Aminobutyric acid uptake by a bacterial system with neurotransmitter binding characteristics. Proc. Natl. Acad. Sci. USA 1989. [Google Scholar] [CrossRef]
- Planamente, S.; Vigouroux, A.; Mondy, S.; Nicaise, M.; Faure, D.; Moréra, S. A conserved mechanism of GABA binding and antagonism is revealed by structure-function analysis of the periplasmic binding protein Atu2422 in Agrobacterium tumefaciens. J. Biol. Chem. 2010. [Google Scholar] [CrossRef]
- Whitfield, J.H.; Zhang, W.H.; Herde, M.K.; Clifton, B.E.; Radziejewski, J.; Janovjak, H.; Henneberger, C.; Jackson, C.J. Construction of a robust and sensitive arginine biosensor through ancestral protein reconstruction. Protein Sci. 2015. [Google Scholar] [CrossRef]
- Tannous, B.A.; Grimm, J.; Perry, K.F.; Chen, J.W.; Weissleder, R.; Breakefield, X.O. Metabolic biotinylation of cell surface receptors for in vivo imaging. Nat. Methods 2006. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Scheerer, P.; Hofmann, K.P.; Choe, H.W.; Ernst, O.P. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 2008. [Google Scholar] [CrossRef] [PubMed]
- Scheerer, P.; Park, J.H.; Hildebrand, P.W.; Kim, Y.J.; Krauß, N.; Choe, H.W.; Hofmann, K.P.; Ernst, O.P. Crystal structure of opsin in its G-protein-interacting conformation. Nature 2008. [Google Scholar] [CrossRef] [PubMed]
- Vilardaga, J.P.; Bünemann, M.; Krasell, C.; Castro, M.; Lohse, M.J. Measurement of the millisecond activation switch of G protein-coupled receptors in living cells. Nat. Biotechnol. 2003, 21, 807–812. [Google Scholar] [CrossRef]
- Ziegler, N.; Bätz, J.; Zabel, U.; Lohse, M.J.; Hoffmann, C. FRET-based sensors for the human M1-, M3-, and M 5-acetylcholine receptors. Bioorganic Med. Chem. 2011, 19, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Masharina, A.; Reymond, L.; Maurel, D.; Umezawa, K.; Johnsson, K. A Fluorescent Sensor for GABA and Synthetic GABA. J. Am. Chem. Soc. 2012, 134, 19026–19034. [Google Scholar] [CrossRef] [PubMed]
- Maier-Peuschel, M.; Frölich, N.; Dees, C.; Hommers, L.G.; Hoffmann, C.; Nikolaev, V.O.; Lohse, M.J. A fluorescence resonance energy transfer-based M2 muscarinic receptor sensor reveals rapid kinetics of allosteric modulation. J. Biol. Chem. 2010, 285, 8793–8800. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Lefkowitz, R.J. The role of ß-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 2002. [Google Scholar] [CrossRef]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002. [Google Scholar] [CrossRef] [PubMed]
- Barnea, G.; Strapps, W.; Herrada, G.; Berman, Y.; Ong, J.; Kloss, B.; Axel, R.; Lee, K.J. The genetic design of signaling cascades to record receptor activation. Proc. Natl. Acad. Sci. USA 2008, 105, 64–69. [Google Scholar] [CrossRef]
- Nguyen, Q.T.; Schroeder, L.F.; Mank, M.; Muller, A.; Taylor, P.; Griesbeck, O.; Kleinfeld, D. An in vivo biosensor for neurotransmitter release and in situ receptor activity. Nat. Neurosci. 2010, 13, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Joseph, V.; Slesinger, P.A.; Kleinfeld, D. Cell-based reporters reveal in vivo dynamics of dopamine and norepinephrine release in murine cortex. Nat. Methods 2014, 11, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Guntas, G.; Hallett, R.A.; Zimmerman, S.P.; Williams, T.; Yumerefendi, H.; Bear, J.E.; Kuhlman, B. Engineering an improved light-induced dimer (iLID) for controlling the localization and activity of signaling proteins. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Bartels, D.; Vonstein, V.; Meyer, F. Annotation of Bacterial and Archaeal Genomes: Improving Accuracy and Consistency. Chem. Rev. 2007, 107, 3431–3447. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, V.M.; Chen, I.-M.A.; Chu, K.; Pati, A.; Ivanova, N.N.; Kyrpides, N.C. Ten Years of Maintaining and Expanding a Microbial Genome and Metagenome Analysis System. Trends Microbiol. 2015, 23, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.S.; Liu, D.R. Methods for the directed evolution of proteins. Nat. Rev. Genet. 2015, 16, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Subach, F.V.; Piatkevich, K.D.; Verkhusha, V.V. Directed molecular evolution to design advanced red fluorescent proteins. Nat. Methods 2011. [Google Scholar] [CrossRef]
- Chia, T.H.; Levene, M.J. Microprisms for in vivo multilayer cortical imaging. J. Neurophysiol. 2009, 102, 1310–1314. [Google Scholar] [CrossRef]
- Andermann, M.L.; Gilfoy, N.B.; Goldey, G.J.; Sachdev, R.N.S.; Wölfel, M.; McCormick, D.A.; Reid, R.C.; Levene, M.J. Chronic Cellular Imaging of Entire Cortical Columns in Awake Mice Using Microprisms. Neuron 2013, 80, 900–913. [Google Scholar] [CrossRef]
- Ghosh, K.K.; Burns, L.D.; Cocker, E.D.; Nimmerjahn, A.; Ziv, Y.; El Gamal, A.; Schnitzer, M.J. Miniaturized integration of a fluorescence microscope. Nat. Methods 2011, 8, 871–878. [Google Scholar] [CrossRef]
- Yu, K.; Ahrens, S.; Zhang, X.; Schiff, H.; Ramakrishnan, C.; Fenno, L.; Deisseroth, K.; Zhao, F.; Luo, M.H.; Gong, L. The central amygdala controls learning in the lateral amygdala. Nat. Neurosci. 2017, 20, 1680. [Google Scholar] [CrossRef] [PubMed]
- Tillberg, P.W.; Chen, F.; Piatkevich, K.D.; Zhao, Y.; Yu, C.-C.; English, B.P.; Gao, L.; Martorell, A.; Suk, H.J.; Yoshida, F. Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nat. Biotechnol. 2016, 34, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Wassie, A.T.; Zhao, Y.; Boyden, E.S. Expansion microscopy: Principles and uses in biological research. Nat. Methods 2019, 16, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-S.; Vandemark, K.; Mezey, D.; Shultz, N.; Fitzpatrick, D. Functional Synaptic Architecture of Callosal Inputs in Mouse Primary Visual Cortex. Neuron 2018, 1–8. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Family | Indicator a | Ex/Em (nm) b | Brightness vs. EGFP (%) c | pKa,apo/pKa,sat | ΔF/F (%) d | Kd (nM) e | koff (s−1) f | ΔF/F/SNR per AP | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Slice or Culture | In Vivo | ||||||||||
| Calcium indicators | |||||||||||
| NTnC | NTnC | 505/518 | 163 | 6.09/6.08 | 100 | 192 | 0.8 | 0.027/18 | ND | [38] | |
| iYTnC2 | 499/518 | 16 | 7.4/8.5 | 450 | 331 | 1.12 | 0.0015/ND | ND | [41] | ||
| YTnC | 495/516 | 17 | 5.2/6.3 | 290 | 410 | 0.96 | 0.008/ND | ND | [39] | ||
| dYTnC2 | 496/518 | ND | ND | 1900 | 2700 | ND | ND | ND | [unpublished] | ||
| FGCaMP | FGCaMP® | apo | 402/516 | 56 | 6.56/7.0 | 590 | 400 | 1.2 | ND | ND | [33] |
| sat | 493/516 | 104 | 6.2/7.33 | 1370 | 460, 4400 | ||||||
| CaMP | GCaMP6s | 497/515 | 124 | 9.77/6.20 | 6220 | 227 | 0.69–1.12 | 0.022–0.28/7.6–183 | 0.25/ND | [26] | |
| GCaMP6f | 497/515 | 109 | 8.77/6.34 | 5080 | 492 | 3.93 | 0.18/101 | 0.15/ND | [26] | ||
| jGCAMP7s | 497/515 | 103 | 7.69/6.36 | 3900 | 68 * | 2.86 | 0.657/25 | ND | [37] | ||
| GECO | jRGECO1a | 560/590 | 35 | 8.6/6.3 | 1060 | 148 * | 7.6 | ~0.29/~13 | 0.13/ND | [9,42] | |
| K-GECO1 | 565/590 | 82 | ND | 1100 | 165 * | ND | ~0.26/~11 | ND | [43] | ||
| CAMPARI | CAMPARI2 | G | 502/516 | 268 | ND | 7.8 | 199 * | 1.43 | ND | ND | [40] |
| R | 562/577 | 126 | ND | ||||||||
| Phytochrome-based | GAF-CaMP | 635/672 | 21, 69 * | 3.0; 9.0/ 3.0; >10.0 | 52 | 15 | ND | ND | ND | [44] | |
| NIR-GECO1 | 678/704 | ND | 6.0/4.7 | 85 | 215 | 1.93 | 2.5/25 | ND | [45] | ||
| Bioluminecent | LUCI-GECO1BRET | donor | 460 | ND | 8.15/6.07 | 406 | 285 * | ND | ND | ND | [46] |
| acceptor | 497/515 | ||||||||||
| Potassium indicators | |||||||||||
| GEPI | KIRIN1FRET | donor | 433/475 | 104 | 3.2 | 130 | 1660 | ND | ND | ND | [47] |
| acceptor | 515/530 | ND | ND | ||||||||
| KIRIN1-GRFRET | donor | 505/515 | 251 | 6.2 | 20 | 2560 | ND | ND | ND | ||
| acceptor | 559/600 | 128 | 5.3 | ||||||||
| GEPIIFRET | donor | 457/475 | ND | ND | 220 | 420–26,000 | 0.75 | ND | ND | [48] | |
| acceptor | 515/527 | ||||||||||
| GINKO1 | 502/514 | 22 | ND | 250 | 420 | ND | ND | ND | [47] | ||
| Sensor | Ex/Em (nm) | ΔF/FAP (%) | SNR | On Kinetics a (ms) | Off Kinetics a (ms) | Number of Simultaneously Imaged Neurons In Vivo | Ref |
|---|---|---|---|---|---|---|---|
| Ace2N-4aa-mNeon | 506/517 | −6.5 | ND | 0.37 (58)/5.5 | 0.5 (60)/5.9 | 2 in primary visual cortex L2/3 | [84] |
| ASAP3 | 485/510 | −10 b | 19 b | 0.94 (72)/7.2 | 3.8 (76)/16 | 1 in primary visual cortex L1 and L2/3 | [81] |
| Voltron525 | 525/549 | −6.5 b | 27 b | 0.64 (61)/4.1 | 0.78 (55)/3.9 | 46 in primary visual cortex L1 | [82] |
| VARNAM | 558/592 | −4.8 | 12 | 0.9/5.2 | 0.8/4.7 | Aggregated population signal | [80] |
| QuasAr2 | 590/715 | 15 | 8.5 | 0.3 (62)/3.2 | 0.3/(73) 4.0 | 14 in mouse nodose ganglia | [99] |
| paQuasAr3 | 590/715 | 23 b | 28 b | 0.9 (57)/15 | 0.93 (79)/15 | 6 in hippocampus | [60] |
| Archon1 | 590/715 | 53 b | 37 b | 0.61 (88)/8.1 c | 1.1 (88)/13 c | 18 in CA1 hippocampus | [65] |
| Sensor | Ligand | Ex/Em (nm) | ΔF/F (%) | Kd (µM) | On Kinetics (ms) | Off Kinetics (ms) | In Vivo Imaging | Ref |
|---|---|---|---|---|---|---|---|---|
| iGluSnFR | Glu a | 490/510 | 100 | 4.9 | 15 | 92 | Single dendritic spines | [127] |
| SF-iGluSnFR-A184S | Glu a | 490/510 | 69 | 0.6 | 85 | 450 | Single dendritic spines | [130] |
| SF-iGluSnFR-S72A | Glu a | 490/510 | 250 | 34 | 5 | 11 | Single dendritic spines | [130] |
| SF-Venus-iGluSnFR | Glu a | 515/528 | 66 | 2.0 | ND | ND | Single dendritic spines | [130] |
| iGluu | Glu a | 490/510 | 170 | 53 | 0.7 | 3 | Not tested | [129] |
| R-iGluSnFR1 | Glu a | 562/588 | −33 | 18 | ND | ND | Not tested | [131] |
| iGABASnFR | GABA | 485/510 | 10 b | ~9 b | ~25 c | ~60 c | Aggregated neuropil imaging | [132] |
| GlyFS | Gly | 434/477 515/528 (FRET) | 28.3 19.0 | 28 21 | ND | ND | Not tested | [133] |
| dLight1.1 | DA | 490/516 | 230 d | 0.33 d | ND | ND | Aggregated signal | [134] |
| dLight1.2 | DA | 490/516 | 340 d | 0.77 d | 9.5 e | 90 e | Aggregated signal | [134] |
| GRABDA1m | DA | 490/510 | 90 | 0.13 | 80 e | 3100 e | Aggregated signal | [135] |
| GRABDA1h | DA | 490/510 | 90 | 0.01 | 110 e | 17,150 e | Aggregated signal | [135] |
| GACh2.0 | ACh | 490/510 | 90 | 2 | 280 d | 760 d | Single neurons with 15 trails averaging | [136] |
| GRABNE1m | NE | 490/510 | 230 | 1.9 | 72 d | 680 d | Aggregated signal | [137] |
| GRABNE1h | NE | 490/510 | 150 | 0.093 | 36 d | 1890 d | Not tested | [137] |
| iTango2 | DA | 488/507 554/581 | 900 | ND | ~105-106 | NA | Single neurons | [138] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piatkevich, K.D.; Murdock, M.H.; Subach, F.V. Advances in Engineering and Application of Optogenetic Indicators for Neuroscience. Appl. Sci. 2019, 9, 562. https://doi.org/10.3390/app9030562
Piatkevich KD, Murdock MH, Subach FV. Advances in Engineering and Application of Optogenetic Indicators for Neuroscience. Applied Sciences. 2019; 9(3):562. https://doi.org/10.3390/app9030562
Chicago/Turabian StylePiatkevich, Kiryl D., Mitchell H. Murdock, and Fedor V. Subach. 2019. "Advances in Engineering and Application of Optogenetic Indicators for Neuroscience" Applied Sciences 9, no. 3: 562. https://doi.org/10.3390/app9030562
APA StylePiatkevich, K. D., Murdock, M. H., & Subach, F. V. (2019). Advances in Engineering and Application of Optogenetic Indicators for Neuroscience. Applied Sciences, 9(3), 562. https://doi.org/10.3390/app9030562