Solvent Effects in Highly Efficient Light-Induced Molecular Aggregation



Abstract
1. Introduction
2. Materials and Methods
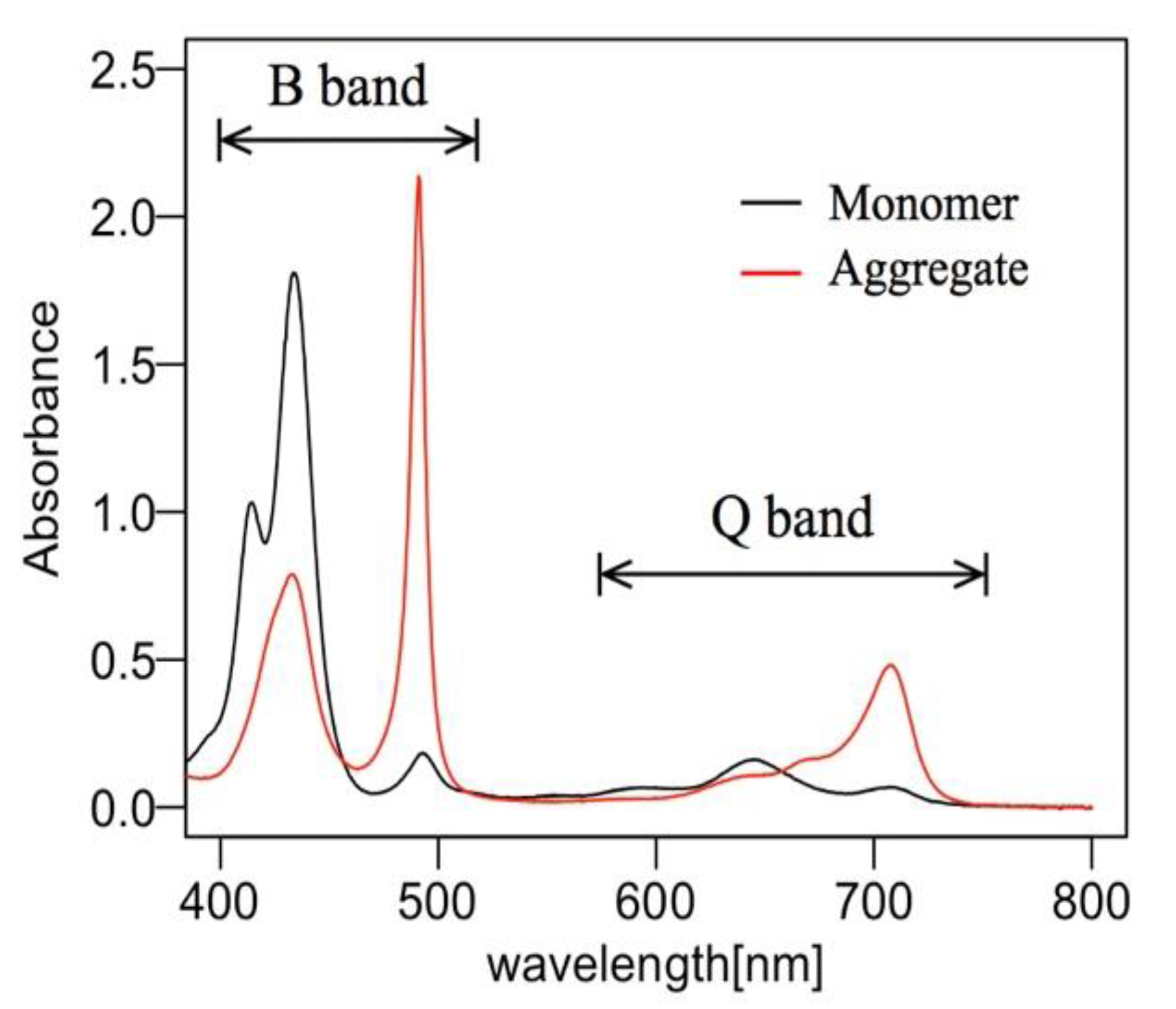
2.1. J-Aggregate
2.2. Sample Preparation
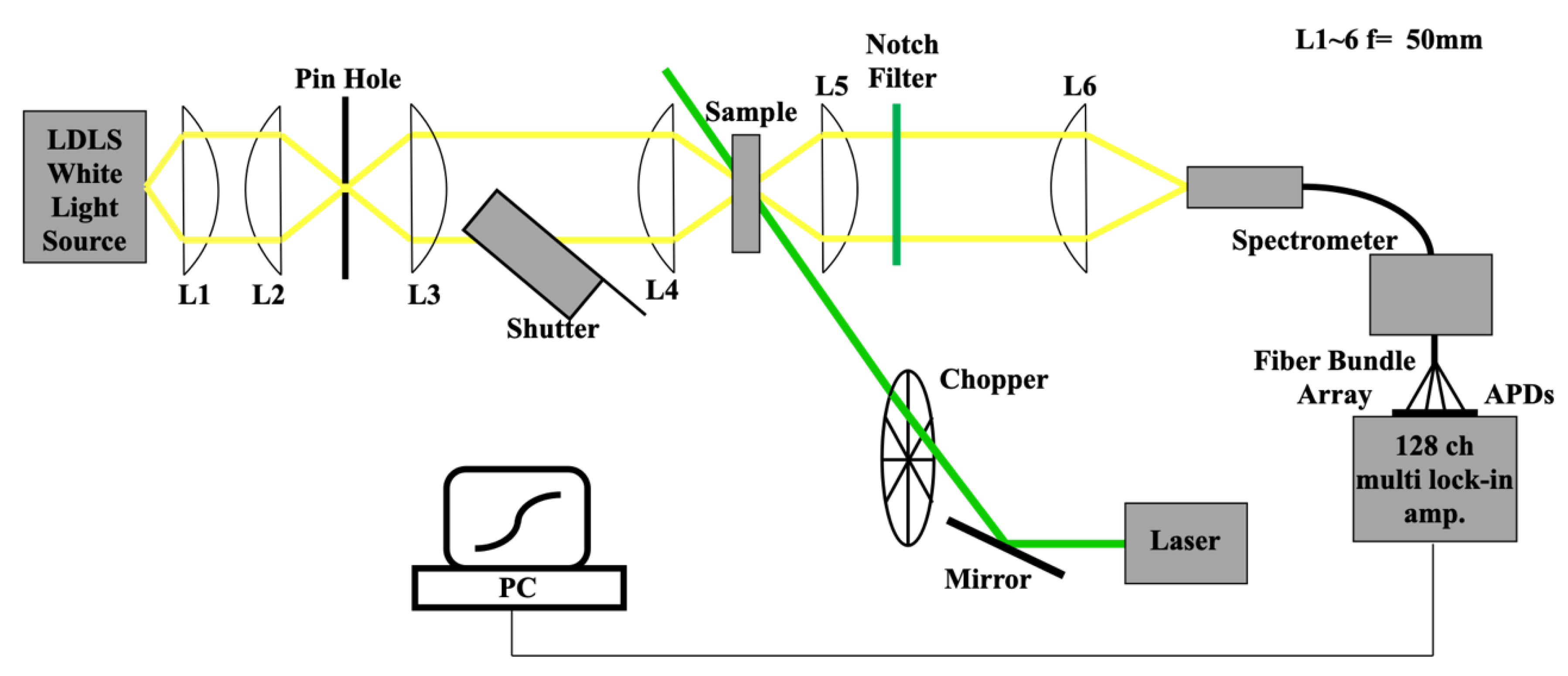
2.3. Experimental Set-up
3. Results and Discussion
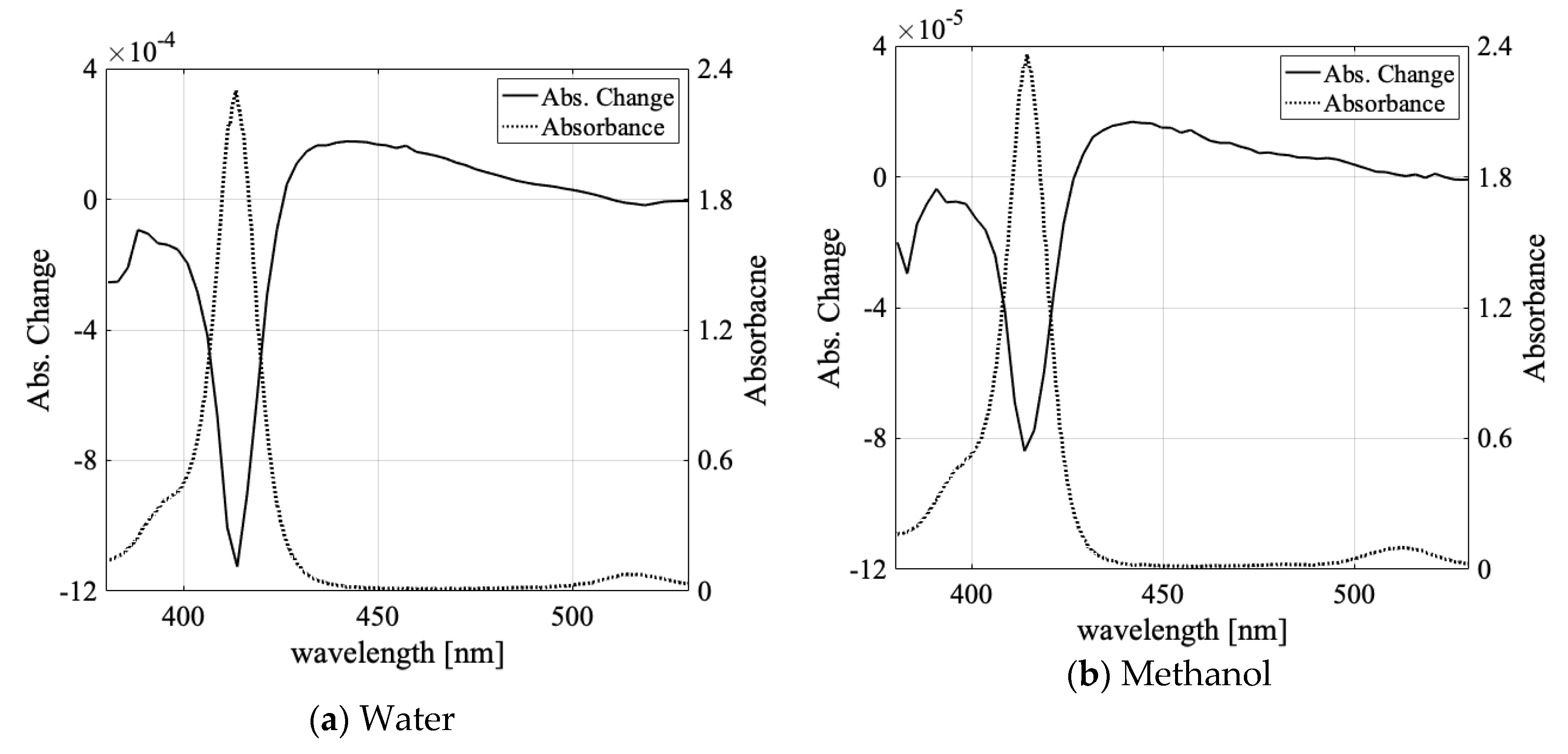
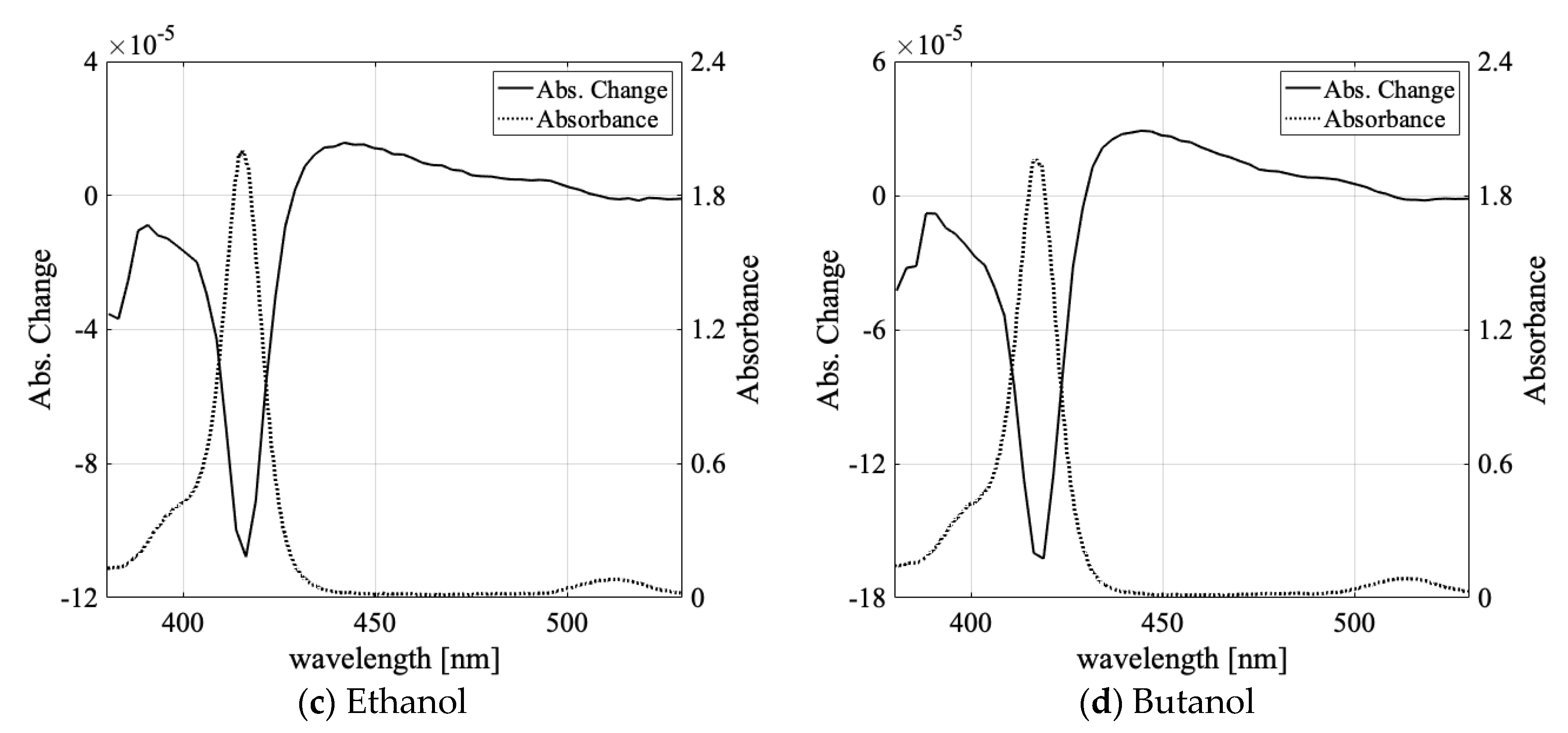
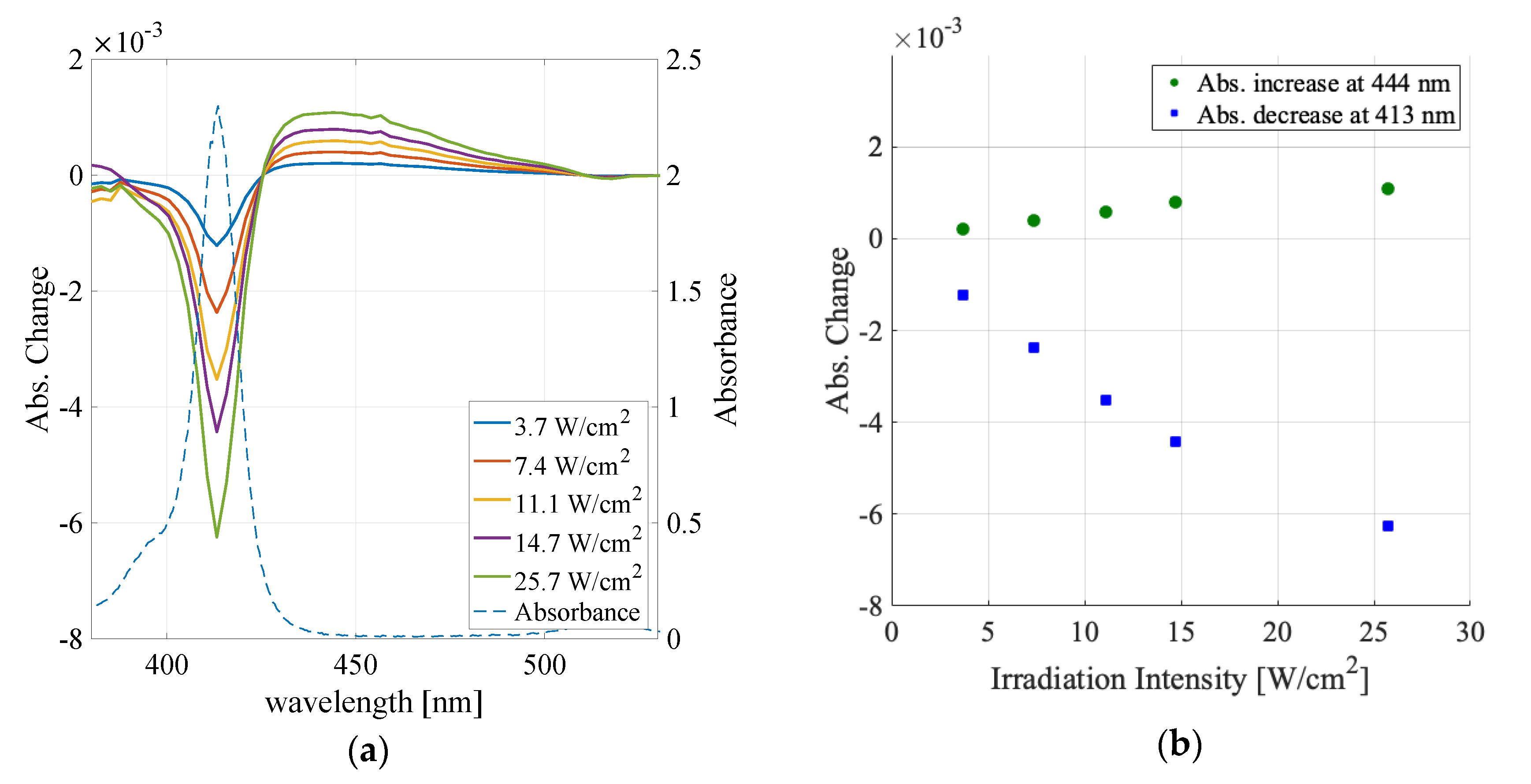
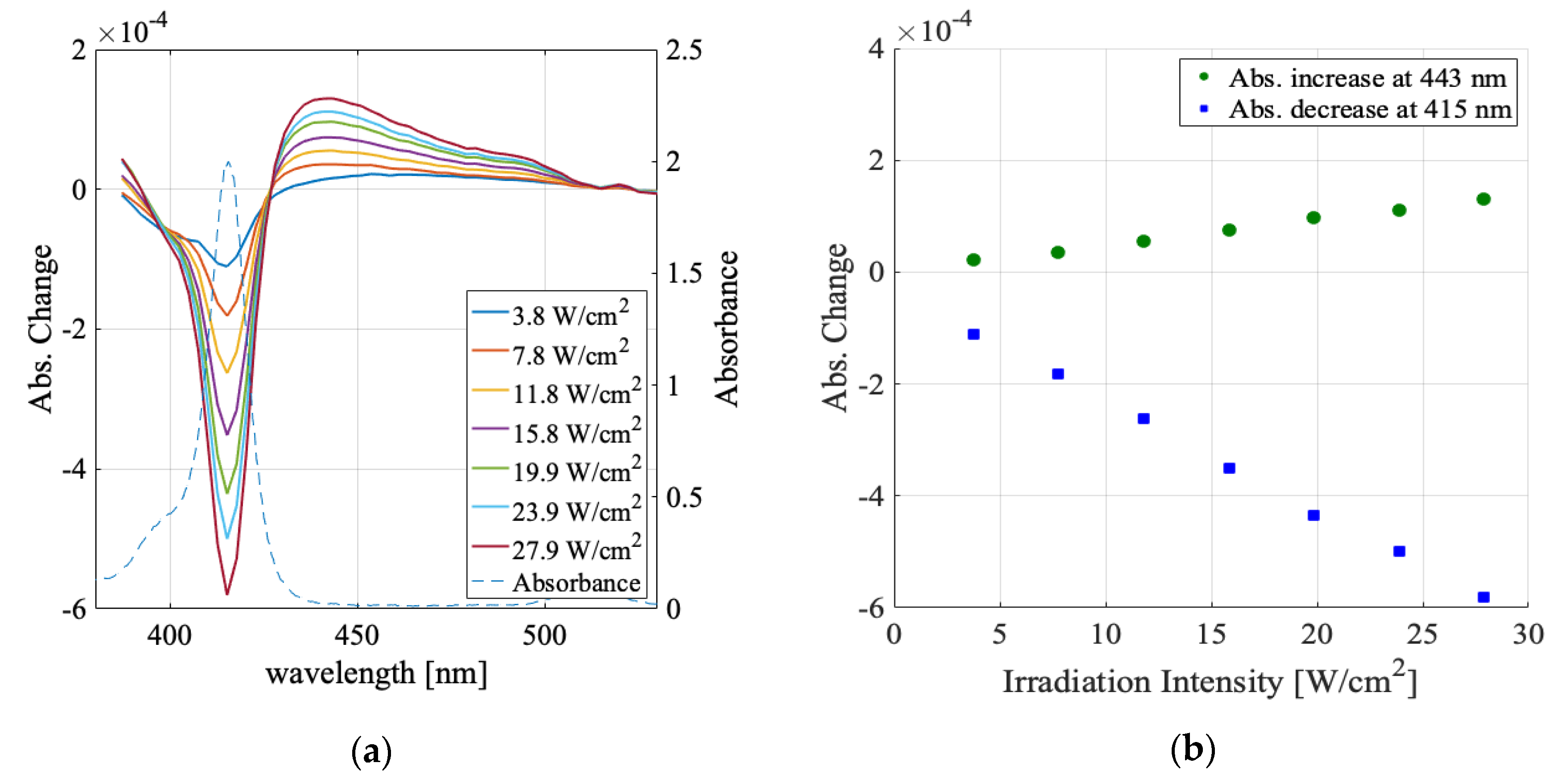
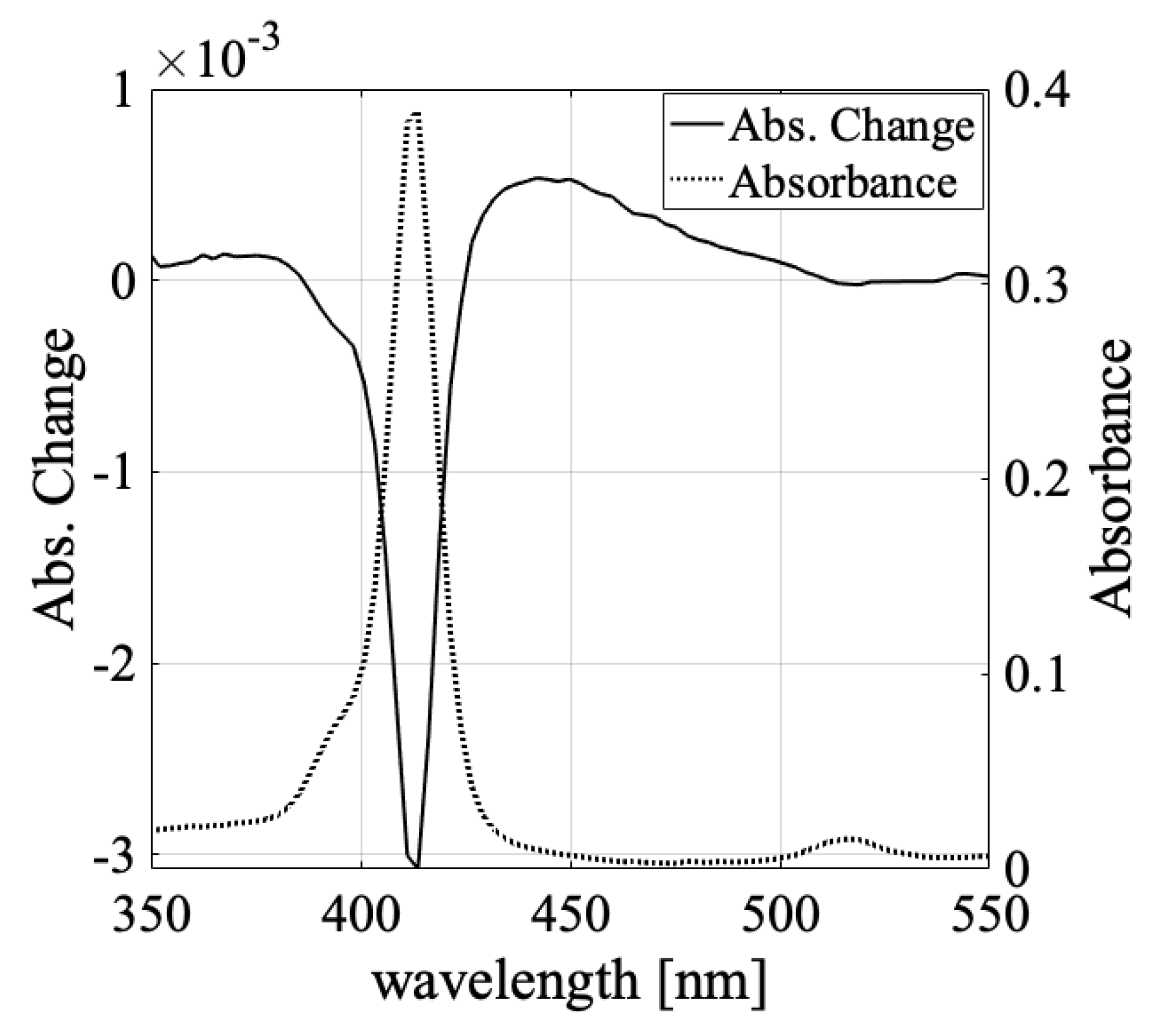
3.1. Solvent Dependence of Absorption Change Spectra
3.2. Irradiation Intensity Dependence
3.3. Absorbance Change in the Shorter Wavelength Region (Light-Induced Formation of H- and J-Aggregates)
3.4. Contribution by Keesom Interaction
3.5. Other Effects (Surface and Thermal Convection Effect)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ashkin, A. Acceleration and Trapping of Particles by Radiation Pressure. Phys. Rev. Lett. 1970, 24, 156–159. [Google Scholar] [CrossRef]
- Ashkin, A.; Dziedzic, J.M.; Smith, P.W. Continuous-wave self-focusing and self-trapping of light in artificial Kerr media. Opt. Lett. 1982, 7, 276. [Google Scholar] [CrossRef] [PubMed]
- Ashkin, A.; Dziedzic, J.M.; Yamane, T. Optical trapping and manipulation of single cells using infrared laser beams. Nature 1987, 330, 769–771. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.C.; Sheet, M.E. Optical tweezers in cell biology. Trends Cell Biol. 1992, 2, 116–118. [Google Scholar] [CrossRef]
- Svoboda, K.; Schmidt, C.F.; Schnapp, B.J.; Block, S.M. Direct observation of kinesin stepping by optical trapping interferometry. Nature 1993, 365, 721–727. [Google Scholar] [CrossRef]
- Hajizadeh, F.; Reihani, S.N. Optimized optical trapping of gold nanoparticles. Opt. Express 2010, 18, 551. [Google Scholar] [CrossRef]
- Pang, Y.; Gordon, R. Optical Trapping of 12 nm Dielectric Spheres Using Double-Nanoholes in a Gold Film. Nano Lett. 2011, 11, 3763–3767. [Google Scholar] [CrossRef]
- Xiao, F.; Ren, Y.; Shang, W.; Zhu, W.; Han, L.; Lu, H.; Mei, T.; Premaratne, M.; Zhao, J. Sub-10 nm particle trapping enabled by a plasmonic dark mode. Opt. Lett. 2018, 43, 3413. [Google Scholar] [CrossRef]
- Arita, Y.; Tkachenko, G.; McReynolds, N.; Marro, N.; Edwards, W.; Kay, E.R.; Dholakia, K. Invited Article: Optical trapping of ultrasmooth gold nanoparticles in liquid and air. APL Photonics 2018, 3, 070801. [Google Scholar] [CrossRef]
- Osborne, M.A.; Balasubramanian, S.; Furey, W.S.; Klenerman, D. Optically Biased Diffusion of Single Molecules Studied by Confocal Fluorescence Microscopy. J. Phys. Chem. B 1998, 102, 3160–3167. [Google Scholar] [CrossRef]
- Chirico, G.; Fumagalli, C.; Baldini, G. Trapped Brownian Motion in Single- and Two-Photon Excitation Fluorescence Correlation Experiments. J. Phys. Chem. B 2002, 106, 2508–2519. [Google Scholar] [CrossRef]
- Inaba, K.; Imaizumi, K.; Katayama, K.; Ichimiya, M.; Ashida, M.; Iida, T.; Ishihara, H.; Itoh, T. Optical manipulation of CuCl nanoparticles under an excitonic resonance condition in superfluid helium. Phys. Status Solidi B 2006, 243, 3829–3833. [Google Scholar] [CrossRef]
- Iida, T.; Ishihara, H. Theoretical Study of the Optical Manipulation of Semiconductor Nanoparticles under an Excitonic Resonance Condition. Phys. Rev. Lett. 2003, 90, 057403. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Ishihara, H. Two-color laser manipulation of single organic molecules based on nonlinear optical response. Eur. Phys. J. B 2013, 86, 98. [Google Scholar] [CrossRef]
- Juan, M.L.; Righini, M.; Quidant, R. Plasmon nano-optical tweezers. Nat. Photonics 2011, 5, 349–356. [Google Scholar] [CrossRef]
- Gao, D.; Ding, W.; Nieto-Vesperinas, M.; Ding, X.; Rahman, M.; Zhang, T.; Lim, C.; Qiu, C.-W. Optical manipulation from the microscale to the nanoscale: Fundamentals, advances and prospects. Light Sci. Appl. 2017, 6, e17039. [Google Scholar] [CrossRef]
- Yoo, D.; Gurunatha, K.L.; Choi, H.-K.; Mohr, D.A.; Ertsgaard, C.T.; Gordon, R.; Oh, S.-H. Low-Power Optical Trapping of Nanoparticles and Proteins with Resonant Coaxial Nanoaperture Using 10 nm Gap. Nano Lett. 2018, 18, 3637–3642. [Google Scholar] [CrossRef]
- Ecarnot, A.; Magno, G.; Yam, V.; Dagens, B. Ultra-efficient nanoparticle trapping by integrated plasmonic dimers. Opt. Lett. 2018, 43, 455. [Google Scholar] [CrossRef]
- Lin, L.; Wang, M.; Peng, X.; Lissek, E.N.; Mao, Z.; Scarabelli, L.; Adkins, E.; Coskun, S.; Unalan, H.E.; Korgel, B.A.; et al. Opto-thermoelectric nanotweezers. Nat. Photonics 2018, 12, 195–201. [Google Scholar] [CrossRef]
- Garetz, B.A.; Aber, J.E.; Goddard, N.L.; Young, R.G.; Myerson, A.S. Nonphotochemical, Polarization-Dependent, Laser-Induced Nucleation in Supersaturated Aqueous Urea Solutions. Phys. Rev. Lett. 1996, 77, 3475–3476. [Google Scholar] [CrossRef]
- Garetz, B.A.; Matic, J.; Myerson, A.S. Polarization Switching of Crystal Structure in the Nonphotochemical Light-Induced Nucleation of Supersaturated Aqueous Glycine Solutions. Phys. Rev. Lett. 2002, 89, 175501. [Google Scholar] [CrossRef] [PubMed]
- Adachi, H.; Takano, K.; Hosokawa, Y.; Inoue, T.; Mori, Y.; Matsumura, H.; Yoshimura, M.; Tsunaka, Y.; Morikawa, M.; Kanaya, S.; et al. Laser Irradiated Growth of Protein Crystal. Jpn. J. Appl. Phys. 2003, 42, L798–L800. [Google Scholar] [CrossRef]
- Adachi, H.; Murakami, S.; Niino, A.; Matsumura, H.; Takano, K.; Inoue, T.; Mori, Y.; Yamaguchi, A.; Sasaki, T. Membrane Protein Crystallization Using Laser Irradiation. Jpn. J. Appl. Phys. 2004, 43, L1376–L1378. [Google Scholar] [CrossRef]
- Okutsu, T.; Furuta, K.; Terao, M.; Hiratsuka, H.; Yamano, A.; Ferté, N.; Veesler, S. Light-Induced Nucleation of Metastable Hen Egg-White Lysozyme Solutions. Cryst. Growth Des. 2005, 5, 1393–1398. [Google Scholar] [CrossRef]
- Veesler, S.; Furuta, K.; Horiuchi, H.; Hiratsuka, H.; Ferté, N.; Okutsu, T. Crystals from Light: Photochemically Induced Nucleation of Hen Egg-White Lysozyme. Cryst. Growth Des. 2006, 6, 1631–1635. [Google Scholar] [CrossRef]
- Tanaka, Y.; Yoshikawa, H.; Masuhara, H. Two-Photon Fluorescence Spectroscopy of Individually Trapped Pseudoisocyanine J-Aggregates in Aqueous Solution. J. Phys. Chem. B 2006, 110, 17906–17911. [Google Scholar] [CrossRef]
- Tanaka, Y.; Yoshikawa, H.; Asahi, T.; Masuhara, H. Laser microfixation of highly ordered J aggregates on a glass substrate. Appl. Phys. Lett. 2007, 91, 041102. [Google Scholar] [CrossRef]
- Sugiyama, T.; Adachi, T.; Masuhara, H. Crystallization of Glycine by Photon Pressure of a Focused CW Laser Beam. Chem. Lett. 2007, 36, 1480–1481. [Google Scholar] [CrossRef]
- Sugiyama, T.; Adachi, T.; Masuhara, H. Crystal Growth of Glycine Controlled by a Focused CW Near-infrared Laser Beam. Chem. Lett. 2009, 38, 482–483. [Google Scholar] [CrossRef]
- Yuyama, K.; George, J.; Thomas, K.G.; Sugiyama, T.; Masuhara, H. Two-Dimensional Growth Rate Control of L -Phenylalanine Crystal by Laser Trapping in Unsaturated Aqueous Solution. Cryst. Growth Des. 2016, 16, 953–960. [Google Scholar] [CrossRef]
- Liu, T.-H.; Yuyama, K.; Hiramatsu, T.; Yamamoto, N.; Chatani, E.; Miyasaka, H.; Sugiyama, T.; Masuhara, H. Femtosecond-Laser-Enhanced Amyloid Fibril Formation of Insulin. Langmuir 2017, 33, 8311–8318. [Google Scholar] [CrossRef] [PubMed]
- Yuyama, K.; Chang, K.-D.; Tu, J.-R.; Masuhara, H.; Sugiyama, T. Rapid localized crystallization of lysozyme by laser trapping. Phys. Chem. Chem. Phys. 2018, 20, 6034–6039. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T. J-Aggregates; World Scientific: Singapore, 1996; ISBN 978-981-02-2737-1. [Google Scholar]
- Kobayashi, T. J-Aggregates: Volume 2; World Scientific: Singapore, 2012; ISBN 978-981-4365-74-1. [Google Scholar]
- Yoon, S.M.; Hwang, I.-C.; Kim, K.S.; Choi, H.C. Synthesis of Single-Crystal Tetra(4-pyridyl)porphyrin Rectangular Nanotubes in the Vapor Phase. Angew. Chem. Int. Ed. 2009, 48, 2506–2509. [Google Scholar] [CrossRef] [PubMed]
- Campione, M.; Capitani, G.C.; Raimondo, L.; Sassella, A. Porphyrin Nanowires with Epitaxially Locked Uniaxial Orientation. J. Phys. Chem. C 2015, 119, 18210–18215. [Google Scholar] [CrossRef]
- Park, Y.; Hong, M.; Koo, J.Y.; Lee, M.; Lee, J.; Moon, D.J.; Sohn, S.H.; Joo, T.; Lim, W.T.; Lim, H.; et al. Reverse Anti-solvent Crystallization Process for the Facile Synthesis of Zinc Tetra(4-pyridyl)porphyrin Single Crystalline Cubes. Sci. Rep. 2017, 7, 2582. [Google Scholar] [CrossRef]
- Ohno, O.; Kaizu, Y.; Kobayashi, H. J-aggregate formation of a water-soluble porphyrin in acidic aqueous media. J. Chem. Phys. 1993, 99, 4128–4139. [Google Scholar] [CrossRef]
- Aggarwal, L.P.F.; Borissevitch, I.E. On the dynamics of the TPPS4 aggregation in aqueous solutions. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2006, 63, 227–233. [Google Scholar] [CrossRef]
- Nakata, K.; Kobayashi, T.; Tokunaga, E. Electric field-controlled dissociation and association of porphyrin J-aggregates in aqueous solution. Phys. Chem. Chem. Phys. 2011, 13, 17756. [Google Scholar] [CrossRef]
- Kato, N.; Saito, K.; Serata, T.; Aida, H.; Uesu, Y. Morphology and thermochromic phase transition of merocyanine J-aggregate monolayers at the air-water and solid-water interfaces. J. Chem. Phys. 2001, 115, 1473–1484. [Google Scholar] [CrossRef]
- Udal’tsov, A.V.; Kazarin, L.A.; Sweshnikov, A.A. Self-assembly of large-scale aggregates of porphyrin from its dimers and their absorption and luminescence properties. J. Mol. Struct. 2001, 562, 227–239. [Google Scholar] [CrossRef]
- Bielejewski, M.; Łapiński, A.; Luboradzki, R.; Tritt-Goc, J. Solvent Effect on 1,2-O-(1-Ethylpropylidene)-α-d-glucofuranose Organogel Properties. Langmuir 2009, 25, 8274–8279. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Stepanenko, V.; Kaiser, T.E.; Würthner, F.; Scheblykin, I.G. Reorganization of perylene bisimide J-aggregates: From delocalized collective to localized individual excitations. Nanoscale 2012, 4, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Villari, V.; Mineo, P.; Scamporrino, E.; Micali, N. Role of the hydrogen-bond in porphyrin J-aggregates. RSC Adv. 2012, 2, 12989. [Google Scholar] [CrossRef]
- Kobayashi, T.; Misawa, K. Hierarchical structure of one-dimensional J-aggregates. J. Lumin. 1997, 72–74, 38–40. [Google Scholar] [CrossRef]
- Wanless, E.J.; Davey, T.W.; Ducker, W.A. Surface Aggregate Phase Transition. Langmuir 1997, 13, 4223–4228. [Google Scholar] [CrossRef]
- Shirakawa, M.; Nakata, K.; Suzuki, M.; Kobayashi, T.; Tokunaga, E. Nonlinear Absorption Spectroscopy of Porphyrin J-aggregates in Aqueous Solution: Evidence for Control of Degree of Association by Light-Induced Force. J. Phys. Soc. Jpn. 2017, 86, 044703. [Google Scholar] [CrossRef]
- Gouterman, M. Spectra of porphyrins. J. Mol. Spectrosc. 1961, 6, 138–163. [Google Scholar] [CrossRef]
- Gouterman, M.; Wagnière, G.H.; Snyder, L.C. Spectra of porphyrins: Part II. Four orbital model. J. Mol. Spectrosc. 1963, 11, 108–127. [Google Scholar] [CrossRef]
- Chen, D.-M.; He, T.; Cong, D.-F.; Zhang, Y.-H.; Liu, F.-C. Resonance Raman Spectra and Excited-State Structure of Aggregated Tetrakis(4-sulfonatophenyl)porphyrin Diacid. J. Phys. Chem. A 2001, 105, 3981–3988. [Google Scholar] [CrossRef]
- El-Hachemi, Z.; Escudero, C.; Acosta-Reyes, F.; Casas, M.T.; Altoe, V.; Aloni, S.; Oncins, G.; Sorrenti, A.; Crusats, J.; Campos, J.L.; et al. Structure vs. properties—Chirality, optics and shapes—In amphiphilic porphyrin J-aggregates. J. Mater. Chem. C 2013, 1, 3337. [Google Scholar] [CrossRef]
- Vlaming, S.M.; Augulis, R.; Stuart, M.C.A.; Knoester, J.; van Loosdrecht, P.H.M. Exciton Spectra and the Microscopic Structure of Self-Assembled Porphyrin Nanotubes. J. Phys. Chem. B 2009, 113, 2273–2283. [Google Scholar] [CrossRef] [PubMed]
- Iwai, S.; Tanaka, M.; Mitsunaga, M.; Kobayashi, T.; Tokunaga, E. Excited-state absorption spectra for optically forbidden f-f transitions in an Eu3+: Y2SiO5 crystal and Eu3+ aqueous solution. J. Opt. Soc. Am. B 2008, 25, 1046. [Google Scholar] [CrossRef]
- Ishino, H.; Iwai, S.; Iwamoto, S.; Okumura, T.; Kobayashi, T.; Tokunaga, E. Nonlinear absorption microspectroscopy of single perylene nanocrystals with a multichannel double lock-in amplifier. Opt. Rev. 2010, 17, 337–340. [Google Scholar] [CrossRef]
- Friend, J.N.; Hargreaves, W.D. XC. Viscosity and the hydrogen bond: Hydroxyl and ortho effects. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1945, 36, 731–756. [Google Scholar] [CrossRef]
- Farjtabar, A.; Gharib, F. Solvent Effect on Protonation Constants of 5,10,15,20-Tetrakis(4-sulfonatophenyl)porphyrin in Different Aqueous Solutions of Methanol and Ethanol. J. Solut. Chem. 2010, 39, 231–244. [Google Scholar] [CrossRef]
- Suzuki, M.; Nakata, K.; Kuroda, R.; Kobayashi, T.; Tokunaga, E. Electrooptic Kerr effect of porphyrin H-aggregates in polymer films: Polymer specific spectral blue shift. Chem. Phys. 2016, 469–470, 88–96. [Google Scholar] [CrossRef]
- Keesom, W.H. The second viral coefficient for rigid spherical molecules, whose mutual attraction is equivalent to that of a quadruplet placed at their centre. KNAW Proc. 1915, 18, 636–646. [Google Scholar]
- Maitland, G.C.; Rigby, M.; Smith, E.B.; Wakeham, W.A. Intermolecular Forces—Their Origin and Determination. Berichte der Bunsengesellschaft für Physikalische Chemie 1983, 87, 291–292. [Google Scholar] [CrossRef]
- Mortazavi-Manesh, S.; Ghotbi, C.; Taghikhani, V. A new model for predicting activity coefficients in aqueous solutions of amino acids and peptides. J. Chem. Thermodyn. 2003, 35, 101–112. [Google Scholar] [CrossRef]
- Nishikawa, Y.; Morishige, K.; Shigematsu, T.; Nishikawa, Y. Determination of acid-base dissociation constant of porphyrins and their spectroscopic behavior. Bunseki Kagaku 1988, 37, 284–291. [Google Scholar] [CrossRef][Green Version]
- Roxworthy, B.J.; Bhuiya, A.M.; Vanka, S.P.; Toussaint, K.C. Understanding and controlling plasmon-induced convection. Nat. Commun. 2014, 5, 3173. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, D.J. Introduction to Electrodynamics, 4th ed.; Always Learning; Pearson: Boston, MA, USA, 2013; ISBN 978-0-321-85656-2. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Water | Methanol | Ethanol | Butanol | |
|---|---|---|---|---|
| Concentration (µmol/L) | 59 | 62 | 52 | 54 |
| The amount of NaOH (µL) | 200 | 200 | 400 | 400 |
| Water | Methanol | Ethanol | Butanol | |
|---|---|---|---|---|
| Absorbance Increase | ||||
| Absorbance Decrease |
| Water | Methanol | Ethanol | Butanol | |
|---|---|---|---|---|
| Hydroxyl group [mol/cm3] | 0.111 | 0.025 | 0.017 | 0.011 |
| A | B | |
|---|---|---|
| Absorbance increase at 444 nm | 0.0018 | 0.037 |
| Absorbance decrease at 413 nm | −0.0101 | 0.038 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shirakawa, M.; Kobayashi, T.; Tokunaga, E. Solvent Effects in Highly Efficient Light-Induced Molecular Aggregation. Appl. Sci. 2019, 9, 5381. https://doi.org/10.3390/app9245381
Shirakawa M, Kobayashi T, Tokunaga E. Solvent Effects in Highly Efficient Light-Induced Molecular Aggregation. Applied Sciences. 2019; 9(24):5381. https://doi.org/10.3390/app9245381
Chicago/Turabian StyleShirakawa, Masayuki, Takayoshi Kobayashi, and Eiji Tokunaga. 2019. "Solvent Effects in Highly Efficient Light-Induced Molecular Aggregation" Applied Sciences 9, no. 24: 5381. https://doi.org/10.3390/app9245381
APA StyleShirakawa, M., Kobayashi, T., & Tokunaga, E. (2019). Solvent Effects in Highly Efficient Light-Induced Molecular Aggregation. Applied Sciences, 9(24), 5381. https://doi.org/10.3390/app9245381